

Abstract
Synapse formation requires the organization of presynaptic active zones, the synaptic vesicle release sites, in precise apposition to postsynaptic neurotransmitter receptor clusters; however, the molecular mechanisms responsible for these processes remain unclear. Here, we show that P/Q-type and N-type voltage-dependent calcium channels (VDCCs) play essential roles as scaffolding proteins in the organization of presynaptic active zones. The neuromuscular junction of double knock-out mice for P/Q- and N-type VDCCs displayed a normal size but had significantly reduced numbers of active zones and docked vesicles and featured an attenuation of the active-zone proteins Bassoon, Piccolo, and CAST/Erc2. Consistent with this phenotype, direct interactions of the VDCC β1b or β4 subunits and the active zone-specific proteins Bassoon or CAST/Erc2 were confirmed by immunoprecipitation. A decrease in the number of active zones caused by a loss of presynaptic VDCCs resembled the pathological conditions observed in the autoimmune neuromuscular disorder Lambert–Eaton myasthenic syndrome. At the synaptic cleft of double knock-out mice, we also observed a decrease of the synaptic organizer laminin β2 protein, an extracellular ligand of P/Q- and N-type VDCCs. However, the transcription level of laminin β2 did not decrease in double knock-out mice, suggesting that the synaptic accumulation of laminin β2 protein required its interaction with presynaptic VDCCs. These results suggest that presynaptic VDCCs link the target-derived synapse organizer laminin β2 to active-zone proteins and function as scaffolding proteins to anchor active-zone proteins to the presynaptic membrane.
Introduction
Neurotransmission requires the sensing of action potentials by presynaptic voltage-dependent calcium channels (VDCCs) and the resultant release of neurotransmitters at active zones. Active zones contain proteinaceous structures that exhibit electron-dense projections or filamentous structures extending from presynaptic membranes on electron micrographs, and they align precisely with postsynaptic specializations (Couteaux and Pécot-Dechavassine, 1970; Landis et al., 1988; Siksou et al., 2007). The cytomatrix of active zone accumulates specifically at active zones and includes Bassoon, Bruchpilot, CAST/Erc2, Munc13, Piccolo, Rim1, and SYD-2 (Cases-Langhoff et al., 1996; Wang et al., 1997; Betz et al., 1998; tom Dieck et al., 1998; Ohtsuka et al., 2002; Yeh et al., 2005; Kittel et al., 2006). The deletion of these molecules causes a loss or enlargement of active zones (Augustin et al., 1999; Zhen and Jin, 1999; Schoch et al., 2002; Dick et al., 2003; Kittel et al., 2006; Fouquet et al., 2009; Kaeser et al., 2009; Mukherjee et al., 2010). However, the molecular mechanism responsible for organizing active zones in precise alignment with postsynaptic specializations is not fully understood.
Knock-out mice analyses of a few synaptic organizers have revealed no defects in active zones (Neuroligin, Neurexin, LRRTM1/2) (Missler et al., 2003; Varoqueaux et al., 2006; Linhoff et al., 2009). Other mutants caused widespread defects in presynaptic/postsynaptic differentiation; thus, active-zone defects could be either primary or secondary phenotypes (FGF22, TSP1/2, Drosophila neurexin or wingless) (Umemori et al., 2004; Christopherson et al., 2005; Eroglu et al., 2009).
However, knock-out mice for laminin β2 (a synaptic organizer) or VDCCs showed phenotypes that were preferentially related to active zones. At neuromuscular junctions (NMJs), we observed a reduced number of active zones in knock-out mice for P/Q-type VDCC or laminin β2 (Nishimune et al., 2004). Importantly, fewer active zones are present at human NMJs in Lambert–Eaton myasthenic syndrome (LEMS), which is characterized by the production of autoantibodies against the P/Q-type VDCC (Fukuoka et al., 1987). At photoreceptor synapses, deletions of VDCC (α1F), laminin β2, or Bassoon caused a dissociation of the ribbons from presynaptic membranes (Libby et al., 1999; Dick et al., 2003; Mansergh et al., 2005). These active-zone phenotypes suggest that laminin β2 and VDCCs play essential roles in organizing active zones.
We have previously shown that the direct interaction of muscle-derived laminin β2 and presynaptic P/Q-type VDCC is required for the organization of active zones at NMJs (Nishimune et al., 2004). Although P/Q-type VDCC knock-out mice formed significantly fewer active zones at NMJs, some active zones still formed. In the knock-out mice, a compensatory upregulation of N-type VDCCs at NMJs has been reported (Urbano et al., 2003). Previously, we have shown that N-type VDCCs bind directly to laminin β2 in vitro, which suggests a compensation for the organization of the active zone by N-type VDCCs in P/Q-type VDCC knock-out mice. These findings prompted us to investigate double knock-out (DKO) mice that lack both P/Q- and N-type VDCCs to determine the role of presynaptic VDCCs in organizing active zones at NMJs without compensation effects.
Materials and Methods
Animals.
The generation of the following genetically engineered mouse strains has been previously described: Cacna1a −/− (Jun et al., 1999) and Cacna1b −/− mice (Ino et al., 2001). In these studies, the absence of mRNAs and proteins for Cacna1a and Cacna1b genes was confirmed by Northern blot analysis and electrophysiological study by whole-cell recordings. The Cacna1a −/− Cacna1b −/− embryos were obtained by mating double heterozygote mice (Cacna1a +/− Cacna1b +/−). For control animals, littermate wild-type or single heterozygote (Cacna1a +/− or Cacna1b +/−) mice were used. All the mouse lines used in this study were maintained on a C57BL/6 background, and both sexes were used for the analyses. All animal studies have been approved by the authors' institutional review board.
Antibodies.
The antibodies were as follows: Bassoon (SAP7F407; Enzo Life Sciences), pan-CAST (anti-ELKS; E4531; Sigma-Aldrich; we confirmed that this antibody also recognizes mouse CAST/Erc2 by staining cDNA transfected HEK293 cells), choline acetyltransferase (AB143; Millipore Bioscience Research Reagents), Flag (M2; Sigma-Aldrich), hemagglutinin (HA) (3F10; Roche), laminin β2 (a gift from R. Timple and T. Sasaki, Max-Planck-Institut für Biochemie, Martinsried, Germany), Myc (9E10; Sigma-Aldrich), neurofilament (SMI312, Covance; NF200, Sigma-Aldrich), Piccolo (142 002; Synaptic Systems), SV2 (Developmental Studies Hybridoma Bank), synapsin I (106 001; Synaptic Systems), synaptophysin (18-0130; Zymed), VDCC β1 subunit (N7/18; Neuromab), VDCC β4 subunit (N10/7; Neuromab), Alexa Fluor 488- and 568-conjugated secondary antibodies, and Alexa Fluor 594- or 647-conjugated α-bungarotoxin (Invitrogen).
Immunohistochemical analysis.
The methods used for the immunohistochemical analyses have been described previously (Nishimune et al., 2004, 2008). In brief, sternocleidomastoid and tibialis muscles were fixed in 4% paraformaldehyde/PBS, cryoprotected in 20% sucrose/PBS, frozen in optimal cutting temperature compound (Sakura), and cut using a cryostat. Longitudinal sections were cut at 30 μm and blocked in 2% BSA/2% normal goat serum/0.1% Triton/PBS. Sections were stained sequentially with primary antibodies, wash solution (PBS), and a mixture of Alexa Fluor (488 or 568)-conjugated secondary antibodies and Alexa Fluor (594 or 647)-conjugated α-bungarotoxin. After washing, the sections were mounted in Vectashield (Vector Laboratories).
For whole-mount immunohistochemistry staining, diaphragms were fixed in 2% paraformaldehyde/PBS, washed in PBS, permeabilized in 0.5% Triton X-100/0.1 m glycine/PBS, and blocked in 2% BSA/2% normal goat serum/0.5% Triton/PBS. Tissues were incubated sequentially with primary antibodies (anti-neurofilament and anti-SV2), wash solutions (PBS), and a secondary antibody (Alexa Fluor 488-conjugated anti-mouse IgG1) together with Alexa Fluor 594-conjugated α-bungarotoxin. After washing, the diaphragms were mounted with Vectashield (Vector Laboratories).
For spinal cord immunohistochemistry staining, mouse embryos were frozen in powdered dry ice immediately after umbilical cord amputation. The embryos were cryosectioned transversally at 20 μm, allowed to dry, and then fixed in 4% paraformaldehyde/PBS for 20 min at room temperature. Serial sections were split for analyses by immunohistochemistry and in situ hybridization. For immunohistochemistry, sections were blocked in 2% BSA/2% normal goat serum/0.1% Triton/PBS. Sections were stained sequentially with primary antibodies, wash solution (PBS), and a mixture of Alexa Fluor (488 or 568)-conjugated secondary antibodies and 4′,6-diamidino-2-phenylindole (DAPI). After washing, the sections were mounted in Vectashield (Vector Laboratories).
No staining was observed when primary antibodies, Alexa Fluor-conjugated α-bungarotoxin or DAPI were omitted. Epifluorescent images were obtained using a Nikon Eclipse 80i microscope [objective lens: 4×, 20×, numerical aperture (NA) 0.75; 40×, NA 1.30]. Sequentially scanned confocal stacks of 0.5 μm/step were obtained using a Nikon C1Si confocal microscope (objective lens: Plan Apo, 100×, NA 1.49). Z-stack projection and image analysis were performed using MetaMorph software, version 7.0 (Molecular Devices). The levels of the images were adjusted, and the images were combined to generate merged color images and cropped in Adobe Photoshop.
Image analysis.
Light microscopic analysis of presynaptic active zones has been described previously (Nishimune et al., 2004). In brief, the Bassoon-positive area (Bsn area), signal intensity (total Bsn signal), and area of α-bungarotoxin-labeled acetylcholine receptors (BTX area) were measured using MetaMorph from maximal projected confocal images (1024 × 1024 file size). We measured the anti-Bassoon antibody-staining signal overlying the α-bungarotoxin-labeled endplates by applying the same threshold value to all images and using the integrated morphometry analysis function in MetaMorph.
For the quantification of synapse size and acetylcholine receptor density, the area and signal intensity of α-bungarotoxin-labeled acetylcholine receptor clusters were measured using MetaMorph from maximal projected confocal images. Each molecule of the Alexa dye-conjugated α-bungarotoxin is labeled with approximately one fluorophore, and it binds irreversibly to acetylcholine receptors (Haugland, 2005). Thus, fluorescently labeled α-bungarotoxin allows the quantification of synapse size and density of acetylcholine receptors within a synapse (Ravdin and Axelrod, 1977; Akaaboune et al., 1999).
For the quantification of SV2 at the NMJ using MetaMorph, we restricted the measurement to presynaptic terminals overlying α-bungarotoxin-labeled acetylcholine receptor clusters. We measured the integrated signal intensity of anti-SV2 antibody staining in maximal projected confocal images by applying the same threshold value to all images. The average signal intensity was determined by dividing the total signal intensity of SV2 at a NMJ by the area of acetylcholine receptor clusters. Synaptophysin and synapsin I signals were measured by the same method.
Colocalization analysis was performed on maximal projected confocal images using ImageJ (W. Rasband, National Institutes of Health, Bethesda, MD; http://rsb.info.nih.gov/ij/) and JACoP plug-in (Bolte and Cordelières, 2006). The degree of overlap between signals of active-zone proteins was analyzed by Pearson's coefficient with Costes' automatic thresholding and randomization (Bolte and Cordelières, 2006). Pearson's coefficient provides a value (r) ranging from −1 to 1, with 1 standing for complete colocalization, 0 for absence of correlation, and −1 for exclusion between two channels. We tested this analysis method using images of NMJs stained with an anti-neurofilament antibody and α-bungarotoxin that exhibit minimal overlap. The r value was −0.06 ± 0.02 (n = 6 NMJs), which showed the absence of correlation and the validity of this analysis method.
For the quantification of the nerve branching pattern, diaphragms stained by whole-mount immunohistochemistry (neurofilament plus SV2 and α-bungarotoxin) were used. The phrenic nerve splits into three nerve trunks as it reaches the hemi-diaphragm (Misgeld et al., 2002). The two main nerve trunks turn in opposite directions running perpendicularly to myotubes in the hemi-diaphragm, and the third trunk innervates the crus. The two main nerve trunks were defined as primary branches. Small groups of axons that leave these primary branches were defined as secondary branches, and additional bifurcations were defined as tertiary branches and then quaternary branches. Epifluorescent images of hemi-diaphragms were magnified in ImageJ, and branches were counted by labeling them using the Cell Counter plug-in. Entire branches of the phrenic nerve were quantified in two separate quadrants of diaphragms.
For the quantification of the cell density in the lumbar spinal cord, DAPI-stained nuclei in spinal cord transverse sections were counted in the dorsal horn (laminae I–V), intermediate zone (lamina VII), and ventral horn (laminae VII–IX). Motor neuron density was quantified by counting anti-choline acetyltransferase-stained cell bodies in the ventral horn using epifluorescent images of spinal cord transverse sections.
The signal intensity of cytosolic Bassoon in the motor neurons was measured within the circumference of the motor neuron cell bodies in the lumbar spinal cord transverse sections, as defined by the anti-choline acetyltransferase staining patterns, avoiding the DAPI-stained nuclei. The average signal intensity within each motor neuron was measured and normalized to the mean value of the motor neuron signal intensity in the control sections.
The signal intensity of synaptic laminin β2 was measured in the endplates defined by the α-bungarotoxin staining pattern in the longitudinal muscle sections. The average signal intensity for each endplate was measured and normalized to the mean value of the synaptic signal intensity in the control sections.
Ultrastructural analysis.
The methods used for electron microscopy analysis have been described previously (Fernández-Chacón et al., 2004; Nishimune et al., 2004). In brief, sternocleidomastoid muscles were fixed in 5% glutaraldehyde/4% paraformaldehyde in PBS, washed, refixed in 1% OsO4, dehydrated, and embedded in resin. Ultrathin sections were stained with lead citrate and uranyl acetate and systematically scanned using a transmission electron microscope. All NMJ profiles encountered were micrographed and counted. Three animals per genotype were analyzed, with 23–32 NMJs per animal measured. Two DKO and control embryo pairs were littermates, and one pair was obtained from two different litters. An active zone was defined as a protrusion of electron-dense material from the presynaptic membrane, where synaptic vesicles aggregated. The electron-dense material extending from the presynaptic membrane may reflect the precipitation of active-zone proteins caused by the chemical fixation method, because a meshwork of fine filaments is observed at the active zone of samples prepared by a rapid freezing and freeze substitution fixation method (Landis et al., 1988). Docked vesicles were defined as synaptic vesicles that were morphologically attached to the presynaptic membrane.
In situ hybridization.
The methods used for in situ hybridization have been described previously (Nishimune et al., 2000, 2005). In brief, mouse embryos were frozen in powdered dry ice immediately after umbilical cord amputation. The embryos were cryosectioned at 20 μm, allowed to dry, and then fixed in 4% paraformaldehyde/PBS for 20 min at room temperature. Serial sections were split for analyses by immunohistochemistry and in situ hybridization. Hybridization with digoxygenin (DIG)-labeled RNA probes, washing, and visualization with alkaline phosphatase-labeled anti-DIG antibody were performed under standard conditions. DIG-labeled RNA antisense probes were the rat Chat 2.2 kbp cDNA fragment (a gift from T. Jessell, Columbia University, New York, NY) and the mouse Bassoon 1.6 kbp cDNA fragment from IMAGE clone 6415760 (ATCC). No signal was observed when the DIG-labeled probes were omitted.
Electromyography.
Gestation day 18.5 embryos were dissected from the uterus, and a tail biopsy was performed for genotyping material. Supramaximal stimulation (typically 36 mA) was delivered to the phrenic nerve at the cervical plexus level, and electromyograms were recorded from the diaphragms by needle electrodes using TEAC Synergy N2, a two-channel EMG/NCS system (VIASYS Healthcare). A single reliable compound muscle action potential was recorded repeatedly ∼200 ms after the stimulus. Thus, we recorded compound muscle action potentials between 150 and 250 ms after stimulation by measuring the peak amplitude from the baseline level. Stimulation was delivered in a blinded fashion without genotype information, and recordings were categorized later by genotype according to the tail biopsy.
Quantitative reverse transcription-PCR.
Six embryos per genotype were analyzed for the expression level of laminin β2 mRNA (lamb2). The total RNA of embryonic day 18.5 (E18.5) hindlimbs was extracted using Trizol reagent (Invitrogen) with DNase treatment (QIAGEN; RNase-free DNase set). First-strand cDNA was synthesized from 5 μg of total RNA using Superscript III (Invitrogen). SYBR Green real-time PCRs were run in triplicate in 384-well reaction plates using the 7900 HT Sequence Detection System (Applied Biosystems). Blank controls were included in parallel for each master mix. A reaction volume of 10 μl was used for the PCRs with SYBR Green PCR master mix (Applied Biosystems 4309155), primers at the concentrations indicated below, and 1 μl of 1:10 diluted cDNA. The cycling conditions were as follows: an initial denaturation at 95°C for 10 min, followed by 40 cycles of denaturation at 95°C for 15 s, and annealing/extension at 60°C for 1 min. These cycles were followed by a melting-curve analysis ranging from 60 to 95°C to determine whether primer dimers were present. No primer dimers were detected under the conditions used. The lamb2 mRNA level was quantified relative to three different housekeeping genes using the comparative CT method. To ensure that the ΔΔCT calculation was valid for the used primers, the amplification efficiency was examined. The absolute value of the slope of the log input amount versus ΔCT was confirmed to be <0.1 for lamb2 compared with the three reference genes, which demonstrated the validity of the ΔΔCT calculation method for these primer sets (ABI, User Bulletin 2, Relative Quantification of Gene Expression). The mouse primers for the reference genes were obtained from the following publications and used at the indicated concentrations: glyceraldehyde-3-phosphate dehydrogenase (gapdh) (300 nm), TATA box binding protein (tbp) (900 nm) (Wang et al., 2010), and 18S ribosomal RNA (S18) (900 nm) (Pernot et al., 2010). The Primer Express3 software was used to design forward (716–736 bp) and reverse (793–815 bp) primers for mouse lamb2 (NM_008483), spanning exons 7 and 8; these primers were used at a concentration of 900 nm. The amplicon size was confirmed by gel electrophoresis.
Coimmunoprecipitation.
Coimmunoprecipitations using HEK293 cell lysates were performed as described previously (Takao-Rikitsu et al., 2004). In brief, expression plasmid vectors encoding the VDCC subunits α1A (P/Q-type; rat Cacna1a; NM_012918), β1b (rat Cacnb1; NM_145121), and β4 (mouse Cacnb4; NM_146123; Origene MC 201619), and EGFP (enhanced green fluorescent protein) (Clontech) were transfected into HEK293 cells. A cDNA encoding the C-terminal cytosolic domain of the rat α1A subunit (449 aa) was subcloned from rat α1A (5290–6636 bp) and expressed as a Myc-tagged fusion protein. A cDNA encoding the C-terminal domain of mouse Bassoon (Bsn) (1053 aa) was subcloned from a cDNA clone mKIAA0434 (353–3514 bp; Kazusa DNA Research Institute) and expressed as an HA-tagged fusion protein. A cDNA encoding full-length mouse CAST/Erc2 (Erc2) was subcloned from an IMAGE clone 6331997 (314–3322 bp; Open Biosystems) and expressed as a Flag-tagged fusion protein. The proteins were expressed independently in HEK293T cells and were extracted with Triton X-100 lysis buffer [20 mm Tris-Cl, pH 7.5, 150 mm NaCl, 0.5 mm, EDTA, 1 mm DTT, 1% w/v Triton X-100, and an EDTA-free protease inhibitor mixture (Roche)]. After 20 min of centrifugation at 10,000 × g, VDCC β1b and β4, Bassoon, and CAST/Erc2 were collected from the soluble fraction, and an insoluble membrane fraction was sonicated to solubilize the P/Q-type VDCCs (α1A+β1b). Two of the proteins were mixed and incubated for 2 h at 4°C; next, they were incubated with antibodies against the P/Q-type VDCC α1A, β1, β4 subunits or Myc for 1 h at 4°C, followed by an incubation with Dynabeads–protein G (Invitrogen) for 1 h at 4°C. The beads were washed extensively with Triton X-100 lysis buffer and boiled in SDS sample buffer to elute the bound proteins. The coimmunoprecipitated proteins were analyzed by Western blot with an anti-HA antibody or an anti-Flag antibody.
Statistics.
All statistics were performed using GraphPad Prism software, version 5.0c. Significance was assessed by an unpaired t test for comparisons of changes between two groups, and by one-way ANOVA plus Bonferroni's multiple-comparison test for changes between multiple groups. The p values are described in the text and figure legends. All data shown are mean ± SEM. Indicated p values are from unpaired t tests, unless specified as one-way ANOVA.
Results
NMJ innervation in DKO mice for P/Q-, N-type VDCCs
To obtain DKO mice for P/Q-, N-type VDCCs (Cacna1a −/− Cacna1b −/−), we mated double heterozygotes but were unable to identify a live DKO mouse on the day of delivery among 11 litters. However, the genotyping of embryos at gestation day 18.5 revealed 22 DKO embryos for P/Q-, N-type VDCCs among 212 embryos from 30 litters, which was similar to the predicted Mendelian frequency of the genotypes (Table 1). These DKO embryos were phenotypically normal by their outward appearance. These results indicated that DKO embryos for P/Q-, N-type VDCCs develop similarly to their littermates during the embryonic stage but die soon after birth.
Double heterozygote (Cacna1a +/− Cacna1b +/−) mating produced near Mendelian ratio embryos at prenatal stage
First, we analyzed the neurotransmission at NMJs by electromyography because the two deleted VDCCs are essential for neurotransmitter release. We stimulated the phrenic nerve at the cervical plexus and recorded the compound muscle action potential of diaphragms in E18.5 embryos. Reliable compound muscle action potentials with an amplitude of 405.6 ± 63.4 μV (mean ± SEM; n = 34 embryos) were recorded from the control embryos. However, five of six DKO embryos demonstrated no detectable compound muscle action potentials (Fig. 1 A).

DKO mice for P/Q-, N-type VDCCs have minimal electromyogram amplitude but grossly normal muscle innervation. A , Compound muscle action potentials were not detected in five of six DKO embryos (Cacna1a −/− Cacna1b −/−). In E18.5 embryos, phrenic nerves were stimulated at the level of the cervical plexus, and compound muscle action potentials were recorded in diaphragms. Left, Averaged traces of 10 sweeps from one animal for each genotype. Right, Summary histogram of six DKO embryos showing a 95% reduction of the compound muscle action potential amplitude (20.8 ± 20.8 μV; n = 6 embryos) compared with control embryos (405.6 ± 63.4 μV; n = 34 embryos). *p < 0.02. All graphs show mean ± SEM. B , Micrographs of DAPI-stained spinal cord sections showing grossly normal cell distributions in E18.5 DKO embryos. Scale bar, 200 μm. C , Phrenic nerve showing a normal branching pattern in the diaphragm of a DKO embryo at E18.5. Diaphragms were stained for nerves (green; anti-neurofilament plus anti-SV2) and acetylcholine receptors (red; Alexa Fluor 594-labeled α-bungarotoxin) by whole-mount immunohistochemistry. Hemi-diaphragms for DKO and littermate control embryos are shown. Scale bar, 300 μm. D , Representative images of individual NMJs in diaphragms shown in C . All of the acetylcholine receptor clusters were innervated in DKO embryos in a manner similar to littermate controls. Scale bar, 10 μm. E , F , The synaptic vesicle-associated proteins synaptophysin ( E , Syp, green) and synapsin I ( F , SynI, green) were concentrated at NMJs (red; Alexa Fluor 594-labeled α-bungarotoxin) and were present at low levels in the axons (blue; anti-neurofilament) in tibialis muscle of DKO embryos similarly to littermate controls. Similar results were observed in three animals per genotype for the analyses shown in B–F . Scale bars: ( E , F ), 10 μm.
The loss of P/Q- and N-type VDCCs had no effect on the gross cytoarchitecture of the spinal cord and diaphragm at E18.5. In the spinal cord of DKO embryos for P/Q-, N-type VDCCs, the cell density visualized by DAPI and the motor neuron density visualized by an antibody against choline acetyltransferase were indistinguishable from those of the littermate controls (Figs. 1 B, 7 A; Table 2). Next, we analyzed the diaphragms of DKO embryos by whole-mount immunohistochemical staining. The nerve branching pattern in the DKO diaphragms was indistinguishable from those of littermate control embryos (Fig. 1 C). The numbers of branches at primary to quaternary levels were compared separately and no significant difference was found between control and DKO diaphragms (Table 2). Furthermore, the examined DKO endplates were all innervated by motor nerves (Fig. 1 D). Grossly normal innervation patterns of NMJs were also observed in sternocleidomastoid and tibialis muscles. The synaptic vesicle-associated proteins SV2, synaptophysin, and synapsin I accumulated at NMJs of DKOs similarly to controls (Fig. 1 D–F). The average signal intensity per synapse for SV2, synaptophysin, and synapsin I did not show a significant difference between E18.5 DKO embryos and littermate controls (Table 2). These results showed that motor axons lacking P/Q- and N-type VDCCs did not elicit a detectable compound muscle action potential in perinatal stage embryos, but did display normal axon projection, endplate recognition/innervation, and an accumulation of the synaptic vesicle-related proteins at presynaptic terminals without morphologically denervated endplates.
DKO embryos for P/Q-, N-type VDCCs and controls had similar levels of spinal cord neuronal density, phrenic nerve branching pattern, synaptic proteins at NMJs, and NMJ size
Decreased active zones at NMJs in DKO embryos
Next, we examined the active zones in NMJs of DKO embryos at E18.5. Active zones were visualized using an antibody against Bassoon, because Bassoon is specifically concentrated at presynaptic active zones of synapses in the central and peripheral nervous system (tom Dieck et al., 1998; Nishimune et al., 2004). At the NMJ, the anti-Bassoon antibody revealed a punctate staining pattern, in which the arrangement, number, alignment with postsynaptic junctional folds, and developmental increase in number indicated that they were active zones of NMJs (Nishimune et al., 2004). Immunohistochemistry staining of Bassoon revealed discrete small puncta of a relatively uniform size at NMJs in sternocleidomastoid and tibialis muscles of control embryos at E18.5 that was similar to the staining pattern in postnatal NMJs. However, DKO embryos displayed significantly reduced levels of Bassoon at NMJs (Fig. 2 A). The total signal intensity of Bassoon per synapse was reduced 68% (arbitrary intensity unit before normalizing: control, 7.99 ± 1.06, n = 34 NMJs from three embryos; DKO, 2.52 ± 0.55, n = 30 NMJs from three embryos; p < 0.0001) and its stained area was reduced 67% (control, 12.05 ± 1.52% of a synapse, n = 34 NMJs from three embryos; DKO, 3.97 ± 0.83% of a synapse, n = 30 NMJs from three embryos; p < 0.0001) (Fig. 2 B,C). Other active zone-specific proteins (Piccolo and CAST family members) also demonstrated a decreased signal intensity at NMJs in DKO embryos (see Fig. 6 B). However, DKO and littermate control endplates showed no differences in the size of acetylcholine receptor clusters or the density of acetylcholine receptors within a synapse (Table 2). These active-zone phenotypes were not attributable to denervation of these endplates, because all of the endplates were innervated in DKO embryos, as shown in Figure 1. Furthermore, the decrease in active-zone numbers was also confirmed by transmission electron microscopy. The ultrastructure of the NMJ profiles in DKO embryos showed a significantly reduced number of electron-dense active zones per profile (control, 1.12 ± 0.14, n = 78 NMJ profiles from three embryos; DKO, 0.54 ± 0.08, n = 79 NMJ profiles from three embryos; p = 0.0007; the sample sizes (n) are the same for all the ultrastructure data) and docked synaptic vesicles per profile (control, 2.59 ± 0.35; DKO, 1.53 ± 0.20; p = 0.0094) (Fig. 2 D–F). However, the NMJ profiles showed no differences in the nerve–muscle apposition size (control, 1.11 ± 0.09 μm; DKO, 1.04 ± 0.08 μm; p = 0.563), the total synaptic vesicle number per profile (control, 89.47 ± 8.36; DKO, 73.33 ± 7.03; p = 0.141), or the number of postsynaptic junctional folds per profile (control, 0.24 ± 0.06; DKO, 0.11 ± 0.05; p = 0.105) (Fig. 2 D,G–J). Thus, the lack of presynaptic P/Q- and N-type VDCCs caused a preferential reduction in the number of active zones.

NMJs of DKO embryos for P/Q-, N-type VDCCs have fewer presynaptic active zones. A , The active-zone protein Bassoon was present at a significantly lower level in the NMJs of DKO embryos for P/Q-, N-type VDCCs (Cacna1a −/− Cacna1b −/−) compared with littermate controls at E18.5. Active zones and acetylcholine receptors of tibialis muscle were visualized by fluorescent immunohistochemistry using an anti-Bassoon antibody (green) and Alexa Fluor 594-labeled α-bungarotoxin (red), respectively. B , C , DKO mice showed decreases in the total Bassoon signal and in the Bassoon-stained area. The measurements are from three embryos per genotype, for a total of 34 NMJs for control and 30 NMJs for DKO. D , Transmission electron micrographs showing a reduced number of active zones in the NMJs of sternocleidomastoid muscle of DKO embryos compared with littermate controls at E18.5. The white arrowheads indicate electron-dense material at active zones, and the red asterisks were placed on the right side of docked synaptic vesicles. The bottom panels show higher magnification views of representative images of different synapses. E–H , Quantification of synaptic ultrastructures revealed a significantly decreased number of active zones ( E ) and docked vesicles ( F ) in the NMJs of DKO embryos compared with littermate controls. However, DKO and control embryos displayed a NMJ apposition of similar size ( G ) and a similar total number of synaptic vesicles per profile ( H ). I , Transmission electron micrographs showing postsynaptic junctional folds in NMJ profiles of sternocleidomastoid muscle of DKO embryos and littermate control embryos at E18.5. The white arrows indicate the junctional folds. J , Quantification of junctional folds in NMJ profiles of DKO embryos compared with littermate controls. DKO embryos displayed a trend of fewer junctional folds, but did not show a significant difference compared with controls by an unpaired t test (p = 0.105). The counts ( E–H , J ) are from three embryos for each genotype, for a total of 78 NMJ profiles in control and 79 NMJ profiles in DKO. The asterisk (*) indicates the following: B , C , p < 0.001; E , p < 0.05; F , p < 0.01. Scale bars: A , 5 μm; D , I , 200 nm.
The reduced active-zone phenotype is not attributable to a lack of synaptic activity because mice lacking choline acetyltransferase have normal numbers of active zones at the NMJ without detectable neurotransmission (Misgeld et al., 2002). In NMJs of Drosophila, alterations of neuronal activities in a sodium channel mutant or a potassium channel mutant did not cause an abnormal localization of Bruchpilot, suggesting that changes in the activity by itself do not alter the number of active zones at the NMJs (Graf et al., 2009). Furthermore, the reduced active-zone phenotype is less likely to be attributable to a lack of Ca2+ influx through VDCCs because laminin β2 induces presynaptic differentiation in motor neurons even when VDCCs are blocked by toxins specific for P/Q- and N-type VDCCs (ω-agatoxin IVa, ω-conotoxin GVIA) (Nishimune et al., 2004). The immunohistochemical and ultrastructural changes of the active zones in DKO mice were much stronger than those observed in single knock-out mice for either P/Q-type VDCCs, in which there is a 35% reduction of the area stained by Bassoon per synapse (Nishimune et al., 2004), or for N-type VDCCs (32% reduction; described below), which suggests a role for both of these presynaptic channels in the organization of active zones.
Attenuated presynaptic active zones at synapses lacking N-type VDCCs
DKO mice were not viable; thus, we examined single knock-out mice for the N-type VDCC (Cacna1b −/−) to analyze the role of laminin β2 binding of N-type VDCCs in organizing active zones in postnatal NMJs. These knock-out mice have been generated previously and show functional disorders of the sympathetic nervous system; however, the development of NMJs has not been examined (Ino et al., 2001). The null mutant mice are viable for more than a year (Ino et al., 2001). We chose to analyze the NMJs of null mutants at postnatal days 0 and 4 when the motor nerve terminals contain a mixture of P/Q- and N-type VDCCs and at postnatal days 10 and 21 after the terminals have switched to possess solely P/Q-type VDCCs (Rosato Siri and Uchitel, 1999). We examined the diaphragm muscles of N-type VDCC knock-out mice by whole-mount immunohistochemistry to visualize the entire innervation pattern of the phrenic nerves. The gross branching pattern of phrenic nerves in the diaphragms of knock-out mice was indistinguishable from that in the littermate control mice among the ages examined (Fig. 3 A). In homozygous mutant mice, the clustering of acetylcholine receptors and the endplate bandwidth were similar to those in control mice. NMJs formed in these mutants, with vesicle-rich nerve terminals juxtaposed with postsynaptic acetylcholine receptors (Fig. 3 B). Each endplate was innervated by multiple axons at postnatal days 0 and 4 and was singly innervated at postnatal days 10 and 21, which suggested that synapse elimination proceeded normally (Fig. 3 B,C). At these ages, denervated NMJs were not observed. However, acetylcholine receptor clusters in mutant mice showed a 30% reduction in size at postnatal day 4 (control, 161.5 ± 7.49 μm2, n = 32 NMJs from three mice; Cacna1b −/−, 113.1 ± 5.69 μm2, n = 31 NMJs from three mice; p < 0.0001). The reduction in synapse size was smaller but similar to that of postnatal P/Q-type VDCC knock-out mice, which demonstrated a 40% decrease (Urbano et al., 2003). However, these decreases in endplate size are not attributable to reduced synaptic activity because postsynaptic development is accelerated in Chat −/− mice that lack neurotransmission (Misgeld et al., 2002). The endplate size does not change between embryonic day 17.5 to postnatal day 0, and exhibits a 50% increase by postnatal day 2 (Misgeld et al., 2002). The postsynaptic development could be delayed in the N-type VDCC knock-out mice at postnatal day 4, which may have caused the 30% decrease in endplate size.

N-type VDCC knock-out mice have normal innervation of NMJs. A , Diaphragm muscle of control mice (left) and N-type VDCC knock-out mice (right; Cacna1b −/−) showed similar innervation patterns at postnatal day 4. The diaphragms were stained by whole-mount immunohistochemistry using anti-neurofilament and anti-SV2 antibodies to reveal nerve morphology (green) and with Alexa Fluor 594-labeled α-bungarotoxin to detect the postsynaptic acetylcholine receptors (red). B , Higher magnifications showing innervated NMJs in the muscle presented in A . Endplate innervation was confirmed in three mice per genotype, for a total of 32 NMJs for control and 31 NMJs for Cacna1b −/−. C , Immunohistochemistry images showing a single axon innervating NMJs at postnatal day 10 in N-type VDCC single knock-out mice and littermate controls. Diaphragms were stained for nerves (green; anti-neurofilament plus anti-SV2) and acetylcholine receptors (red; Alexa Fluor 594-labeled α-bungarotoxin) by whole-mount immunohistochemistry. Together with Figure 3 B showing multiple innervation of NMJs at postnatal day 4, these results suggest that synapse elimination proceeded normally in this mutant. Scale bars: A , 500 μm; B , C , 10 μm.
Next, we examined active zones in the NMJs of mice lacking the N-type VDCC at postnatal day 4 because motor nerve terminals use N-type VDCCs for synaptic transmission before the first week of age and because we previously showed that the presynaptic organizer laminin β2 binds to N-type VDCC in vitro (Rosato Siri and Uchitel, 1999; Nishimune et al., 2004). A similar punctate staining pattern of Bassoon was observed at the NMJs of tibialis muscles of N-type VDCC knock-out mice at postnatal day 4, but there was a 30% decrease in Bassoon signal intensity (arbitrary intensity unit before normalizing: control, 5.15 ± 0.35, n = 32 NMJs from three mice; Cacna1b −/−, 3.64 ± 0.29, n = 31 NMJs from three mice; p = 0.0015) and a 32% decrease in Bassoon-stained area (control, 8.13 ± 0.54% of a synapse, n = 32 NMJs from three mice; Cacna1b −/−, 5.54 ± 0.41% of a synapse, n = 31 NMJs from three mice; p = 0.0003) after normalization to the NMJ size (Fig. 4). The reduced level of Bassoon signal was similar to that observed in P/Q-type VDCC knock-out mice (Nishimune et al., 2004). These results suggest that N-type VDCCs play key roles in controlling the size of the postnatal neuromuscular synapse and the organization of active zones during perinatal stages.

N-type VDCC knock-out mice have fewer presynaptic active zones at NMJs. A , The active-zone protein Bassoon was present at significantly lower levels at NMJs in N-type VDCC knock-out mice (Cacna1b −/−) compared with littermate control mice at postnatal day 4. Active zones and acetylcholine receptors at tibialis muscle were visualized by fluorescent immunohistochemistry using an anti-Bassoon antibody (green) and Alexa Fluor 594-labeled α-bungarotoxin (red), respectively. The bottom panels show pseudocolored images of the Bassoon signal, with warmer colors representing stronger signals. Endplate size was measured using these high-magnification images and revealed a 30% reduction in the knock-out mice. B , C , N-type VDCC mutant mice displayed a decrease in total Bassoon signal ( B ) and in the Bassoon-stained area ( C ) after normalization for differences in synapse size, in tibialis muscle. The quantifications are from three mice per genotype, for a total of 32 NMJs for control and 31 NMJs for Cacna1b −/−. The asterisk (*) indicates the following: B , p < 0.01; C , p < 0.001. Scale bar, 5 μm.
Direct interactions of VDCC β subunits with active-zone proteins
To determine how the extracellular interaction of laminin β2 and VDCCs is linked to the cytosolic active-zone protein Bassoon, we tested whether the P/Q-type VDCC and Bassoon formed a complex by immunoprecipitation. We coexpressed the P/Q-type VDCC subunit α1A (Cacna1A) with β1b (Cacnb1) or β4 (Cacnb4), because the β subunits are required to mask the endoplasmic reticulum retention signal for cell surface expression of the α subunits and these β subunits accumulate at the NMJs (Bichet et al., 2000; Pagani et al., 2004). When the P/Q-type VDCC (α1A+β1b or α1A+β4 subunits) was immunoprecipitated using an anti-α1A antibody, Bassoon coimmunoprecipitated with the VDCC (Fig. 5 A). To determine the Bassoon binding subunit, we independently expressed recombinant proteins of the α1A subunit C-terminal cytosolic domain because this domain binds to the active-zone protein Bruchpilot in Drosophila, or full-length VDCC β1b or β4 subunits because these subunits bind to the active-zone protein Rim1 (Kiyonaka et al., 2007; Fouquet et al., 2009). Bassoon coimmunoprecipitated with the VDCC β1b subunit, but not with the C-terminal cytosolic domain of the α1A subunit (Fig. 5 B). The C-terminal domain of the α1A subunit or the GFP did not coimmunoprecipitate with the VDCC β1b subunit, which demonstrated the specificity of the VDCC β1b subunit–Bassoon interaction. Bassoon also coimmunoprecipitated with the VDCC β4 subunit (Fig. 5 B). These results demonstrate that Bassoon can bind directly to the β1b and β4 subunits of presynaptic VDCCs.

VDCC β subunits form protein complexes with Bassoon and CAST/Erc2. A , Left, Bassoon coimmunoprecipitated with the P/Q-type VDCC complexes (α1A+β1b, α1A+β4 subunits) using an anti-α1A antibody. Right, CAST/Erc2 coimmunoprecipitated with the P/Q-type VDCC complex (α1A+β1b subunits) using an anti-α1A antibody. B , Bassoon coimmunoprecipitated with the VDCC β1b subunit. The Myc-tagged C-terminal cytosolic domain of the α1A subunit did not bind to Bassoon. Bassoon was also coimmunoprecipitated with the VDCC β4 subunit. C , CAST/Erc2 coimmunoprecipitated with the VDCC β1b subunit. The Myc-tagged C-terminal cytosolic domain of the α1A subunit did not bind to CAST/Erc2.
Fouquet et al. (2009) demonstrated a direct interaction of Drosophila Bruchpilot (Brp; ELKS/Rab6-interacting protein/CAST family member) and Cacophony (Cac; ortholog of the P/Q-type VDCC pore forming α subunit). In rodents, CAST/Erc2 (Erc2, ELKS/Rab6-interacting protein/CAST family member) localizes at NMJs (Juranek et al., 2006). Thus, we assessed the interaction of the rodent P/Q-type VDCC (α1A+β1b subunits) and the active zone-specific protein CAST/Erc2 by immunoprecipitation. When the P/Q-type VDCC (α1A+β1b subunits) was immunoprecipitated using an anti-α1A antibody, CAST/Erc2 coimmunoprecipitated with the VDCC (Fig. 5 A). Similarly to Bassoon, CAST/Erc2 coimmunoprecipitated with the VDCC β1b subunit but not with a recombinant protein of the C-terminal cytosolic-domain of the α1A subunit (Fig. 5 C). These results show that, unlike the binding of Drosophila Bruchpilot to the C-terminal domain of Cacophony, mammalian CAST/Erc2 binds to the VDCC β1b subunit.
These direct interactions of the VDCC β subunits and Bassoon or CAST/Erc2 may allow presynaptic VDCCs to anchor multiple active-zone proteins to the presynaptic membrane. An antibody against pan-CAST family members (CAST/Erc2 and ELKS) stained the NMJs of control animals with a light-and-dark pattern that resembled the active zones stained using anti-Piccolo and anti-Bassoon antibodies (Fig. 6 A,B). Colocalization of these active-zone proteins was indicated by the significantly higher Pearson's coefficients of overlapping signals compared with that obtained from proteins with minimal overlap (Fig. 6 C). Importantly, these pan-CAST and Piccolo signals decreased significantly in DKO embryos for P/Q, N-type VDCCs, similar to the staining observed for Bassoon (Fig. 6 B). These results suggest that the anchoring of active-zone proteins by direct interactions with the presynaptic VDCC is important for organizing the presynaptic active zones.
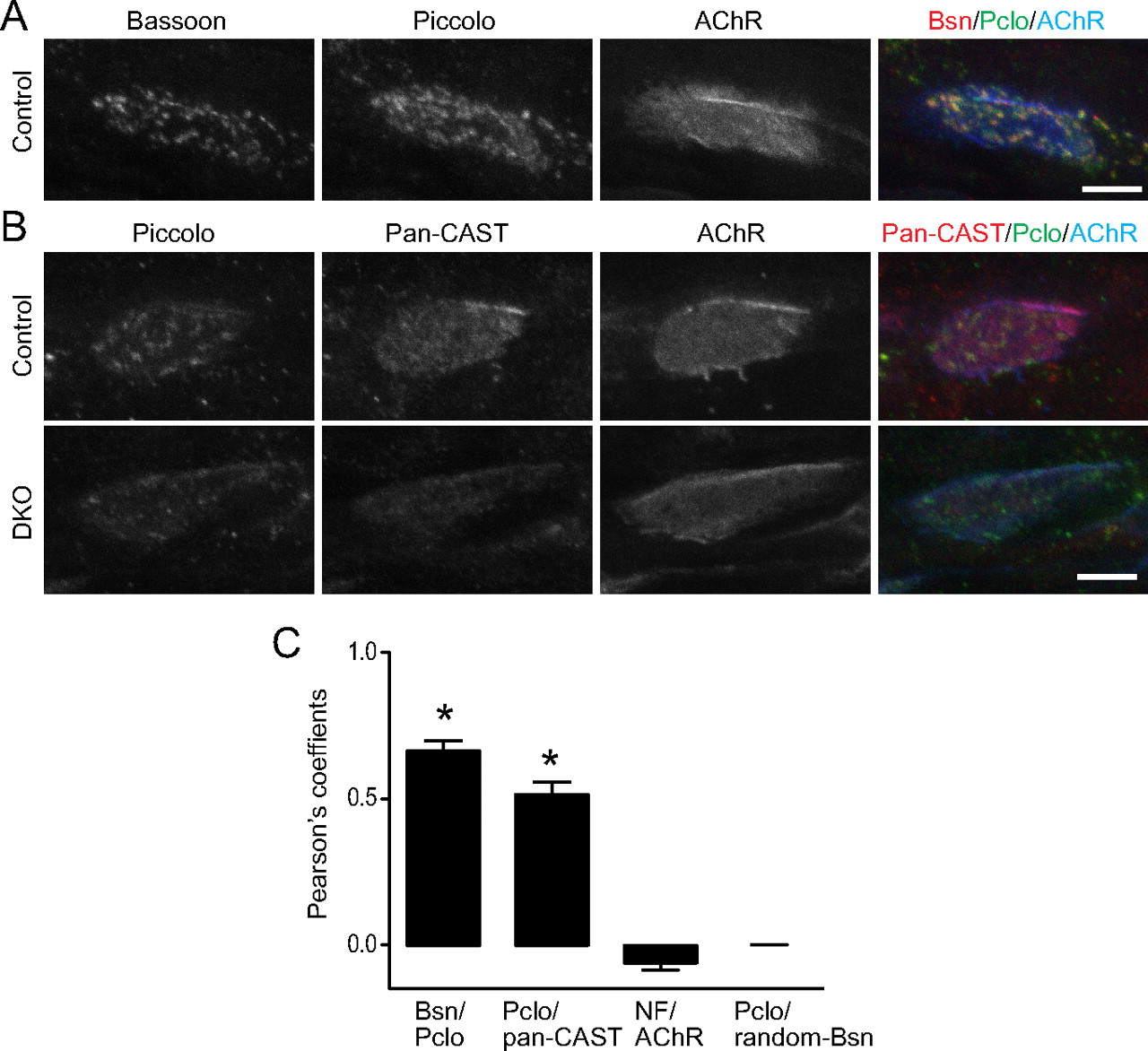
A , In control embryos, Piccolo (Pclo) showed a punctate staining pattern similar to the active-zone marker Bassoon (Bsn) at the acetylcholine receptor (AChR) cluster visualized by fluorescence immunohistochemistry. B , The pan-CAST (CAST/Erc2 and ELKS) antibody stained control NMJs diffusely, with stronger intensity observed at active zones, which were visualized by the Piccolo staining pattern. The pan-CAST staining pattern was not as uniform as that of AChR visualized by Alexa Fluor 594-labeled α-bungarotoxin. Importantly, the pan-CAST and the Piccolo signals were present at lower levels at NMJs of DKO embryos at E18.5. Similar results were obtained for three animals per genotype. C , Colocalization analysis of active-zone proteins at the NMJ by the Pearson's coefficients (r). Active-zone proteins (Bassoon, Piccolo, and pan-CAST) at NMJs were double labeled by immunohistochemistry. Colocalization of these active-zone proteins was indicated by the significantly higher Pearson's coefficients r value for Bassoon (Bsn)/Piccolo (Pclo) (0.66 ± 0.03) or Piccolo/pan-CAST (0.52 ± 0.04) compared with the r value for neurofilament (NF)/AChR (−0.06 ± 0.02) that showed minimal overlap. A Piccolo image was compared against 200 randomized images of a Bassoon image (random-Bsn) and we obtained Pearson's coefficient r value of 0.0 ± 0.0010, which showed the absence of correlation between Bassoon and Piccolo by chance. The analyses are from six NMJ images per pair of antigens. A significant difference was detected using one-way ANOVA (p < 0.0001). A post hoc analysis showed significance for the r values of Bassoon/Piccolo and Piccolo/pan-CAST compared with that of neurofilament/acetylcholine receptor or Piccolo/randomized-Bassoon images (asterisks). Scale bars: A , B , 5 μm.
Normal Bassoon expression levels in DKO motor neuron cell bodies
The attenuated accumulation of Bassoon protein at the presynaptic terminals of DKO NMJs may be caused by a loss of anchoring of Bassoon protein or by a decreased expression level of Bassoon in the motor neuron cell body. We used in situ hybridization to analyze the relative expression levels of Bassoon (bsn) mRNA in motor neuron cell bodies. This method was chosen over quantitative reverse transcription (RT)-PCR detection of mRNA purified from the spinal cord because it allows the specific detection of signals from motor neurons without contaminating effects from other neurons in the spinal cord. Furthermore, the method simultaneously revealed Bassoon expression levels in interneurons in the spinal cord that form circuits with motor neurons. A side-by-side comparison of DKO and control spinal cords on the same slide revealed similar levels of bsn signal among interneurons in the spinal cord of E18.5 embryos (Fig. 7 A). To specifically compare the level of bsn signal in motor neurons, their cell bodies were identified by their location in the ventral horns and by their large nuclei stained with DAPI, which are darker than those of interneurons and glial cells. We also used a probe against choline acetyltransferase (Chat) in an adjacent section and confirmed the location of motor neuron cell bodies by in situ hybridization. We detected similar levels of bsn signal in the motor neurons of DKO mice compared with littermate control mice (Fig. 7 A,B).

DKO motor neuron cell bodies express normal levels of Bassoon. A , Similar levels of Bassoon (Bsn) mRNA were detected by in situ hybridization in lumbar spinal cord neurons of DKO embryos for P/Q-, N-type VDCCs and littermate controls at E18.5. A probe against choline acetyltransferase (Chat) was used in adjacent sections to confirm the location of motor neuron cell bodies. B , At higher magnification, similar levels of dark alkaline phosphate reaction product from in situ hybridization can be seen in motor neurons in the ventral horns of different spinal cord sections. DAPI staining was used to visualize the large nuclei of motor neurons. The red arrowheads indicate the cell bodies of motor neurons. Similar results were obtained in two animals. C , Similar levels of cytosolic Bassoon protein were detected by immunohistochemistry in the motor neuron cell bodies of lumbar spinal cord from DKO embryos for P/Q-, N-type VDCCs and littermate controls at E18.5. Anti-choline acetyltransferase (ChAT) antibody and DAPI staining were used to identify motor neuron cell bodies and their nuclei. The dotted lines show representative examples of the circumference of the motor neuron cell bodies used for the measurement of the cytosolic Bassoon signal. D , The Bassoon signal intensity was measured in the cytosolic region, excluding the nuclei, of motor neurons and normalized to control levels. The Bassoon signal intensities of control and DKO motor neurons demonstrated no differences by an unpaired t test (mean arbitrary intensity unit before normalizing to control ± SEM: control, 32.15 ± 1.29, n = 89 motor neurons from 2 embryos; DKO, 31.79 ± 1.02, n = 84 motor neurons from 2 embryos; p = 0.828). Scale bars: A , 200 μm; B , C , 50 μm.
Next, we analyzed the expression levels of Bassoon protein in motor neuron cell bodies in E18.5 embryos by immunohistochemistry. The sections were triple-labeled with antibodies against Bassoon and choline acetyltransferase and with DAPI to identify the location of motor neurons in the ventral horns. Coincident with the transcription levels, we detected similar levels of anti-Bassoon staining in the motor neuron cell bodies of DKO mice compared with controls (Fig. 7 C,D). These results show that the lack of P/Q- and N-type VDCCs did not alter the transcription or translation levels of the active-zone protein Bassoon in motor neurons.
Attenuated laminin β2 protein accumulation at NMJs in DKO embryos
Finally, we analyzed the levels of laminin β2 protein at NMJs in DKO embryos, because P/Q-type and N-type VDCCs are presynaptic receptors for laminin β2 (Nishimune et al., 2004). At the NMJs of control embryos at E18.5, laminin β2 colocalized with acetylcholine receptor clusters and was present at a higher concentration at the synaptic cleft basal lamina compared with the extrasynaptic area (Fig. 8 A). Notably, DKO embryos demonstrated a 55% decrease in the level of laminin β2 protein at NMJs (arbitrary intensity unit before normalizing: control, 57.02 ± 2.36, n = 142 NMJs from three embryos; DKO, 25.91 ± 0.86, n = 127 NMJs from three embryos; p < 0.0001) (Fig. 8 A,B). However, the transcription level of laminin β2 mRNA was not significantly different between E18.5 DKO embryos and littermate controls by quantitative RT-PCR (Fig. 8 C). Relative expression levels of lamb2 normalized against gapdh did not show a statistically significant difference between DKO and control embryos (control, 1.160 ± 0.108; DKO, 1.425 ± 0.105; n = 6 embryos; p = 0.110). Similar results showing no differences between the groups were obtained after normalization to S18 (control, 0.708 ± 0.075; DKO, 0.884 ± 0.107; n = 6 embryos; p = 0.208) and tbp (control, 1.398 ± 0.134; DKO, 1.732 ± 0.353; n = 6 embryos; p = 0.397). Previous reports using microarray or immunohistochemistry analyses showed that the level of laminin β2 mRNA or protein does not change as a result of a lack of synaptic transmission (Chat −/− mice), denervation, or reinnervation (Sanes et al., 1986, 1990; Patton et al., 1999; Misgeld et al., 2002; Kostrominova et al., 2005; Batt et al., 2006; Zhou et al., 2006). These results suggest that the detected change in the laminin β2 protein level in DKO embryos is not attributable to decreased synaptic transmission. We previously showed that P/Q- and N-type VDCCs are specific receptors for synaptic laminin β2 (Nishimune et al., 2004). Together, the current results suggest that the P/Q- and N-type VDCCs at motor nerve terminals play key roles as receptors in accumulating/retaining laminin β2 protein at the synaptic cleft of NMJs.

Laminin β2 protein levels are decreased at NMJs of DKO embryos for P/Q-, N-type VDCCs. A , Immunohistochemistry staining showed that laminin β2 was present at higher levels in synaptic compared with extrasynaptic regions of muscle fibers in control embryos at E18.5. The synaptic laminin β2 level was significantly decreased in littermate DKO embryos. Scale bar, 5 μm. B , The synaptic laminin β2 signal intensity showed a 55% decrease in DKOs. Measurements were obtained for three animals per genotype, for a total of 142 NMJs for control and 127 NMJs for DKO. *p < 0.0001. C , Laminin β2 mRNA was present at similar levels in DKO embryos and littermate controls. Six embryos per group were analyzed by quantitative real-time PCR, and the results were normalized to gapdh. Relative expression levels are indicated by a scatterplot with the mean ± SEM indicated by whiskers. A significant difference was not detected using an unpaired t test (p = 0.1097).
Discussion
Synapse formation requires the association of active zones with presynaptic VDCCs and precise alignment of active zones with postsynaptic specializations. A few transsynaptic molecular mechanisms of synaptogenesis have been identified, but little is known about the mechanism responsible for organizing and anchoring presynaptic active zones (Graf et al., 2009; Klassen et al., 2010). Here, we showed that presynaptic P/Q- and N-type VDCCs play essential roles in active-zone organization. The DKO mice for P/Q-, N-type VDCCs exhibited defects preferentially in the active zone. To organize synapses, the postsynaptic muscle cells express, secrete, and concentrate laminin β2 in the synaptic cleft (Hunter et al., 1989; Noakes et al., 1995; Miner et al., 2006). Laminin β2 binds specifically to the presynaptic P/Q- and N-type VDCCs that are concentrated at motor nerve terminals (Nishimune et al., 2004). The VDCC β subunits bind to the active zone-specific proteins Bassoon, CAST/Erc2, and Rim1 (Kiyonaka et al., 2007; present study). These results suggest that the VDCCs link an extracellular synaptogenic cue to cytosolic active zone-specific proteins and anchor them to organize the active zones.
Role of synaptic transmission and calcium channel activity in active-zone formation
The molecular mechanism responsible for the initial phase of synapse formation does not seem to require synaptic transmission. Mice lacking choline acetyltransferase (Chat −/−) display no synaptic transmission at NMJs but have a normal number of active zones at the motor nerve terminals (Misgeld et al., 2002). Even in the absence of synaptic transmission, morphologically normal synapses with active zones form in mice lacking munc13 or munc18 (Verhage et al., 2000). Our results partially coincide with these findings because the NMJs of DKO embryos showed target recognition, nerve terminal formation, and an accumulation of synaptic vesicles and vesicle-related proteins without detectable electromyogram amplitudes. However, the motor nerve terminals of DKO mice lacked an accumulation of several active zone-specific proteins. This difference between Chat −/− mice and VDCC DKO mice suggests that the active-zone defect is not attributable to reduced synaptic transmission but is instead attributable to the lack of P/Q- and N-type VDCC proteins.
The extent to which ion channel activity of VDCCs is required for active-zone formation is unclear. However, laminin β2 was able to induce presynaptic differentiation in cultured motor neurons despite P/Q- and N-type VDCC blockade by specific toxins, providing evidence for dispensability of Ca2+ influx into nerve terminals for active-zone formation (Nishimune et al., 2004). Thus, a loss of VDCC-associated scaffolding mechanisms, rather than ion channel activity, may be more important in reducing active zones in VDCC DKO mice.
Presynaptic VDCCs and active zones
In Drosophila, VDCC α2δ-3 subunit is required for NMJ development, but its requirement for active-zone organization is not clear (Ly et al., 2008; Kurshan et al., 2009). The Drosophila active zone-specific protein Bruchpilot binds directly to the P/Q-type VDCC homolog Cacophony. In Drosophila mutants for Bruchpilot, the accumulation of Cacophony at NMJs decreased to ∼50% of the control level, but the remaining Cacophony accumulates at active zone-like structures (Kittel et al., 2006; Fouquet et al., 2009). These findings suggest that VDCCs may be one of the first presynaptic proteins to localize active-zone proteins.
In mice, the accumulation of VDCCs at presynaptic terminals requires the cytosolic domains of VDCCs (Synprint region, C terminus domain), but how VDCCs affect active-zone organization has not been analyzed (Maximov and Bezprozvanny, 2002; Mochida et al., 2003). VDCC α2δ-1 subunit is involved in excitatory synaptogenesis, but its requirement for active-zone organization is not clear (Eroglu et al., 2009). Synapse organizer α-neurexins were proposed to accumulate VDCCs at brainstem synapses from the analysis of α-neurexin triple knock-out mice (Missler et al., 2003). However, direct physical interactions of VDCCs and neurexins have not been described. The presynaptic protein calcium/calmodulin-dependent serine protein kinase (CASK) may link VDCCs to neurexins (Hata et al., 1996; Maximov et al., 1999). However, CASK does not seem to participate in the active-zone formation because CASK knock-out neurons exhibit overall normal electrical properties and synapse morphology without an active-zone phenotype (Atasoy et al., 2007).
We previously showed that VDCCs can accumulate at NMJs by an extracellular interaction with laminin β2 (Nishimune et al., 2004). The present results show direct interactions among the VDCC subunits β1b and β4 and the active-zone proteins Bassoon and CAST/Erc2. In DKO embryos for P/Q-, N-type VDCCs, these active-zone proteins were significantly attenuated at NMJs. These results suggest that presynaptic VDCCs organize active zones by anchoring active-zone proteins at presynaptic terminals. A physical protein–protein interaction is required to anchor the active-zone structure, as shown by the interaction of Bassoon and ribbon-specific protein RIBEYE at the photoreceptor synapse (tom Dieck et al., 2005). However, a loss of a single active zone-specific protein can be compensated for by multiple cytosolic interactions. For example, knock-out mice for Rim1α or ELKS2α show no ultrastructural morphological defects at synapses in the CNS, although the NMJs and retinal photoreceptor synapses in these mice remain to be assessed (Schoch et al., 2002; Kaeser et al., 2009). But combining knock-out for Piccolo and knockdown of Bassoon in mice demonstrated a decreased number of docked vesicles (Mukherjee et al., 2010). Our current results and previous reports show that VDCCs bind directly to Bassoon, CAST/Erc2, Rim, and Piccolo (Shibasaki et al., 2004; Kiyonaka et al., 2007; Uriu et al., 2010). Furthermore, CAST/Erc2 binds directly to Bassoon and Rim (Takao-Rikitsu et al., 2004; Wang et al., 2009). Importantly, Bassoon, Piccolo, and Rim are found in protein complexes containing VDCCs in vivo (Carlson et al., 2010; Müller et al., 2010). Thus, VDCCs appear to be essential for anchoring these active-zone proteins to the presynaptic membrane to organize the active zones. A unitary assembly model has been proposed for the formation of active zones, but the molecular mechanisms responsible for anchoring the content of such transport vesicles remain unknown (Zhai et al., 2001; Shapira et al., 2003). We propose that the extracellular protein interactions of laminin β2 and presynaptic P/Q- and N-type VDCCs may comprise a molecular mechanism to anchor VDCCs and active-zone proteins in alignment with the postsynaptic specialization.
Accumulation of laminin β2 at NMJs
The DKO mice demonstrated normal transcription levels of laminin β2 but significantly reduced levels of protein accumulation at NMJs. In contrast, mice lacking choline acetyltransferase (Chat −/−) display no synaptic transmission at NMJs but show a concentration of laminin β2 protein at NMJs (Misgeld et al., 2002). Similarly, the mRNA or protein level of laminin β2 at NMJs does not seem to change in response to denervation or reinnervation (Sanes et al., 1986, 1990; Patton et al., 1999; Kostrominova et al., 2005; Batt et al., 2006; Zhou et al., 2006). These reports suggest that reduced protein levels of laminin β2 at the DKO synaptic cleft are independent of the reduced neurotransmission or muscle activity.
The mechanism to concentrate laminin β2 protein at the NMJ and myotendinous junctions is currently unknown. The transcription of laminin β2 does not seem to be specific to synaptic nuclei (Kishi et al., 2005; Jevsek et al., 2006). Developmentally, laminin β2 protein is concentrated at higher levels over the acetylcholine receptor clusters than extrasynaptic areas at E15, when innervation is beginning to develop (Patton et al., 1997; Sanes and Lichtman, 1999, 2001). Importantly, laminin β2 protein is concentrated specifically at NMJs and myotendinous junctions when rat lamb2 is transgenically overexpressed throughout the muscle in a laminin β2 knock-out mouse background (Miner et al., 2006). Thus, potential mechanisms for the accumulation of laminin β2 at the NMJ include its selective translation, secretion, or stabilization in the extracellular space. Our data suggest that the interaction of laminin β2 with presynaptic VDCC proteins is required to maintain laminin β2 protein in the synaptic cleft.
Implications for LEMS
Autoantibodies against P/Q-type VDCC are found in 75–85% of LEMS patients, suggesting that this channel is essential for the etiological mechanism of this disease (Flink and Atchison, 2003). The passive transfer of patient IgGs into mice causes LEMS-like symptoms with a prominent decrease in presynaptic active zones (Fukunaga et al., 1983; Fukuoka et al., 1987). In these mice, the reduction in quantal content of neurotransmission (64%) is correlated with the reduced number of active-zone structures (72%), which suggests that the active-zone structure plays essential roles in the reduction of neurotransmission (Fukunaga et al., 1983; Lang et al., 1983; Kim, 1985; Fukuoka et al., 1987; Smith et al., 1995).
The molecular mechanism described in this study may play an essential role in organizing mammalian NMJs. The presynaptic VDCC proteins play essential roles in organizing active zones by functioning, at least in part, as scaffolding proteins linking an extracellular synapse organizer directly to cytosolic active zone-specific proteins. Importantly, LEMS patients carry antibodies against the 11th extracellular laminin-binding domain of the P/Q-type VDCC α subunit (Takamori et al., 2000). Thus, the decrease in presynaptic active zones in LEMS patients may be caused by autoantibodies that block the interaction of presynaptic VDCCs and the synapse organizer laminin β2.
Footnotes
-
This work was supported by National Institutes of Health–National Center for Research Resources Grants P20RR016475 and P20RR024214 and The Whitehall Foundation (H.N.), and by Kansas Intellectual and Developmental Disabilities Research Center Grant P30 NICHD HD 002528. We thank H. S. Shin for knock-out mice, K. Si for plasmids, R. Timple and T. Sasaki for antibody, L. Christenson, B. Fegley, D. Warren, and E. White-Grindley for technical assistance, and J. R. Sanes and M. J. Werle for comments on this manuscript. The monoclonal antibody SV2 developed by Dr. K. M. Buckley was obtained from the Developmental Studies Hybridoma Bank maintained by the National Institute of Child Health and Human Development and the Department of Biological Sciences, University of Iowa (Iowa City, IA).
-
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Hiroshi Nishimune, University of Kansas Medical Center, Department of Anatomy and Cell Biology, 3901 Rainbow Boulevard, MS 3051, HLSIC Room 2073, Kansas City, KS 66160. hnishimune{at}kumc.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}