

Abstract
Immunization against amyloid-β (Aβ) can reduce amyloid accumulation in vivo and is considered a potential therapeutic approach for Alzheimer's disease. However, it has been associated with meningoencephalitis thought to be mediated by inflammatory T-cells. With the aim of producing an immunogenic vaccine without this side effect, we designed CAD106 comprising Aβ1–6 coupled to the virus-like particle Qβ. Immunization with this vaccine did not activate Aβ-specific T-cells. In APP transgenic mice, CAD106 induced efficacious Aβ antibody titers of different IgG subclasses mainly recognizing the Aβ3–6 epitope. CAD106 reduced brain amyloid accumulation in two APP transgenic mouse lines. Plaque number was a more sensitive readout than plaque area, followed by Aβ42 and Aβ40 levels. Studies with very strong overall amyloid reduction showed an increase in vascular Aβ, which atypically was nonfibrillar. The efficacy of Aβ immunotherapy depended on the Aβ levels and thus differed between animal models, brain regions, and stage of amyloid deposition. Therefore, animal studies may not quantitatively predict the effect in human Alzheimer's disease. Our studies provided no evidence for increased microhemorrhages or inflammatory reactions in amyloid-containing brain. In rhesus monkeys, CAD106 induced a similar antibody response as in mice. The antibodies stained amyloid deposits on tissue sections of mouse and human brain but did not label cellular structures containing APP. They reacted with Aβ monomers and oligomers and blocked Aβ toxicity in cell culture. We conclude that CAD106 immunization is suited to interfere with Aβ aggregation and its downstream detrimental effects.
Introduction
Amyloid-β (Aβ) immunotherapy has emerged as a potential therapy for Alzheimer's disease (AD) (Klafki et al., 2006; Citron, 2010). Both active immunization against Aβ and passive transfer of monoclonal Aβ antibodies were shown to attenuate amyloid plaque formation in the brains of APP transgenic mouse models (Schenk et al., 1999; Bard et al., 2000). This treatment also diminished the amyloid-associated pathology (Lombardo et al., 2003; Oddo et al., 2004; Brendza et al., 2005; Buttini et al., 2005) and could improve learning deficits (Janus et al., 2000; Morgan et al., 2000). However, Aβ immunization of APP transgenic mice was shown to potentially change vascular amyloid and increase cerebral microhemorrhages (Pfeifer et al., 2002; Wilcock et al., 2004, 2007; Racke et al., 2005; Schroeter et al., 2008). Proposed mechanisms of Aβ immunotherapy include Fc receptor-mediated phagocytosis of amyloid (Bard et al., 2000), direct interference with Aβ aggregation (Bacskai et al., 2002; McLaurin et al., 2002) by antibodies in the brain, and peripheral sequestration of Aβ, enhancing its net cerebral efflux (DeMattos et al., 2001). Experimental data are not entirely consistent with these mechanisms (Levites et al., 2006; Winkler et al., 2010).
In humans, evidence for a reduction of the amyloid load by Aβ immunotherapy was obtained from AD patients immunized with aggregated Aβ1–42 and the adjuvant QS21 (AN-1792). Histopathological examination showed neocortical areas devoid of amyloid plaques in antibody responders but not in control patients (Nicoll et al., 2003; Ferrer et al., 2004; Masliah et al., 2005; Holmes et al., 2008). However, cerebral amyloid angiopathy may have been (transiently) increased together with microhemorrhages (Boche et al., 2008). Neurofibrillary tangles were not reduced. Clinically, modest improvements were observed in some memory tests, while other measures did not change significantly after 1 year (Gilman et al., 2005). Recent reports of patients followed for several years are conflicting as they describe either no change in disease progression (Holmes et al., 2008) or a reduction in functional decline (Vellas et al., 2009). Studies with Aβ monoclonal antibody bapineuzumab indicated an amyloid reduction detected by PET imaging (Rinne et al., 2010) and possible beneficial cognitive and functional outcomes but also vasogenic edema in some patients (Salloway et al., 2009).
Early clinical trials involving immunization with Aβ1–42 were stopped due to the development of aseptic meningoencephalitis in 6% of the treated patients (Orgogozo et al., 2003). Autopsy studies of affected patients demonstrated a T-cell-mediated autoimmune response (Nicoll et al., 2003; Ferrer et al., 2004) presumably due to the use of full-length Aβ, which contains T-cell epitopes in the C-terminal region of the molecule (Monsonego et al., 2003). Consequently, different N-terminal Aβ peptides were used ending between amino acids 11 and 28 (Ghochikyan et al., 2006; Maier et al., 2006; Muhs et al., 2007; Petrushina et al., 2008).
In the present report, we describe the active Aβ immunotherapy CAD106 designed to avoid an activation of Aβ-specific T-cells. CAD106 contains only the short Aβ1–6 peptide but was efficacious in reducing the amyloid accumulation in APP transgenic mice without evidence of unwanted side effects and is currently tested in AD patients (Phase II).
Materials and Methods
APP transgenic mice.
The transgenic mice used express human APP751 under control of the murine Thy-1 promoter. In APP23 mice (Sturchler-Pierrat et al., 1997), transgenic APP contains the “Swedish” (K670N/M671L) mutation, whereas both the Swedish and the “London” (V717I) mutation are combined in APP24 (Abramowski et al., 2008). Expression is neuronal, largely brain-specific, and exceeds endogenous mouse APP expression by ∼7-fold (APP23) or 3.5-fold (APP24). APP23 deposit mostly compact amyloid plaques starting at ∼6 months of age in the neocortex, followed by hippocampus, thalamus (∼9 months), and caudate–putamen (∼17 months), but later also develop vascular amyloid deposits and microhemorrhages (Calhoun et al., 1998; Phinney et al., 1999; Winkler et al., 2001; Bondolfi et al., 2002). APP24 mice show an increased Aβ42/Aβ40 ratio due to the London mutation and develop mostly diffuse amyloid deposits starting at ∼8 months in neocortex (hippocampus, ∼10 months; thalamus, ∼13 months; caudate–putamen, ∼14 months). They contain less vascular amyloid than APP23. All mice were hemizygous for the transgene, on the C57BL/6 background, and female with the exception of study 3, in which males were used. The experiments were performed at Novartis Pharma AG (Basel, Switzerland) in compliance with protocols approved by the Animal Care and Use Committees of the Kanton Basel, Switzerland.
Immunization, bleeding, and tissue sampling of mice.
Mice were immunized subcutaneously with 25 μg of CAD106, 25 μg of Qβ, 100 μg of Aβ1–42 (Bachem AG) or PBS (vehicle) per mouse. For the dose–response study, female C57BL/6 mice (10 per group) were injected with 25, 75, 225 μg of CAD106 per mouse. While CAD106 and Qβ were administered in PBS, Aβ1–42 was injected with complete Freund's adjuvant (first injection), incomplete Freund's adjuvant (second to fourth injections), or PBS (fifth to eighth injections). For passive immunization, each mouse received 500 μg of β1 antibody (Schrader-Fischer and Paganetti, 1996) intraperitoneally. To obtain sera, animals were bled via the tail vein before the first immunization (preimmune serum) and during the study. At the end of the experiment, mice were bled and brains were collected. One hemisphere was fixed in 4% formaldehyde solution. For biochemical analyses, the forebrain was prepared from the second hemisphere by removal of olfactory bulb, cerebellum, and brainstem, immediately frozen on dry ice, and stored at −80°C.
Determination of Aβ antibody titers.
Antibody titers were measured by standard ELISA on plates coated with the Aβ1–40 peptide (0.1 μg/well). Serum dilutions from 1:100 to 1:10,000,000 were used and detected with goat anti-mouse IgG (H+L)-alkaline phosphatase (Jackson ImmunoResearch; 115-055-146) or rabbit anti-monkey IgG (whole molecule)-alkaline phosphatase (Sigma-Aldrich; A-1929) and p-nitro-phenylphosphate (Sigma-Aldrich; N-2765) as substrate. Mouse monoclonal antibody β1 was used for the calibration curve. For monkey sera, a chimeric β1 version with a human Fc part was used. Titers were expressed in equivalents of micrograms per milliliter of the β1 antibody. The lower limit of detection was 0.1 μg/ml.
Cytokine determination by Elispot assay (T-cell activation).
Female BALB/c mice (six per group) were immunized three times with Aβ1–42/Freund's adjuvant as positive control (QS21 as in AN-1792 was not available) or nonadjuvanted CAD106 as used throughout the study. Spleen cells were prepared 10 d after final dose of vaccine. Plated cells were stimulated in vitro for cytokine release as described by Cribbs et al. (2003) using 20 μg/ml Aβ1–40, 40–1, 1–6-GGC, and 6–20 (Bachem AG). Positive spots were quantified by mouse IFN-γ Elispot kit (ALP) (Mabtech; 3321-2A) according to the manufacturer's recommendations.
Cytokine and chemokine quantification in brain.
Supernatants (100,000 × g; 15 min; 4°C) from brain homogenates in TBS (Abramowski et al., 2008) were prepared and cytokine or chemokine protein levels measured using a multiplexed particle-based flow cytometric cytokine assay (Vignali, 2000). Kits were purchased from Bio-Rad and R&D Systems. The procedures closely followed the manufacturer's instructions. The analysis was conducted using a conventional flow cytometer (Guava EasyCyte Plus; Millipore).
Determination of Aβ in forebrain.
Brain samples were homogenized, formic acid was extracted, and Aβ40 and Aβ42 were determined as described (Abramowski et al., 2008) using ELISAs and electrochemiluminescence-linked immunoassays (Meso Scale Discovery).
Immunohistochemistry.
Brain histology and assessment of cerebral amyloid plaque load were done as described previously (Sturchler-Pierrat et al., 1997; Abramowski et al., 2008) using paraffin embedding and microwave-treated sections stained with Aβ antiserum NT12, biotinylated anti-rabbit IgG secondary antibody (Vector Laboratories; BA1000), and the avidin–biotin peroxidase technique (ABC-Elite kit; Vector Laboratories; PK6100) with DAB (Roche Diagnostics; 11718096001) as substrate. To analyze different Aβ isoforms, fibrillar Aβ deposits, or blood vessel basement membranes, brain sections processed as above were stained with the following antibodies: rabbit polyclonal antibody to Aβ (NT12), rabbit antiserum to collagen type IV (Fitzgerald Industries; RDI-600-401-106), and mouse monoclonal antibodies to the C termini of Aβ40 (25H10) and Aβ42 (29C12; Nano-Tools). Aβ antigenicity was enhanced by microwave heating; sections for collagen staining were pretreated with 0.1% Pronase (20 min at 37°C). Indirect tyramide signal amplification was used for fluorescence visualization (TSA Plus kit NEL 741/TSA NEL 701; PerkinElmer). To test for lymphocytes, microwave-treated sections were stained with a rat anti-CD45R/B220 antibody (PharMingen; 550286) for B-cells followed by biotinylated anti-rat IgG (BA 4000; Vector Laboratories) and the ABC system. T-cells were labeled with a rat anti-CD3 antibody (Serotec; MCA1477), anti rat secondary antibody (DAKO; P0450), and the tyramide signal amplification system. Sagittal brain sections from three different anatomical levels, 100–150 μm apart, were evaluated by microscopical inspection. The amyloid plaque load was quantified using an imaging system (Abramowski et al., 2008), while Aβ-positive neocortical vessels and T- or B-cells were counted manually. Microhemorrhages were evaluated after Perl's Berlin blue staining (Carson, 1996) using 12 sections per brain to visualize ferric iron in hemosiderin as previously described (Winkler et al., 2001; Pfeifer et al., 2002).
Immunization of rhesus monkeys.
Four male rhesus monkeys (10–15 years) were initially immunized with 50 μg of CAD106 subcutaneously, followed by immunizations with 25 μg of CAD106 intramuscularly at days 28, 54, and 110. Blood was collected at various times for antibody titer determination. The study was conducted at the Novartis Forschungsinstitut (Vienna, Austria) in compliance with the Animal Care Committee-approved animal license and standard operation procedures. Antisera obtained after the third and fourth immunizations from the two monkeys with the highest titers were pooled and affinity-purified on a SulfoLink resin (Pierce; 20401) coupled with Aβ1–6-GGC according to the manufacturer's instructions.
Aβ oligomer preparation and Aβ toxicity assay.
Aβ oligomers were produced according to Lambert et al. (2001) from monomerized Aβ1–42 (Bachem) and photochemically cross-linked as described by Bitan et al. (2001). Different dilutions of the centrifuged oligomer preparation and the Aβ antibodies were added into the culture medium of PC12 cells. After 24 h, cell viability was assessed by MTT (Shearman et al., 1995).
Statistical analyses.
Effects on amyloid plaque load and the corresponding total Aβ concentrations were analyzed with the nonparametric Mann–Whitney test as these data were not normally distributed. Student's t test was used to evaluate differences in APP, secreted APP (sAPP) or C-terminal fragments, Aβ cytotoxicity, and microhemorrhage number and severity, and the paired t test for T-cell stimulation assays. Animal number comparisons (microhemorrhages, T- or B-cells) were analyzed with Fisher's exact test. Two-tailed tests were used, and p < 0.05 was considered significant.
Results
To avoid activating Aβ-specific T-cells, we chose the Aβ1–6 peptide (DAEFRH) as antigen, which is shorter than typical T-cell epitopes (Rammensee et al., 1993; Rötzschke and Falk, 1994; Rammensee, 1995) and resides outside the region of Aβ reacting with T-cells (Monsonego et al., 2003). It was extended by a spacer (GGC) and covalently conjugated to the virus-like particle (VLP) derived from Escherichia coli RNA phage Qβ. Each VLP contains ∼350–550 Aβ peptides. The VLP carrier provides both a scaffold for ordered presentation of the antigen and T-helper cell epitopes (Bachmann et al., 1993). Screening of the Aβ1–6 sequence including the spacer GGC against the SYFPEITHI database (http://www.syfpeithi.de/) comprising >4000 peptide sequences known to bind class I and class II MHC molecules did not identify any obvious or typical T-cell epitopes (data not shown).
Immunization with CAD106 does not activate Aβ-specific T-cells in vivo
A dose–response study conducted in old wild-type mice immunized three times with CAD106 indicated that saturation of the Aβ antibody response was achieved with dose levels between 75 and 225 μg (data not shown). The 25 μg dose was close to but not within the plateau and therefore was chosen for all further studies in mice.
The presence of activated Aβ-specific T-cells was checked for following three immunizations of mice with Aβ1–42/Freund's adjuvant or CAD106. Ten days after the last injection, when all animals had developed Aβ antibody titers, isolated spleen cells were stimulated with different Aβ peptides and interferon-γ-secreting T-cell spots quantified (Fig. 1). All CAD106-immunized mice showed background values regardless of the peptide used for stimulation. In contrast, vaccination with Aβ1–42 resulted in a threefold to fourfold increase in the number of Aβ-reactive T-cells after in vitro stimulation with Aβ1–40 or Aβ6–20, both of which contain T-cell epitopes (Cribbs et al., 2003; Monsonego et al., 2003). As expected, no stimulation was found with the reverse peptide (Aβ40–1) as well as the Aβ antigen of CAD106 (Aβ1–6-GGC), confirming the lack of a T-cell epitope on this peptide. Immunization with CAD106 stimulated Qβ-reactive T-cells, which is required for T-cell help (data not shown).

Analysis of Aβ-specific T-cells in mice immunized with CAD106 or Aβ1–42. Wild-type mice were immunized three times with CAD106 (n = 6) or Aβ1–42/Freund's adjuvant (n = 6). Ten days thereafter, spleen cells were isolated and stimulated with DMSO (background) or Aβ peptides 1–40 (relevant antigen), 40–1 (reverse peptide), 1–6-GGC (Aβ epitope of CAD106), and 6–20 (Aβ fragment with T-cell epitope). Interferon-γ-secreting spots were quantified to determine T-cell stimulation. Mean number of spots ± SEM per 250,000 cells is given. *p < 0.05 versus DMSO (paired t test).
Generation of Aβ-specific antibodies in APP transgenic mice
Antibody responses were determined in all animals used for studies on amyloid deposition. Mice in the test groups received CAD106, while those in the control groups were administered PBS (Table 1). Aβ antibody titers in nonimmunized control animals were below the limit of detection. In contrast, all CAD106-immunized mice developed Aβ-specific IgG at least 10-fold above the detection limit. Median titers in the immunized groups were in the 10–110 μg/ml range from the third injection onward and, as expected for active immunization, varied somewhat throughout the studies. No difference in antibody response was apparent between the APP23 and APP24 transgenic mice and wild-type mice (Table 1) (data not shown).
Efficacy studies with CAD106 in APP transgenic mice
In CSF, Aβ antibodies were either not measurable or just above the limit of detection (data not shown). As minor contaminations with blood during mouse CSF sampling cannot be excluded, these results indicate only a low level of Aβ antibodies in the CSF compartment.
The antibody response to CAD106 showed a classical IgM to IgG shift (data not shown). Thereafter, Aβ-specific antibodies of all IgG subclasses were found with some variation in subclass ratio noted between animals. With few exceptions, the subclass pattern of each animal remained relatively constant with time (data not shown). The antibodies recognized Aβ amino acids 3–6, (E)FRH, as minimal epitope (data not shown).
Reduction of amyloid deposition after CAD106 immunization of APP24 mice
To first test CAD106 in a preventive setting, APP24 mice were immunized before the onset of plaque formation, at 7.5 months of age (study 1). At the end of the study (15.7 months), plaque number and relative area covered by plaques in the neocortex were reduced compared with PBS-treated animals by 80 and 78%, respectively (Table 1). Larger reductions up to 100% were found in thalamus and caudate–putamen (Table 2). For comparison, a group of mice was immunized in parallel with aggregated Aβ1–42 in Freund's adjuvant. Tenfold lower Aβ antibody titers were obtained (data not shown) in agreement with a reduced response against Aβ1–42 in C57BL/6 mice (Das et al., 2003), and amyloid deposition in the cerebral cortex and hippocampus was unaffected. However, despite the lower titers, there was still a remarkable reduction in plaque load in the thalamus (number, −62%; area, −79%) and caudate–putamen (number, −79%; area, −84%). These reductions were slightly lower than those observed with CAD106 in the corresponding regions.
Plaque load reduction in different brain regions of APP transgenic mice after treatment with CAD106
In the second study, a first cohort of APP24 mice (study 2a) was treated from 9.5 until 19.5 months starting right after the onset of amyloid deposition in the neocortex. The reduction in plaque number and area was comparable with the first study (Tables 1, 2). This cohort also included a group of animals immunized with the carrier Qβ in the absence of Aβ sequences. No effect on amyloid plaque deposition was found (Fig. 2). A second cohort (study 2b) received injections only from 13.5 months onward, at which time a considerable amount of amyloid has already been deposited in cortex and hippocampus, but was also terminated at 19.5 months. Treatment at this advanced stage of plaque formation led to highly significant reductions as well (Tables 1, 2), although the effects were considerably less pronounced. In the neocortex, a 57% reduction of plaque number and a 32% reduction of plaque area were found. Very strong effects were again observed in the caudate–putamen, where amyloid deposition starts latest: 92% reduction of the plaque area and 94% reduction in plaque number. Visual inspection of the brain sections indicated that diffuse amyloid was affected to a larger extent than compact amyloid in all studies (Fig. 3).

Effect of CAD106 or the carrier Qβ on amyloid plaque load in APP24 mice. Animals were immunized for 10 months (9.5–19.5 months of age; study 2a) with CAD106 or Qβ VLP; controls received vehicle (PBS). A, Neocortical plaque number. B, Neocortical plaque area. Plaque number is expressed as plaques per square millimeter, and plaque area in percentage of total cortex area. The boxes define the 25th and 75th percentiles, with a line at the median and error bars defining the 10th and 90th percentiles. ***p < 0.001 CAD106 versus PBS or Qβ (Mann–Whitney rank sum test).

Amyloid deposition in neocortex of APP24 mice after CAD106 or vehicle treatment. Amyloid staining (Aβ antiserum NT12) of neocortex sections from mice treated between 9.5 and 19.5 months (study 2a) with PBS (A) or CAD106 (B). Note the decrease of diffuse parenchymal amyloid plaques, the compact appearance of most remaining plaques, and the increase of vascular amyloid in (B). Cerebral amyloid angiopathy in parenchymal vessels (arrows) and a single meningeal vessel (arrowhead) are shown. Scale bar, 100 μm.
In a further study (3), very old mice with a high amyloid load were treated from 21.5 to 25.7 months, which resulted in a borderline reduction in plaque number (−15%) but not plaque area in neocortex, indicating a small effect on newly formed deposits (Table 1).
CAD106 treatment significantly reduced total forebrain Aβ42 (study 1, −46%; study 2a, −37%; study 2b, −32%) compared with control groups (Table 1). Smaller reductions (−6 to −20%), not always reaching significance, were found for Aβ40 (data not shown). However, immunization of very old mice did not result in a significant reduction of either Aβ peptide (study 3).
Forebrain homogenates from study 2 were also analyzed for an effect of CAD106 immunization on APP and its metabolites. No significant change in the level of full-length APP or the secreted N-terminal fragments sAPPα and sAPPβ or the C-terminal fragments C99 and C83 was found (data not shown). This indicates that the antibodies induced by CAD106 did not alter APP metabolism in vivo, in contrast to their effect on amyloid deposition.
Amyloid lowering effect of CAD106 in APP23 mice
APP23 mice were immunized with CAD106 during preplaque to advanced stage of amyloid deposition (between 2.9 and 18.5 months; study 4). Compared with PBS controls, the number of amyloid plaques and plaque area were lower in all four brain regions examined (Tables 1, 2). Reductions in a similar range (20–40%) were found in the cerebral cortex, hippocampus, and thalamus. There was almost no amyloid deposition in caudate–putamen. In agreement with these histological findings, the overall Aβ42 reduction in forebrain was 33%. No significant effect on Aβ40 was found. As observed in APP24 mice, immunization with Qβ did not affect any of the measures for Aβ deposition (data not shown).
Old mice with very advanced amyloid plaque pathology were used to compare active and passive immunization approaches (study 5). Mice were immunized between 17.5 and 22.7 months of age with either CAD106 or the monoclonal antibody β1 (500 μg/mouse weekly, i.p.). With this dose, the β1 group had approximately five times higher antibody titers until they had declined by study end. In neocortex, both CAD106 and β1 reduced the number of plaques by 32 and 42% (p < 0.00001), respectively, compared with the control group (Fig. 4). The total cortical plaque area was slightly lower with both treatment regimens, amounting to −9% for CAD106 (nonsignificant, p = 0.08) and to −14% for β1 (p < 0.01). Visual inspection indicated that small-sized plaques were reduced, while medium- and large-sized plaque numbers remained unchanged.

Effect of CAD106 or antibody β1 on amyloid plaque load in aged APP23 mice. The animals were treated with CAD106, monoclonal Aβ antibody β1, or vehicle (PBS) from 17.5 to 22.5 months of age (study 5). Neocortical plaque number (A) and plaque area (B) are shown as described in Figure 2. ***p < 0.001, **p < 0.01, #p = 0.08, CAD106 or β1 versus PBS (Mann–Whitney rank sum test).
To confirm this, APP23 mice of similar age (16–22.2 months) were immunized with CAD106 (study 6). Consistent with the results of study 5, a 47% reduction in amyloid plaque number (p < 0.001) was found in the neocortex but the plaque area was not significantly reduced (Table 1). Quantification according to plaque size demonstrated a selective reduction of small Aβ plaques (plaque area, 1–500 μm2) (Fig. 5). These contribute relatively little to the total plaque area, which explains the discrepancy between the effects on plaque number and area. In line with this finding, treatment with CAD106 or β1 did not significantly reduce the amount of Aβ40 and 42 in these very old mice (Table 1).

Size distribution of amyloid plaques in CAD106 and vehicle-treated APP23 mice. Animals were immunized from 16 to 22.2 months of age (study 6). The number of plaques ± SEM in different size groups was determined in the cerebral cortex. Only plaques >1 μm2 were considered to assure a reliable determination. *p < 0.05, ***p < 0.001, CAD106 versus PBS (t test).
Differential effect of CAD106 treatment on vascular Aβ
Inspection of brain sections from CAD106-treated animals indicated an overall lowering of amyloid deposits, while vascular amyloid remained constant or even increased relative to controls (Fig. 3). Quantitative analysis showed an elevation in number of Aβ-containing blood vessel profiles in some but not all studies with CAD106-immunized APP24 mice (Fig. 6A). Only a very minor fraction of the blood vessels was associated with Aβ in APP24 mice (data not shown). This low frequency of vascular Aβ may explain the variation between these studies. Further characterization (study 1) indicated that CAD106 increased the deposition of Aβ42 but not of Aβ40 in the vasculature (Fig. 6B). Moreover, Congo red-stained vessels were not increased. The additional vascular Aβ seemed mainly nonfibrillar, which differs from the fibrillar amyloid typically deposited in blood vessels.

Effect of CAD106 immunization on vascular Aβ in APP24 mice. Aβ deposits in neocortical blood vessels were labeled as described below. The number of positive vessels (mean ± SEM) is given as percentage of vehicle (PBS). A, Total Aβ-containing blood vessels (NT12 antiserum) in different CAD106 studies (1–3) as indicated by the treatment period. B, Blood vessels stained for total Aβ (NT12), Aβ40 (25H10), Aβ42 (29C12), and fibrillar amyloid (Congo red) in animals from study 1. **p < 0.01, *p > 0.05, CAD106 versus PBS (Mann–Whitney rank sum test).
No significant increase in amyloid-containing blood vessels was found in the CAD106-treated APP23 mice, although a nonsignificant elevation was observed after 2.9–18.5 months of treatment (data not shown).
No evidence of increased microhemorrhages following CAD106 immunization of APP24 and APP23 mice
Vascular amyloid is a risk factor for microhemorrhages and an increase has been observed after Aβ immunization (Pfeifer et al., 2002; Wilcock et al., 2004, 2007; Racke et al., 2005). To investigate whether this occurred following immunization with CAD106, brains from the above studies were analyzed by hemosiderin staining and data from the same APP transgenic line were pooled to obtain sufficiently large groups. In APP24 mice, only a single microhemorrhage was found in a Qβ control animal, which is in agreement with the rare occurrence of microbleeds in this line (data not shown). In contrast, aged APP23 mice had more microhemorrhages, but there was neither an increase in frequency nor severity in CAD106-treated animals (Table 3). Separate analysis of the studies also did not show any treatment-related increases.
Microhemorrhage frequency and severity in APP23 mice immunized with CAD106
Proinflammatory cytokines and chemokines
Brain homogenates were also analyzed for the proinflammatory cytokines TNFα and IL-12 (p40) as well as for the chemokines RANTES (regulated on activation normal T cell expressed and secreted) and KC (keratinocyte-derived chemokine)/GRO (growth-related oncogene). IL-1β and IL-6 were below the detection limit. Mice (8 per group) with antibody titers above the median were chosen from studies 1, 2a, 3, 4, and 6. Quantification did not show any changes the CAD106 as compared with the control groups (data not shown).
Evaluation of T-cell and B-cell infiltration
Brains from study animals were analyzed for T- and B-cells to identify potential inflammatory cell infiltration. Perivascular lymphocytes were only occasionally detected in aged, nontransgenic or APP transgenic mice, and immunization with CAD106 did not increase the incidence of these cells (data not shown). However, 1 of 116 CAD106-treated animals had frequent T- and B-cell infiltrates (study 2a). The infiltrates were present throughout the brain and not restricted to amyloid-containing regions. This is in contrast to the described meningoencephalitis associated with brain amyloid in an APP transgenic mouse passively immunized with an Aβ antibody (Lee et al., 2005). The brain homogenate from the animal in our study was analyzed by PCR to identify an infectious agent but no positive result was obtained. Therefore, the relevance of this singular observation remains unclear.
CAD106-induced rhesus monkey antibodies react with different Aβ aggregation states and block its toxicity in vitro
Rhesus monkeys were immunized with CAD106 because they have the same Aβ sequence as humans and their immune system can be considered as closely related. All four immunized monkeys developed Aβ IgG antibodies. The reversibility of the Aβ-specific antibody response was evaluated after the final immunization and was found to decline in all animals with a half-life in the range of 2–4 weeks.
Reactivity of the monkey antibodies with amyloid plaques, APP, and other Aβ-containing polypeptides in brain was investigated by immunohistochemistry. Affinity-purified, pooled antibodies stained plaques on brain sections from APP23 mice and human AD patients (data not shown). No cross-reactivity with cellular brain structures was detected.
To further test reactivity against Aβ oligomers, CAD106-induced monkey Aβ antibodies were tested for their ability to recognize synthetic oligomers on Western blots (Fig. 7A). This analysis demonstrated binding of the antibodies to different higher-molecular-weight Aβ species representing oligomers. Reactivity with oligomers was independent of the method or the Aβ isoform (Aβ1–40 or 1–42) used for preparation (data not shown). Aβ monomers were also recognized.
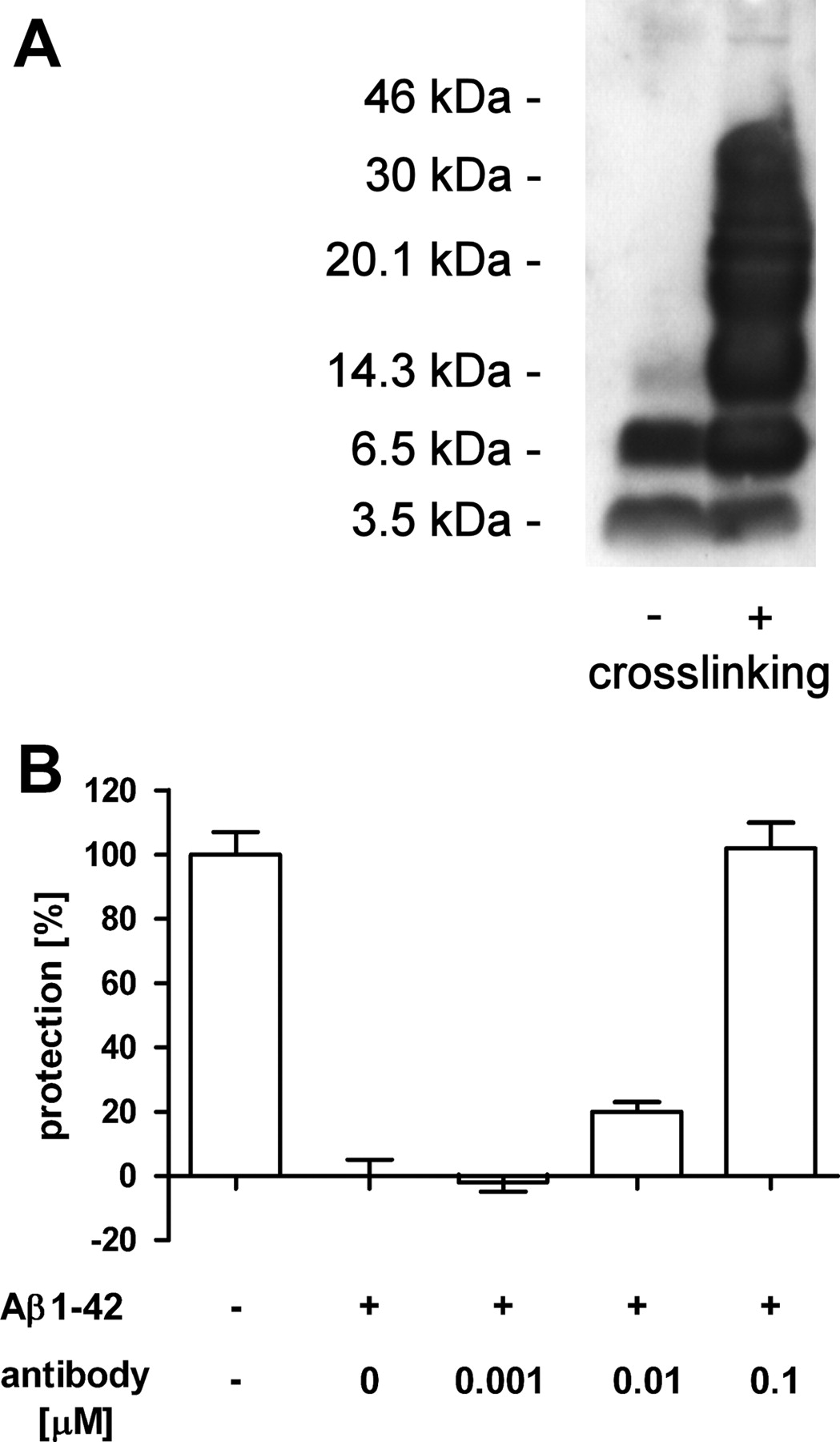
Rhesus monkey antibodies induced by CAD106 react with Aβ oligomers and protect against Aβ toxicity. A, Western blot of SDS-stable Aβ1–42 oligomers without (left lane) or with chemical cross-linking (right lane). CAD106-induced antibodies react with Aβ monomers (3.5 kDa) as well as oligomers of increasing size. No reactivity was observed with preimmune sera (data not shown). B, Aβ1–42 oligomer toxicity assay using PC12 cells (control, nontreated). CAD106 antibodies from monkeys reverted the oligomer-induced toxicity in a concentration-dependent manner. The concentration of Aβ1–42 oligomers was 0.1 μm (normalized for monomeric Aβ). Error bars indicate SEM.
The antibodies were also tested for neutralization of Aβ in an in vitro toxicity assay. As shown in Figure 7B, they blocked Aβ1–42 oligomer-induced toxicity to PC12 cells in a concentration-dependent manner. The IC50 was equal to the antibody titer. Similar results were obtained when cytotoxicity was induced with fibrillar Aβ1–40 (data not shown).
Discussion
In this study, we have characterized the second-generation active Aβ immunotherapy CAD106, a vaccine designed to avoid undesired inflammatory side effects while inducing efficacious antibodies against Aβ. We demonstrated that the induced antibodies interfere with amyloid deposition in cortical and subcortical brain regions, bind to Aβ aggregates, and block their toxicity to cells.
All mice and rhesus monkeys immunized with CAD106 developed Aβ antibody titers regardless of their age. This finding is remarkable considering that Aβ1–6 is a small autoantigen in human APP transgenic mice and monkeys and that CAD106 was used without additional adjuvant. Immunogenicity of the vaccine is provided by its highly repetitive structure, provision of T-cell help, and optimal size for diffusion into the lymph node (Bachmann and Jennings, 2010). The antibodies recognized a short sequence, Aβ3–6, as minimal epitope. Because all main IgG subclasses were generated, the entire range of effector functions is possible for CAD106-induced Aβ antibodies. This seems advantageous as the mechanism of Aβ immunotherapy remains unclear and the IgG subclass requirements are controversial (Klafki et al., 2006). When immunization was stopped, Aβ antibody titers declined regardless of the presence of endogenous Aβ, presumably due to the lack of T-cell help once the vaccine cleared. Discontinuation of CAD106 treatment thus leads to a decreased antibody exposure with a similar kinetic to passive immunotherapy.
Immunization with the VLP Qβ alone did not effect amyloid deposition. This finding together with the absence of Aβ-specific T-cell activation demonstrates that the efficacy of CAD106 is mediated by Aβ-specific antibodies. This conclusion is in accord with previous studies involving passive immunotherapy (Bard et al., 2000) and our data with monoclonal antibody β1. In a direct comparison with CAD106, the higher Aβ antibody titers reached after β1 infusion resulted in slightly larger group effects. Conversely, lower titers obtained with Aβ1–42 immunization led to smaller effects suggesting a titer–response relationship. Nevertheless, a correlation between Aβ antibody titers and the effect on amyloid deposition could not be established. The high intrinsic variation of the amyloid deposition process together with the variation of titer over time did not allow establishing such a relationship.
Our efficacy studies further indicate that the reduction in amyloid accumulation after Aβ immunotherapy with CAD106 is inversely related to Aβ levels in the brain. First, larger effects were generally observed in APP24 mice, which have lower soluble Aβ levels and deposit less amyloid than APP23 (Abramowski et al., 2008). Second, the magnitude of the effect on amyloid (plaque number and area) in both transgenic lines differed between the regions analyzed. The strongest reduction was in the caudate–putamen followed by thalamus, hippocampus, and cerebral cortex. Human APP expression and hence Aβ increase considerably in the reverse order (Sturchler-Pierrat et al., 1997).
A comparison of different readouts for amyloid reduction revealed the largest effect was measured for plaque number followed by plaque area, total forebrain Aβ42, and total forebrain Aβ40 levels. This rank order was most apparent when treatment started at plaque-bearing age, whereas differences vanished for preventive immunizations. Analyses in old, plaque-bearing mice demonstrated a reduction of small plaques, which contribute little to plaque area and Aβ peptide levels. It is conceivable that these small plaques are newly formed deposits and that Aβ immunotherapy with CAD106 or monoclonal antibody β1 has a larger effect on the initiation of amyloid deposition. Further supporting this notion was the observation that larger effects were found when immunization started before amyloid deposition. Others have also described that immunotherapy used in a preventive mode rather than in a therapeutic setting is more efficacious (Das et al., 2001). Similar results were obtained following reduction of Aβ generation with a γ-secretase inhibitor (Abramowski et al., 2008), indicating that de novo amyloid deposition is more sensitive to lowering of free Aβ than its assembly onto preexisting deposits as observed during in vitro assembly studies.
Surprisingly, immunization with CAD106 or monoclonal β1 reduced Aβ42 more strongly than Aβ40 even though the respective antibodies do not discriminate between either isoforms and the two APP transgenic lines used differ in their Aβ42/Aβ40 ratio. In line with this finding, we also showed diffuse amyloid deposits consisting mainly of Aβ42 were principally removed. Additionally, the lack of effect on vascular Aβ may have contributed to this finding as it is dominated by Aβ40.
Analysis of vascular amyloid showed increases in those APP24 mouse studies, in which a strong reduction of total amyloid accumulation was obtained. The relative increase (up to 2.5-fold) must be considered in relation to the rare occurrence of vascular amyloid in these animals. Studies in old APP24 and in APP23 mice, in which amyloid lowering was less, showed no elevation of vascular amyloid, suggesting an inverse relationship between overall amyloid removal and vascular deposition. A mechanistic link is supported by the observed increase of Aβ42 but not Aβ40 and its deposition in diffuse, nonfibrillar form, which is atypical for vascular amyloid. Similar deposition of vascular Aβ42 was observed in humans during postmortem analysis of the active Aβ1–42 immunotherapy study AN1792 (Boche et al., 2008). Both results contrast with the vascular amyloid reduction found after passive immunization with an N-terminal Aβ antibody in PDAPP mice (Schroeter et al., 2008). This transgenic APP line deposits vascular amyloid primarily in meninges, while it was mostly parenchymal in our and the human studies. Differences in the models as well as antibody qualities may have contributed to these different outcomes.
Various APP transgenic mouse studies have demonstrated increases in microhemorrhages following Aβ immunotherapy (Pfeifer et al., 2002; Wilcock et al., 2004, 2007; Racke et al., 2005; Schroeter et al., 2008). These were associated with both, elevation (Pfeifer et al., 2002; Wilcock et al., 2004, 2007; Racke et al., 2005) or lowering (Schroeter et al., 2008) of vascular Aβ. The elevation of vascular Aβ in some CAD106 immunization studies did not lead to an increase in microhemorrhages. The nonfibrillar nature of the additional vascular Aβ, the slow antibody titer increases with active immunotherapy or antibody modifications (Wilcock et al., 2006) may have contributed to this result. In addition, no evidence was found for brain infiltration of lymphocytes or meningoencephalitis. Our expectation that immunization with CAD106 would not induce Aβ-specific T-cells because of the minimal length of the peptide antigen and lack of typical T-cell epitopes was confirmed by experimental analysis.
CAD106-induced antibodies selectively stained amyloid plaques on brain sections from human AD patients and APP transgenic mice. No cross-reactivity was observed with cellular APP or its metabolites. CAD106 treatment did not alter the steady-state level of full-length APP or the metabolites sAPPβ and C99 containing the Aβ1–6 sequence. These observations argue against a significant interaction of the antibodies with APP and its primary metabolites in vivo. Hence, the reduction of Aβ deposition is unlikely to be due to interference with APP processing as observed with a β1 intrabody (Paganetti et al., 2005). Accordingly, our data demonstrate that CAD106-induced antibodies bind to different aggregation states of Aβ including toxic oligomers and fibrils. They further show the antibodies are able to neutralize toxicity induced by these assemblies in a cellular assay.
In conclusion, CAD106 induces efficacious Aβ antibody titers in animals while avoiding potential mechanism-related side effects. The antibodies effectively interfere with amyloid accumulation in brain and protect from Aβ toxicity. CAD106 is currently tested in AD patients as a potentially disease-modifying agent.
Footnotes
-
C.W., K.-H.W., A.C.T., P.F., S.D., L.H.J., G.T.J., R.L., R.O., J.R., M.Z., A. M., M.F.B., and M.S. are or have been employees of either Novartis Pharma AG or Cytos Biotechnology AG.
-
We acknowledge P. Meyer and B. Stumper for immunizing the rhesus monkeys. We also thank our colleagues for many helpful discussions and technical support throughout the study as well as for critical reading of this manuscript.
- Correspondence should be addressed to Dr. Matthias Staufenbiel, Novartis Pharma AG, Forum 1, Novartis Campus, CH-4056 Basel, Switzerland. matthias.staufenbiel{at}novartis.com





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}