

Abstract
In this study, we evaluated whether a cross talk between nuclear factor κB (NF-κB) and Notch may take place and contribute to regulate cell morphology and/or neuronal network in primary cortical neurons. We found that lack of p50, either induced acutely by inhibiting p50 nuclear translocation or genetically in p50−/− mice, results in cortical neurons characterized by reduced neurite branching, loss of varicosities, and Notch1 signaling hyperactivation. The neuronal morphological effects found in p50−/− cortical cells were reversed after treatment with the γ-secretase inhibitor DAPT (N-[N-(3,5-difluorophenacetyl)-1-alanyl 1]-S-phenylglycine t-butyl ester) or Notch RNA interference. Together, these data suggested that morphological abnormalities in p50−/− cortical neurons were dependent on Notch pathway hyperactivation, with Notch ligand Jagged1 being a major player in mediating such effect. In this line, we demonstrated that the p50 subunit acts as transcriptional repressor of Jagged1. We also found altered distribution of Notch1 and Jagged1 immunoreactivity in the cortex of p50−/− mice compared with wild-type littermates at postnatal day 1. These data suggest the relevance of future studies on the role of Notch/NF-κB cross talk in regulating cortex structural plasticity in physiological and pathological conditions.
Introduction
Functional and structural plasticity, often referred to as “neuroplasticity,” is a fundamental brain property involving chemical, electrical, molecular, and cellular responses and leading to connection reorganization within and/or between brain regions. Potentially, a better understanding of the molecular events participating in neuroplasticity may provide relevant information for innovative therapeutic approaches in a variety of neurological disorders, including Alzheimer's disease, depression, autism, and schizophrenia, which have been associated with deregulated neuroplasticity (van Spronsen and Hoogenraad, 2010).
Among others, two important players involved in neuroplasticity regulation are nuclear factor κB (NF-κB) and Notch signaling pathways. NF-κB proteins are ubiquitously expressed transcription factors that play different roles depending on the cellular context in which they act and the dimer subunit composition. Their functional roles include regulation of nerve cell survival and plasticity, immune and inflammatory responses, proliferation, neurogenesis, apoptosis, angiogenesis, and oncogenesis (Grilli and Memo, 1999; Mattson and Meffert, 2006; Kaltschmidt and Kaltschmidt, 2009). NF-κB-dependent transcriptional activity becomes relevant in neurons when axons and dendrites grow and synapses are formed, and remains high under basal conditions in most regions of the adult brain (Mattson, 2005). NF-κB signaling is also known to promote neurite outgrowth and to enhance the size and complexity of neuronal processes in the developing nervous system and in cultured neurons (Gutierrez et al., 2005; Gavaldà et al., 2009; Gutierrez and Davies, 2011). As for many other complex biological processes, the NF-κB signaling pathway is likely to work in concert with others to regulate nerve cell survival and plasticity.
Notch is recognized as a key player in neurodevelopment (Artavanis-Tsakonas et al.,1990; Williams et al., 1995; de la Pompa et al., 1997), whereas in adulthood it appears to be involved in synaptic plasticity regulation and in “morphological maturation” of terminally differentiated neurons (Berezovska et al., 1999; Redmond et al., 2000; Wang et al., 2004). It has been previously shown that Notch activation induces neurite remodeling in different experimental models of neuronal cells by acting on cytoskeletal structures and modulates the expression of genes whose products are responsible for contact-dependent inhibition of dendrite outgrowth (Šestan et al., 1999; Ferrari-Toninelli et al., 2008, 2009).
Together, both Notch and NF-κB signalings may potentially contribute by a reciprocal cross talk interaction to regulate neuronal functional and structural plasticity in physiological and pathological conditions. Numerous cellular contexts have been described wherein interaction of Notch and NF-κB signaling pathways takes place (Bash et al., 1999; Wang et al., 2001; Nickoloff et al., 2002; Espinosa et al., 2003; Shin et al., 2006; Osipo et al., 2008; Cao et al., 2010), but clear indications of their functional cross talk in cortical neurons and its potential relevance in CNS disorders remain to be demonstrated (Ang and Tergaonkar, 2007).
In this study, we evaluated whether a cross talk between NF-κB and Notch may take place and contribute to regulate cell morphology and/or neuronal network in primary cortical neurons. The results obtained from in vitro and in vivo experiments support the idea that modulation of Notch/NF-κB cross talk may represent a potential target for pharmacological treatment of deregulated neuroplasticity.
Materials and Methods
Primary cortical neurons cultures.
NF-κB p50−/− (B6;129P2-Nfkb 1tm 1 Bal/J; The Jackson Laboratory) and wild-type (wt) mice (B6;129PF2; The Jackson Laboratory) were maintained in high-efficiency particulate air-filtered THOREN units (THOREN Caging Systems) at the University of Piemonte Orientale animal facility, were kept three to four per cage, and had ad libitum access to food and water (Denis-Donini et al., 2008). Animal treatments were performed in accordance with the National Institutes of Health guidelines and approved by the local institutional animal care and use committee. To obtain primary cortical neurons, embryonic day 15 (E15) cortices were isolated from p50−/− and wt mice, pooled, mechanically dissociated into a single-cell suspension in Neurobasal Medium (Invitrogen) containing 2% B27 supplement (N-B27 medium; Invitrogen), 500 μm glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin (Sigma-Aldrich), and centrifuged for 5 min at 200 × g. Cells were plated onto poly-d-lysine (Sigma-Aldrich)-coated glass coverslips or dishes (5 × 104 cells/cm2) and cultured in N-B27 medium for 7 d in vitro (DIV). We assessed cell viability using the trypan blue exclusion assay, which provides a measure of cell membrane integrity. At 7 DIV, the medium was removed from the dishes and the attached cells were washed three times with PBS (Sigma-Aldrich). Cells were harvested with trypsin–EDTA (Invitrogen) and resuspended in medium. The cells were mixed 1:1 with 20% trypan blue and counted using a hemocytometer. The number of blue (dead) cells was calculated and subtracted to the total cell number. Four separate experiments were performed from four different cell preparations.
Immunofluorescence and confocal analysis.
Cells were plated at a density of 5 × 104 cells/cm2 in a 24-well plate, grown on glass coverslip (coated with poly-d-lysine; Sigma-Aldrich), and then fixed. Cells were incubated in PBS (Sigma-Aldrich) containing 1% bovine serum albumin (BSA) (Sigma-Aldrich) and 0.2% Triton X-100 overnight at 4°C with the appropriate antibody. After rinses, cells were incubated with the secondary antibody in PBS for 1 h at room temperature. Slice were mounted using the Dako Fluorescent Mounting Medium and examined by Zeiss LSM 510 META confocal laser-scanning microscope (Carl Zeiss). Three-dimensional images were performed with the LSM Image Browser software on z-stack scansion images (1 μm interval sections) taken from fixed cells. For in vivo analysis, postnatal day 1 (P1) mice of either sex were killed, and brains were fixed by immersion in 4% buffered formalin. After overnight postfixation, brains were cryopreserved by dehydration in 30% sucrose solution, mounted in embedding medium, and cut in 15-μm-thick serial coronal brain sections using the cryostat. Brain sections were then incubated with primary antibody in PBS solution containing 3% BSA and 0.3% Triton X-100 at 4°C overnight. After rinses, brain sections were incubated with Alexa Fluor-conjugated (Invitrogen) secondary antibody in PBS/BSA (1%) for 1 h at room temperature. Slice were mounted and examined by confocal microscopy.
Branching study.
βIII-Tubulin-labeled neurons were visualized and digitally acquired using a Zeiss LSM 510 META confocal laser-scanning microscope (Carl Zeiss). To quantify the extent and complexity of neuronal processes, the Sholl profile analysis was undertaken (Sholl, 1953). Briefly, a series of concentric rings with regular radial increments (20 μm) centered in the neuronal soma were traced, bifurcation (Bi) and terminal (Ti) points of processes were counted in each ring and the number (Xi) of processes intersecting each ring was calculated using the iterative equation Xi = Xi − 1 + Bi − Ti (Gutierrez and Davies, 2007).
Antibodies.
The following antibodies were used: monoclonal anti-βIII-Tubulin (Promega) (working dilution: 1:1000 for immunofluorescence); monoclonal anti-GAPDH (Millipore) (working dilution: 1:1000 for immunoblotting); monoclonal anti-Notch1 [which recognizes both Notch1 full-length and Notch intracellular domain (NICD)] (Sigma-Aldrich) (working dilution: 1:100 for immunofluorescence and 1:1000 for immunoblotting); polyclonal anti-Jagged1 (Abcam) (working dilution: 1:500 for immunoblotting, 1:100 for immunofluorescence).
Conjugated CY3, CY2 (Jackson ImmunoResearch Laboratories), and FITCH (Sigma-Aldrich) were used as secondary antibodies.
RNA isolation.
Total RNA was isolated from 7 DIV wt and p50−/− cortical cells (∼2.5 × 106 cells) using the RNeasy kit (QIAGEN) and digested with the RNase-free DNase set (QIAGEN), according to the manufacturer's protocol. Quality of RNA samples was tested by RNA electrophoresis to ensure nucleic acid integrity.
Quantitative real-time PCR.
One microgram of total RNA from wt and p50−/− cortical cells was transcribed into cDNA using murine leukemia virus reverse transcriptase (Promega) and oligo-dT(15–18) as a primer (final volume, 50 μl). Parallel reactions containing no reverse transcriptase were used as negative controls to confirm the removal of all genomic DNA. Murine-specific primers were designed using the Primer3 software (http://frodo.wi.mit.edu) (Rozen and Skaletsky, 2000). The oligonucleotide sequences of the primers (M-Medical) used are as follows: Notch1, forward primer, 5′-TGA GAC TGC CAA AGT GTT GC-3′, reverse primer, 5′-GTG GGA GAC AGA GTG GGT GT-3′; Jagged1, forward primer, 5′-CAG TGC CTC TGT GAG ACC AA-3′, reverse primer, 5′-AGG GGT CAG AGA GAC AAG CA-3′; Hes1, forward primer, 5′-CCC ACC TCT CTC TTC TGA CG-3′, reverse primer, 5′-AGG CGC AAT CCA ATA TGA AC-3′; GAPDH, forward primer, 5′-AAC TTT GGC ATT GTG GAA GG-3′, reverse primer, 5′-ACA CAT TGG GGG TAG GAA CA-3′. Amplification and detection were performed with the iCYCLER iQ Q-RT-PCR Detection System (Bio-Rad); the fluorescence signal was generated by SYBR Green I. Samples were run in triplicate in a 25 μl of reaction mix containing 12.5 μl of 2× SYBR Green Master Mix (Bio-Rad), 12.5 pmol of each forward and reverse primer, and 2 μl of diluted cDNA. Each PCR experiment included serial dilutions of a positive control for construction of the calibration curve, a positive and a negative DNA sample, and water blanks. The PCR program was initiated by 10 min at 95°C followed by 40 cycles, each one of 15 s at 95°C and 1 min at 56–62°C. A subsequent dissociation curve analysis verified the products specificity. Gene expression levels are presented as fold change in target gene expression in the test (p50−/− cells) normalized to the internal control gene (GAPDH) and relative to the calibrator (wt cells). Results were estimated as Ct values; the ΔCt was calculated as the mean of the Ct for the target gene minus the mean of the Ct for the internal control gene. The ΔΔCt represented the mean difference between the ΔCt of the test minus the ΔCt of the calibrator. The N-fold differential expression in the target gene of the test compared with calibrator was expressed as 2−ΔΔCt. Data analysis and graphics were performed using GraphPad Prism 4 software and were the results of a single experiment run in triplicate for each gene.
Western blots.
Protein extracts were prepared from 7 DIV cortical cells from wt and p50−/− embryos. Western blotting was performed using 6–12% SDS polyacrylamide gels with 20–50 μg of protein extract loaded per lane. Nitrocellulose filters were incubated with primary antibodies raised against Notch1 and Jagged1 overnight at 4°C, followed by HRP-conjugated secondary antibodies. Membranes were stripped and reprobed with anti-GAPDH antibody. Densitometric analysis was performed using Quantity One software system (Bio-Rad), and each band was normalized to GAPDH signal in each lane and expressed as percentage.
Cell treatments.
Ten micromolar γ-secretase inhibitor N-[N-(3,5-difluorophenacetyl)-1-alanyl 1]-S-phenylglycine t-butyl ester (DAPT) (Calbiochem/EMD Biosciences) was added to the culture medium of 4 DIV cortical cells, and cells were analyzed after 72 h. The DAPT treatment resulted in 34% reduction in Notch1 protein levels and 48% reduction in NICD protein levels.
siRNA probes targeted to Notch1 receptor were purchased from Dharmacon. The mouse-specific Notch1 interference was performed using an Accell SMARTpool siRNA mixture (DHE-041110-00) containing a mixture of four siRNAs targeting the Notch1 gene (NM_008714). A nontargeting Accell siRNA pool (DHD-001910-10) was used as a control in all siRNA transfection experiments. Primary cortical neurons were transfected with Accell siRNAs in Neurobasal-B27 medium to preserve cell viability. Primary cortical neurons were treated with Accell siRNA probes after 4 DIV. Cells were cultured for 3 additional days after transfection with 1 μm siRNA, according to the manufacturer's instructions, then treated, lysed, and subsequently analyzed for mRNA and protein contents. siRNA treatment effectiveness and specificity were validated by measuring Notch1 mRNA levels and using a scrambled siRNA (siRNA−). The siRNA treatment resulted in 58% reduction in Notch1 mRNA levels, 48% reduction in Notch1 protein levels, and 30% reduction in NICD protein, compared with vehicle-treated cells.
NF-κB SN-50 cell-permeable inhibitor peptide (Calbiochem/EMD Biosciences) was added to the culture medium at the concentration of 10 μm for 48 h. To prove SN-50 specificity, a mutant SN-50 (SN-50 mut) (Calbiochem/EMD Biosciences) was used at the concentration of 10 μm for 48 h.
TNF-α (Invitrogen) was added to the culture medium at the concentration of 10 ng/ml for 24 h.
Chromatin immunoprecipitation analysis.
Chromatin immunoprecipitation (ChIP) analysis was performed essentially as described by Lanni et al. (2010). Protein complexes were cross-linked to DNA in living cells by adding formaldehyde directly to the cell culture medium at 1% final concentration. Chromatin extracts containing DNA fragments with an average size of 300 bp were incubated overnight at 4°C with milk shaking using polyclonal anti-p50 and anti-p65 antibodies (Santa Cruz). DNA–protein complexes were recovered with protein A/G-agarose (Santa Cruz). Before use, protein A/G was blocked with 1 μg/μl sheared herring sperm DNA and 1 μg/μl BSA overnight at 4°C, and then incubated with chromatin and antibodies for 3 h at 4°C. PCR was performed using immunoprecipitated DNA and promoter-specific primers flanking the NF-κB site for Jagged1 (M-Medical) and specific primers for 36B4, used as housekeeping gene (kind gift from Dr. Delbarba, University of Milano, Milano, Italy). Primer sequences used were as follows: Jagged1 promoter, forward primer, 5′-TTC AGG GGT GAT CAA GGA AG-3′, reverse primer, 5′-TGG CAT ACT GGG AAT GTC AA-3′; 36B4, forward primer, 5′-AGG ATA TGG GAT TCG GTC TCT TC-3′, reverse primer, 5′-TCA TCC TGC TTA AGT GAA CAA ACT-3′. Immunoprecipitation with nonspecific Igs (no Ab) was performed as negative controls. PCR products were run on a 2.5% agarose gel and visualized with ethidium bromide staining using UV light. The amount of precipitated chromatin measured in each PCR was normalized with the amount of chromatin present in the input of each immunoprecipitation.
Statistical analysis.
A t test analysis or one-way ANOVA followed by Bonferroni's multiple-comparison test as post hoc analysis was performed. Data are expressed as mean ± SEM of n = 3–5 experiments. A value of p < 0.05 was considered to be statistically significant.
Results
Neurite branching and varicosities are reduced in p50−/− cortical neurons
Previous studies demonstrated that NF-κB signaling is involved in regulation of neurite outgrowth, size, and complexity of cortical neuron arborizations (Gutierrez et al., 2005, 2008; Gavaldà et al., 2009). To gain insight into the role of NF-κB in neuronal differentiation, we have taken advantage of the availability of mice carrying a homozygous deletion of the NF-κB1 gene encoding the p50 subunit (Sha et al., 1995; Denis-Donini et al., 2008). Brain cortical neuroblasts were isolated from E15 wt and p50−/− embryos. Cells were grown on poly-d-lysine-coated glass coverslips and maintained in culture for 7 d. Cells were then stained with an anti-βIII-Tubulin antibody. Immunoreactivity and morphology were analyzed by confocal microscopy. We observed a remarkable difference in neurite branching of wt and p50−/− cultures. As shown in a representative image in Figure 1A, cortical cells from wt mice showed a complex and extended neuritic network, reminiscent of mature and differentiated cortical neurons. Conversely, the p50−/− derived cortical cells displayed short neurites and a poor branching phenotype (Fig. 1A, right panel). We then performed Sholl analysis, a valuable and widely used method for quantifying the extent and complexity of neuronal processes (Sholl, 1953; Gutierrez and Davies, 2007). An overall reduction in the spatial complexity of the neuritic arborizations was demonstrated in p50−/− derived cortical cells (Fig. 1B), with a significant decrease in neurite length and number of bifurcations when compared with wt derived cells.

Neurite branching and varicosities are reduced in p50−/− cortical neurons. Reduced complexity and varicosity density of neuronal processes in p50−/− (KO) compared with wild-type (WT) cortical cells. A, Representative 3D images of cortical neurons from WT (left) and KO mice (right) stained with anti-βIII-Tubulin antibody. Scale bar, 20 μm. B, Graphic representation of branching quantification performed using the Sholl profile analysis. Data are expressed as mean ± SEM. C, Quantification of varicosity loss in KO neurons compared with WT. Varicosity density was measured counting the number of varicosities in 100 μm neurite length. Data are expressed as mean ± SEM. *p < 0.0001.
The presence of varicosities is a well recognized morphological feature of differentiated cortical cells. They are regarded as presynaptic, dynamic structures that are able to remodel their morphology in response to a variety of stimuli (De Paola et al., 2003; Nikonenko et al., 2003; Udo et al., 2005; Umeda et al., 2005; Ferrari-Toninelli et al., 2009). Varicosities appear as membrane swellings of various size. They can represent more stable structures, giving origin to secondary branching neurites, filopodia-like structures, or en passant varicosities, fast-turnover structures containing secretory granules (De Paola et al., 2003). We compared the presence of varicosities in wt and p50−/− cortical cells. Wt cortical cells in culture appeared to have long, thin, and varicose neurites, whereas p50−/− cortical cell neurites were short, thicker, and “smooth,” since they were lacking varicosities. We quantified these differences by measuring the density of varicosities in 100 μm neurite length. Varicosity densities were 7.57 ± 0.68 and 1.96 ± 0.33 in wt and p50−/− derived cortical cells, respectively (Fig. 1C).
Hyperactivation of Notch1 pathway in absence of p50 subunit
The morphological features observed in cortical neurons from p50-deficient mice were highly reminiscent of cortical cells in cultures after Notch pathway stimulation by a synthetic Jagged1 ligand (Ferrari-Toninelli et al., 2008). We therefore investigated by quantitative real-time PCR (Q-RT-PCR) the expression levels of three major participants in the Notch signaling pathway, namely the Notch1 receptor, its ligand Jagged1, and the downstream bHLH family member HES1, a Notch1 target gene commonly used to detect Notch1 pathway activation (Iso et al., 2003), in wt and p50−/− cortical cells. As reported in Figure 2, Notch1 and HES1 mRNA levels were increased by 83 and 73% in p50−/− compared with wt cultures. Remarkably, Jagged1 mRNA levels increased by 17-fold in p50−/− compared with wt cultures. Notch1 and Jagged1 protein expression was then evaluated by immunofluorescence. Cortical cells were double-stained with an anti-Notch1 antibody, which recognizes both full-length Notch and its C-terminal intracellular fragment (NICD), and an anti-βIII-Tubulin antibody. After immunostaining, we analyzed at least 40 fields (15–20 cells/field), derived from three different culture preparations. As shown in Figure 3A, Notch1 immunoreactivity appeared to be increased in p50−/− compared with wt cortical cells, considering both the number of Notch1-positive nuclei and the overall fluorescence intensity (Fig. 3A, merge). The total fluorescence intensity was quantified and expressed as percentage of mean fluorescence intensity. As reported in Figure 3B, fluorescence intensity was increased by twofold in p50−/− compared with wt cultures. Western blot analysis using a specific anti-NICD antibody further supported the finding of an increased Notch pathway activation in p50−/− cortical cells. As reported in Figure 3C, NICD protein levels were significantly increased (approximately +60%) in p50−/− compared with wt derived cortical cells.

Notch1 pathway is upregulated in p50−/− cortical cells. Q-RT-PCR was performed using RNA extracted from WT and KO cortical cells to evaluate Notch1, Jagged1, and HES1 expression levels. Data are expressed as fold change of target gene expression in WT and KO cortical cells, normalized to the internal control gene (GAPDH). Data were analyzed according to the comparative Ct method.

Notch1 protein expression levels are increased in p50−/− cortical cells. A, Representative confocal images of cortical cells stained with anti-βIII-Tubulin (green) and anti-Notch1 (red) antibodies. Pictures show an increase of Notch1-positive staining in KO compared with WT cortical cells. Scale bar, 20 μm. B, Quantification of Notch1 immunostaining fluorescence intensity. Values are expressed as percentage of mean fluorescence intensity (% mean ± SEM) and are from at least 40 cellular fields. *p < 0.001 versus WT. C, Representative immunoblot of WT and KO cortical cell lysates using an anti-Notch1 antibody. Graph: Densitometric analysis of NICD levels measured by Western blot in WT and KO cortical cells. Data are normalized to the GAPDH signal, expressed as mean ± SEM, and are obtained from three experiments run with two different cell preparations. *p < 0.01.
Similarly, to evaluate Jagged1 protein levels, wt and p50−/− cortical cells were double-stained with an anti-Jagged1 and an anti-βIII-Tubulin antibodies (Fig. 4A). Fluorescence intensity of Jagged1 immunostaining was quantified, and data were reported in Figure 4B. Fluorescence intensity was increased by 127% in p50−/− compared with wt cortical cells. Data were confirmed by Western blot analysis: the results from three different experiments demonstrated a significant increase (+269%) in Jagged1 level in p50−/− cultures (Fig. 4C).

Jagged1 protein expression levels are increased in p50−/− cortical cells. A, Representative confocal images of cortical cells stained with anti-βIII-Tubulin (red) and anti-Jagged1 (green) antibodies. Pictures show increased Jagged1 immunostaining in KO compared with WT cortical cells. Scale bar, 20 μm. B, Quantification of Jagged1 immunostaining fluorescence intensity. Values are expressed as percentage of mean fluorescence intensity (% mean ± SEM) and are from at least 40 cellular fields. *p < 0.001. C, Representative immunoblot of cortical cell lysates obtained from WT and KO mice using an anti-Jagged1 antibody. Graph: Densitometric analysis of Jagged1 levels measured by Western blot in WT and KO cortical cells. Data are normalized to the GAPDH signal, expressed as mean ± SEM, and are obtained from three experiments run with two different cell preparations. *p < 0.0001.
Overall, these results demonstrate that Notch1 receptor, NICD, and Jagged1 are significantly upregulated in the absence of p50 subunit.
Notch1 pathway inhibition restores neurite branching and varicosities in p50−/− cortical cells
To investigate whether the observed morphological features of p50−/− cortical neurons were correlated to Notch1 pathway hyperactivation, the Notch1 pathway was inhibited either pharmacologically, using the γ-secretase inhibitor DAPT (Kanungo et al., 2008), or genetically, using the Notch1 siRNA approach (siRNA NOTCH).
Notch pathway inhibition by DAPT or siRNA NOTCH resulted in dramatic changes in p50−/− cortical neuron morphology. In particular, as shown by representative pictures in Figure 5A, 3-d-long treatment with DAPT or siRNA NOTCH (but not vehicle or scrambled siRNA) resulted in increased neurite branching and varicosities in p50−/− cells that became indistinguishable from wt cortical neurons. Varicosity density and branching were quantified in DAPT/siRNA NOTCH-treated p50−/− cells and compared with those of p50−/− vehicle or scrambled siRNA-treated cells. Quantitative data confirmed increased varicosity density (Fig. 5B) and branching (Fig. 5C) in DAPT and siRNA NOTCH-treated p50−/− cortical neurons. Together, these data suggest that morphological abnormalities in p50−/− cortical neurons are dependent on Notch pathway hyperactivation.
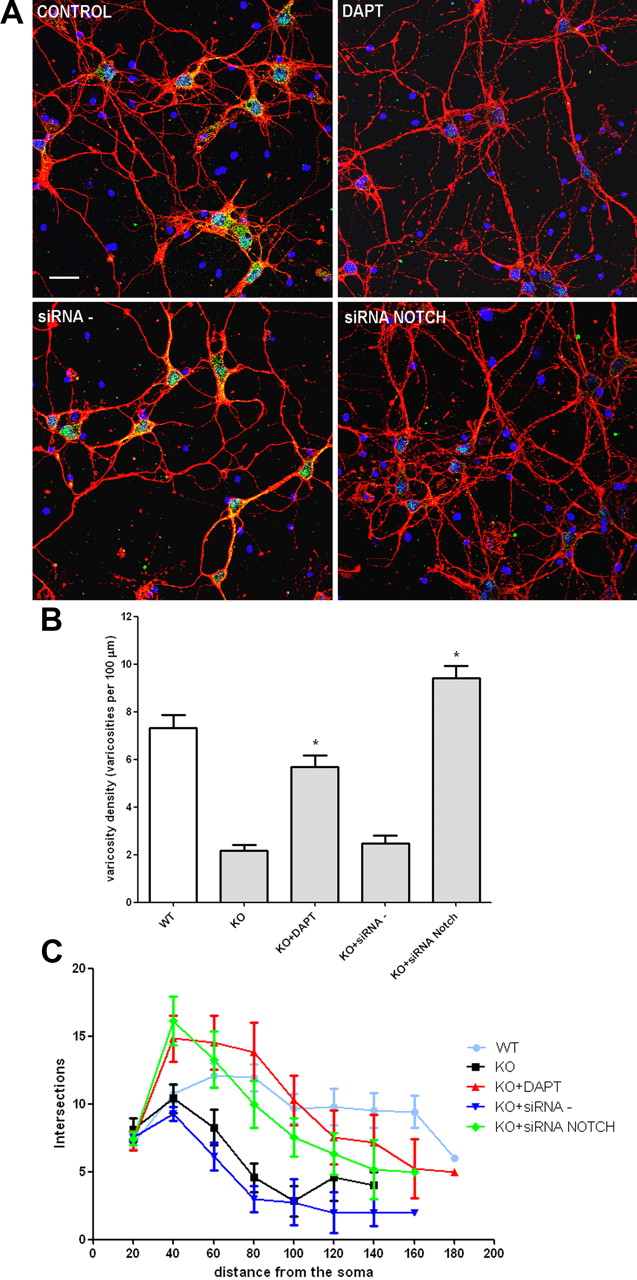
Notch1 blockade restores neurite branching and varicosities in p50−/− cortical neurons. A, Representative confocal images of cortical cells stained with anti-βIII-Tubulin antibody (red), DAPI (blue), and anti-Notch1 antibody (green). KO cortical neurons were exposed to the γ-secretase inhibitor DAPT, the siRNA NOTCH, or the scrambled siRNA probe (siRNA−) for 72 h. Scale bar, 20 μm. B, Quantification of varicosity density in WT and KO neurons after different treatments, as indicated. Varicosity density was measured counting number of varicosities in 100 μm neurite length. Data are expressed as mean ± SEM. *p < 0.001 versus KO. C, Graphic representation of branching quantification performed using the Sholl profile analysis in WT and KO neurons after different treatments, as indicated. Data are expressed as mean ± SEM.
No morphological changes were observed in wt cortical neurons treated with either DAPT or siRNA Notch (data not shown).
p50 NF-κB subunit is directly involved in Notch pathway modulation
NF-κB signaling was acutely manipulated using SN-50 peptide (Lin et al., 1995). Treatment of wt cells with 10 μm SN-50 for 48 h resulted in increased Jagged1 (+191%) and Notch1 (+66%) mRNA levels, compared with vehicle- or mutated SN-50 (SN-50 mut)-treated cells (Fig. 6A). Branching studies by Sholl analysis revealed that SN-50 significantly reduced neurite length and complexity in wild-type neurons (Fig. 6B). Also, varicosities were reduced after SN-50 treatment: varicosity densities were 7.53 ± 0.41, 8.24 ± 0.42, and 3.00 ± 0.26 in vehicle-, SN-50 mut-, and SN-50-treated cortical cells, respectively.

Acute NF-κB signaling manipulation induces Jagged1 upregulation and morphological changes. A, Q-RT-PCR was performed on cDNA from WT cortical cells after 10 μm SN-50 or SN-50 mut treatment at 48 h. Data are presented as fold change of target gene expression normalized to the internal control gene (GAPDH). Data were analyzed according to the comparative Ct method. SN-50 treatment resulted in an upregulation of Jagged1 mRNA expression compared with the vehicle-treated cells (CTR). An SN-50 mutant peptide (SN-50 mut) was used to validate SN-50 specificity. B, Graphic representation of branching quantification performed using the Sholl profile analysis in WT neurons after SN-50 treatment. Data are expressed as mean ± SEM. C, ChIP experiments were performed with anti-p50 and anti-p65 antibodies on WT cortical neurons treated with 10 ng/ml TNF-α for 24 h or 10 μm SN-50 for 48 h. PCR analysis was performed on the immunoprecipitated DNA samples using specific primers for the Jagged1 promoter. A sample representing linear amplification of the total input chromatin (Input) was included as control. Additional controls included immunoprecipitation performed with nonspecific Igs (no Ab). Amplification of 36B4 housekeeping gene was used as control of p50 and p65 binding specificity to the Jagged1 promoter. D, Q-RT-PCR was performed on cDNA from WT cortical cells after 24 h of TNF-α treatment (10 ng/ml). Data are presented as fold change of target gene expression normalized to the internal control gene (GAPDH). Data were analyzed according to the comparative Ct method. TNF-α treatment resulted in an upregulation of Jagged1 mRNA expression compared with the vehicle-treated cells (CTR).
We evaluated the role of p50 in regulating Jagged1 expression in mouse neuronal cortical cells using the ChIP approach. In line with previous data (Johnston et al., 2009), we found that vehicle-treated cells yielded a strong signal for p50 and only a weak p65 signal on the Jagged1 promoter (Fig. 6C). In sharp contrast to control cells, this ratio is reversed in TNF-treated cells, in which we found a weak p50 signal and a strong p65 signal, correlating with the higher transcriptional activity of the Jagged1 gene in TNF-treated cells (Fig. 6D). A similar pattern of results was obtained in cells treated with SN-50. Together, the TNF-α and SN-50 results suggest that, in resting cells, the NF-κB site is likely occupied mostly by p50 homodimers, whereas in TNF-α- or SN-50-treated cells there is a shift toward p65-containing complexes, and this event is associated with enhanced Jagged1 transcription. As it is not entirely clear what the consequences of deletion of p50 throughout embryogenesis may have for NF-κB signaling in cortical neurons, we evaluated p65 protein levels in p50−/− and wt cortical neurons. We found that p50−/− neurons expressed significantly higher protein level (+152%) compared with wt.
Localization of Notch1- and Jagged1-immunoreactive neurons in p50−/− P1 mice cortex
Finally, we investigated whether Notch1 hyperactivation observed in p50−/− embryonal cortical neurons was also observed in vivo. To this purpose, brains from P1 wt and p50−/− mice were collected, fixed, cut in 15-μm-thick coronal sections, and immunostained for Notch1 and Jagged1 proteins. In P1 wt mice, Notch1 immunoreactivity was detected in the basal zone of the motor/somatosensory cortex and gradually decreased in intensity toward the upper cortical layers, as shown in the representative images in Figure 7A, top panel. Conversely, in p50−/− brain sections, Notch1-immunoreactive cells were detected throughout all cortical layers (Fig. 7A, bottom panel). A similar expression pattern was also observed for Jagged1 immunofluorescence in p50−/− compared with wt brains: Jagged1-positive cells were mainly localized in the basal cortical layer in P1 wt brain sections, whereas in p50−/− mice, Jagged1-positive cells were spread throughout the upper layers (Fig. 7B).

In vivo increase of Notch1- and Jagged1-positive neurons in p50−/− mouse cortex. Representative confocal images of cerebral cortex from P1 WT and KO mouse brain sections stained with anti-Notch1 (red) or anti-Jagged1 (red) antibody and DAPI (blue). Inset, Scheme of a P1 mouse brain section, with the indication (red mark) of the region of interest. A, Top panel, Representative images of WT cortex with Notch1-positive neurons localized mainly in the basal layer and few Notch1-stained cells in the upper cortical layers (arrows). Bottom panel, Representative images of KO mouse cortex with Notch1-expressing neurons both in the basal cortical layer and in the whole cortical stratifications (arrows). B, Top panel, Representative images of WT cortex with Jagged1-positive neurons localized in the basal layer. Bottom panel, Representative images of KO mouse cortex with Jagged1-expressing neurons that exceed the basal layer and spread through other cortical layers. WM, White matter. Scale bar, 20 μm.
Discussion
In the CNS, activation of NF-κB has been reported to be involved in neuronal cell differentiation and survival (Maggirwar et al., 1998; Kovács et al., 2004), inflammatory response (Li and Verma, 2002), neurogenesis (Denis-Donini et al., 2005, 2008), proliferation, and apoptosis (Grilli and Memo, 1999; Kucharczak et al., 2003). More recently, emerging data demonstrated its crucial involvement in regulating the growth and complexity of neuronal arborizations (Gutierrez et al., 2005; Gavaldà et al., 2009; Russo et al., 2009; Gutierrez and Davies, 2011) and in synaptic plasticity and memory in the adult brain (Albensi and Mattson, 2000; Kaltschmidt et al., 2006). Nevertheless, the mechanisms by which NF-κB may exert such a role is largely unknown.
p50−/− mice were chosen as an experimental model to investigate the contribution of NF-κB in regulating cortical cell morphology and to identify the intracellular pathway(s) involved in such effects. p50−/− mice have been extensively used in an attempt to clarify the role of NF-κB in developing, adult, and aged nervous system. Indeed, this mouse model has been shown to display accelerated aging (Lu et al., 2006) and a dramatic impairment in hippocampal neurogenesis linked to a selective cognitive deficit in hippocampal-dependent spatial short-term memory (Denis-Donini et al., 2008). Mice lacking p50 subunit also display an increased exploratory activity and reduced anxiety behavior (Kassed and Herkenham, 2004).
In the present study, measuring parameters commonly used to define the stage of neuron differentiation during brain development, we unraveled marked morphological and biochemical differences between wt and p50−/− cortical cells in culture. We demonstrated that neuritic branching and varicosity density were significantly reduced in p50−/− neurons. Our results on the NF-κB role in neurite growth regulation are in line with data from Gutierrez et al. (2007, 2008) in nodose and sensory neurons. These authors reported that different protein subunits contribute to NF-κB-mediated effect on neurites, and, in particular, they described a remarkable inhibitory effect of SN-50, a peptide that interferes with p50 nuclear translocation, on neuritic branching. We now further extend these observations, demonstrating the role of NF-κB on neuronal branching and differentiation also in cortical neurons.
We were interested in investigating the mechanism by which NF-κB is able to act on neuritic morphology. Using primary murine hippocampal cultures, Salama-Cohen et al. (2005) provided evidence that Notch and NF-κB signaling pathways converge at the level of Hes1/5 to regulate dendritic growth. In previous studies, we characterized the effects of Notch1 in remodeling neuronal processes, analyzing its effect on cytoskeleton, varicosities, and neurotransmitter release (Ferrari-Toninelli et al., 2008, 2009). Surprisingly, the reduced varicosity density and neurite ramifications observed in p50−/− neurons strongly resembled the morphology seen in cortical neurons or differentiated SH-SY5Y cells after Notch1 receptor stimulation by its ligand Jagged1. Since the relevance of Notch signaling in development and synaptic transmission, it appeared reasonable to analyze the Notch1 pathway in p50−/− cortical cells.
Notch1 is well known for its involvement in important cellular processes like neurogenesis (Artavanis-Tsakonas et al., 1999), cell fate determination (Louvi and Artavanis-Tsakonas, 2006), and neural stem cell maintenance in the adult brain (Imayoshi et al., 2010). As recently reviewed by Ables et al. (2011), several studies suggested that Notch signaling may serve important functions in the regulation of neurite outgrowth and plasticity. Both Šestan et al. (1999) and Berezovska et al. (1999) found that Notch1 activation inhibits neurite outgrowth or causes their retraction, whereas Notch1 signaling inhibition promotes neurite extension. Redmond et al. (2000) demonstrated that NICD translocates to the nucleus during neuronal differentiation where it regulates the expression of genes whose products influence dendritic morphology. Here, we collected evidence indicating that the Notch signaling is hyperactivated in p50−/− cortical neurons. Remarkably, Jagged1 transcript was increased by 17-fold in p50−/− cells compared with wt cells, suggesting that it might be directly affected by the lack of p50 subunit. We manipulated acutely NF-κB signaling in wt neurons. Specifically, wt cortical cultures were treated with SN-50 and resulted in a significant increase of both Jagged1 and Notch1 mRNA levels. SN-50-treated cells also displayed a significant reduction in neurite branching and varicosity density. Using the ChIP approach, we found that, in resting cells, the NF-κB site within Jagged1 promoter is occupied mostly by p50 homodimers, whereas in TNF-α or SN-50 treated cells there is a shift toward p65-containing complexes, which correlates with enhanced Jagged1 transcription. This is in line with the data by Johnston et al. (2009) demonstrating that, in human endothelial cells, Jagged1 transcription is repressed by p50 homodimers. Our data suggest that, in wt culture system, p50-containing dimers may act as repressor of Jagged1 gene expression. A vast array of information is available in the literature (Zhong et al., 2002; Driessler et al., 2004; Grundström et al., 2004) describing the specific role of p50 homodimers as transcriptional repressors within the family.
We propose Jagged1 as one of the major players in Notch pathway hyperactivation. Its interaction with the receptor may indeed result in massive translocation of NICD to the nucleus and thereby in increased transcription of the Notch target gene HES1. HES1 is a Notch1-responsive bHLH family member implicated in controlling the maintenance of undifferentiated cells and timing of cell differentiation (Akimoto et al., 2010). Since different Notch receptors and ligands have been described, we cannot exclude the possible involvement of other members of the Notch pathway in this effect. However, in our in vitro model the increased NICD nuclear levels and HES1 expression unequivocally testify for a Notch pathway hyperactivity.
We were then interested in evaluating the possible functional link between morphological features and Notch pathway activity. To test this hypothesis, the Notch pathway was inhibited and morphology of cortical neurons was studied. Two different approaches were undertaken: a pharmacological treatment with the γ-secretase inhibitor DAPT, and Notch1 gene silencing. DAPT is a well known Notch1 pharmacological inhibitor (Sastre et al., 2001; Geling et al., 2002; Crawford and Roelink, 2007), preventing Notch1 intracellular cleavage, which is required for NICD nuclear translocation and signaling activation. DAPT exerts a strong inhibitory effect on Notch1 activation, but it has poor selectivity, because of the high number of γ-secretase substrates (Lleó, 2008). siRNA NOTCH is a Notch1 gene interference tool that specifically downregulates Notch1 transcription. Both experimental treatments resulted effective in inhibiting the Notch1 signaling. Interestingly, treatment of p50−/− cortical cells with either DAPT or siRNA NOTCH was able to recover varicosities loss and increase neurite branching. These findings indicated that the morphological alterations present in cortical neurons lacking the NF-κB p50 subunit are dynamic and reversible. More importantly, these results also suggested that Notch hyperactivation mediated the morphological features observed in the absence of the NF-κB p50 subunit.
Notch effects on neurite plasticity might be relevant to the generation of highly refined neuronal circuits (Luo and O'Leary, 2005). Axonal and dendrite architecture is indeed responsible for polarization and migration of neuronal cells and crucial for correct lamination during cortex development. The analysis of p50−/− and wt mouse brains, at postnatal day 1, revealed an altered distribution of Notch1- and Jagged1-positive cells in the cortex. In control brains, Notch1- and Jagged1-positive cells were uniformly localized in the basal layer of the cortex, with few cells in the upper layers. Conversely, p50−/− mice showed an increased number of both Notch1- and Jagged1-expressing cells, with a distribution not only in basal but also in upper layers. In this regard, it should be noted that the Notch pathway has been found to be implicated in cortical neuron migration and in cortical layering, mainly through the cross talk with the Reelin signaling pathway (Gaiano, 2008; Hashimoto-Torii et al., 2008). Although young and adult p50−/− mouse brains do not show overt structural alterations, our data suggest that future studies will be needed to address the possibility that in absence of p50 very subtle defects may occur in neuronal networks and in particular in cortex and that they may potentially contribute to behavioral alterations.
In summary, we found that lack of p50, either induced acutely by inhibiting p50 nuclear translocation in wt cortical cells or genetically in p50−/− mice, results in cortical neurons characterized by reduced neurite branching, loss of varicosities, and Notch1 signaling hyperactivation. The neuronal morphological effects caused by lack of p50 were reverted by inhibition of Notch signaling.
A direct link between Notch and NF-κB pathways is also supported by ChIP data showing that activation of Jagged1 gene transcription is associated with removal of p50 homodimers bound to the NF-κB-responsive element in the Jagged1 promoter region.
These data demonstrate that a cross talk between NF-κB and Notch1 signaling in primary neuronal cortical cells does exist. Our data also support a role for this interaction in the regulation of cortical neuron remodeling.
Footnotes
This work was supported by grants from the Italian Ministry of Education, University and Research (PRIN 2007) and Regione Lombardia (Network-Enabled Drug Design). We thank Dr. Giovanna Cenini for critical discussions and comments on this manuscript, and Dr. Annamaria Lanzillotta and Dr. Valeria Bortolotto for technical support.
- Correspondence should be addressed to Prof. Maurizio Memo, Department of Biomedical Sciences and Biotechnologies, University of Brescia, Viale Europa 11, 25123 Brescia, Italy. memo{at}med.unibs.it





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}