

Abstract
The infiltration of monocytes into the lesioned site is a key event in the inflammatory response after spinal cord injury (SCI). We hypothesized that the molecular events governing the infiltration of monocytes into the injured cord involve cooperativity between the upregulation of the chemoattractant stromal cell-derived factor-1 (SDF-1)/CXCL12 in the injured cord and matrix metalloproteinase-9 (MMP-9/gelatinase B), expressed by infiltrating monocytes. SDF-1 and its receptor CXCR4 mRNAs were upregulated in the injured cord, while macrophages immunoexpressed CXCR4. When mice, transplanted with bone marrow cells from green fluorescent protein (GFP) transgenic mice, were subjected to SCI, GFP+ monocytes infiltrated the cord and displayed gelatinolytic activity. In vitro studies confirmed that SDF-1α, acting through CXCR4, expressed on bone marrow-derived macrophages, upregulated MMP-9 and stimulated MMP-9-dependent transmigration across endothelial cell monolayers by 2.6-fold. There was a reduction in F4/80+ macrophages in spinal cord-injured MMP-9 knock-out mice (by 36%) or wild-type mice, treated with the broad-spectrum MMP inhibitor GM6001 (by 30%). Mice were adoptively transferred with myeloid cells and treated with the MMP-9/-2 inhibitor SB-3CT, the CXCR4 antagonist AMD3100, or a combination of both drugs. While either drug resulted in a 28–30% reduction of infiltrated myeloid cells, the combined treatment resulted in a 45% reduction, suggesting that SDF-1 and MMP-9 function independently to promote the trafficking of myeloid cells into the injured cord. Collectively, these observations suggest a synergistic partnership between MMP-9 and SDF-1 in facilitating transmigration of monocytes into the injured spinal cord.
Introduction
Spinal cord injury (SCI) elicits an intensive local neuroinflammatory response, characterized in part by the infiltration of blood-borne monocytes into the damaged tissue. Studies of macrophage depletion have revealed an improvement in neurologic recovery after SCI (Popovich et al., 1999), suggesting that these cells largely contribute to secondary pathogenesis (Popovich et al., 2002). However, the events that signal the influx of monocytes into the injured spinal cord have yet to be fully elucidated.
Here we consider synergism between SDF-1 (CXCL12), its receptor, CXCR4, and matrix metalloproteinase-9 (MMP-9) in the homing and transmigration of blood-borne monocytes into the injured cord. CXCR4 is highly expressed on hematopoietic progenitor cells, lymphocytes, monocytes, and neutrophils (Förster et al., 1998; Cole et al., 1999). SDF-1, acting through CXCR4 and/or possibly CXCR7 (Zabel et al., 2009), is the principal chemokine that regulates trafficking of hematopoietic stem and progenitor cells (Lapidot et al., 2005; Jin et al., 2006). SDF-1 is increased in microvascular endothelial cells and astrocytes in the ischemic brain (Ceradini et al., 2004; Hill et al., 2004; Li and Ransohoff, 2008) and in multiple sclerosis-related lesions (Miller et al., 2005; Calderon et al., 2006; Krumbholz et al., 2006; McCandless et al., 2008). Moreover, SDF-1/CXCR4 ligation mediates homing of stem progenitor cells (Ceradini et al., 2004) and bone marrow-derived cells, especially monocytes (Hill et al., 2004), to the ischemic brain. In vitro studies further demonstrate that SDF-1, acting on CXCR4+ monocytes, leads to decreased β2 integrin/LFA-1-dependent binding activity of monocytes, thus enabling their migration across cytokine-activated endothelium (Malik et al., 2008).
How myeloid cells transmigrate across the blood–spinal cord barrier after SCI is an important question. Quiescent monocytes express low levels of MMP-9 (Bar-Or et al., 2003). However, this protease is upregulated in response to a variety of events, including those related to proinflammatory stimuli and cell–cell contact (Opdenakker et al., 2001). The CD95L-CD95 system, well known for signaling to apoptosis in a variety of cellular contexts (Bouillet and O'Reilly, 2009), is also involved in mediating recruitment of peripheral myeloid cells into the injured spinal cord. Based on in vitro studies, migration of myeloid cells across matrices was shown to be MMP dependent (Letellier et al., 2010). These interesting findings raise the question of which MMPs are involved in this pathway in vivo.
In this study, we address how MMP-9 (gelatinase B) directs transmigration of blood-borne monocytes into the injured spinal cord. We show SDF-1/CXCR4 and MMP-9 act synergistically to support the migration of blood-borne monocytes into the injured spinal cord. As blood-borne monocytes are determinants of injury and reparative processes (Yona and Jung, 2010), there is a need to understand the signaling pathways that govern their migration into the injured cord, with a long-term goal of their selective modulation to enhance neurologic recovery.
Materials and Methods
Animals.
These studies were approved by the Institutional Animal Care and Use Committee at the University of California San Francisco and in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals. Transgenic mice, expressing GFP under the control of a chicken β-actin promoter (Persons et al., 1998) and c-fms-EGFP mice (Sasmono et al., 2003), were on an FVBn background. MMP-9-knock-out (KO) and wild-type (WT) littermates (Vu et al., 1998) were bred on an FVBn background. KO and WT littermates, used for the in vivo studies, were generated by breeding heterozygous mice. Homozygous females and males of respective genotypes were bred for the in vitro studies. Genotypes were identified by the PCR using tail tissue and specific oligonucleotide primers (Vu et al., 1998; Ducharme et al., 2000).
Spinal cord injury.
Adult female mice (4–6 months of age) were anesthetized and subjected to a moderate spinal cord contusion injury as we have described previously (Noble et al., 2002). Briefly, a laminectomy was performed at the T9 vertebral level, and a 3 g weight was dropped 5 cm onto the exposed dura mater. Postoperative care included subcutaneous administration of antibiotics and manual expression of the bladder twice daily.
Quantitative real-time reverse transcriptase-PCR.
Total RNA was extracted from naive mice and at 3 and 7 d after injury (5 animals/group) from a 5 mm length of cord, centered over the epicenter, or a matched segment of the uninjured cord using Trizol (Invitrogen) and following the vendor-recommended protocol. RNA was further purified using a Qiagen column (Qiagen) with DNase treatment. Each of the cDNAs was individually analyzed on a 96-well plate coated with primers of CXCR4 and SDF-1 (SABiosciences). Quantitative real-time reverse transcriptase-PCR (RT2qPCR) was performed on an ABI PCR Machine 7900 (Applied Biosystems). The level of gene expression was calculated using the ΔΔCt method and normalized to housekeeping genes using the available SABiosciences software (SABiosciences). For each gene, fold changes were calculated as the difference in gene expression between injured and uninjured. Significant upregulation is defined as a value greater than twofold with a p value of ≤0.05.
In situ zymography.
The injured spinal cords were removed quickly without fixation and frozen. Sections 2 μm in thickness were cut on a cryostat and incubated in 0.05 m Tris-HCl, 0.15 m NaCl, 5 mm CaCl2, and 0.2 mm NaN3, pH 7.6, containing 40 μg/ml FITC-labeled DQ gelatin (Invitrogen), at 37°C for 1 h. After a brief rinse, sections were coverslipped and observed by fluorescence microscopy.
Generation of GFP chimeric mice.
WT mice were sublethally irradiated with 7.5 Gy from a 137Cs-ray source. The following day, bone marrow-derived cells were obtained from GFP-transgenic mice in which GFP is under control of a chicken β-actin promoter, at 8 weeks of age by flushing the femora and tibiae with PBS supplemented with 2% fetal bovine serum (FBS). Red blood cells were lysed in 0.15 m ammonium chloride and 0.01 m potassium bicarbonate solution on ice. The number of viable cells, defined by an absence of trypan blue staining, was quantified using a hemocytometer, and 1–2 × 106 cells/mouse were then injected into the tail vein of irradiated WT mice.
Two months after transplantation, the rate of chimerism in recipient mice was confirmed by counting the number of GFP+ cells in blood smears using a fluorescence microscope (Optiphot EF-D3; Nikon) equipped with a SPOT camera (SPOT Imaging solutions). The rate of chimerism, estimated to be >90%, was confirmed by flow cytometry (FACSCalibur; Becton Dickinson).
Immunocytochemistry.
Mice were deeply anesthetized and perfused with 4% paraformaldehyde (PFA) in 0.1 m PBS, pH 7.4. The spinal cord was removed and postfixed for 4 h, then transferred into sucrose (20% in PBS) for cryoprotection. Frozen sections (20 μm in thickness) were cut on a cryostat. For cultured cells, they were replated on an eight-well chamber in serum-free medium and cultured overnight. Cultures were then fixed by 4% PFA.
Sections were then incubated with primary antibody rat anti-mouse F4/80 (1:200, Invitrogen), rat anti-mouse CD11b (1:100, AbD Serotec), chicken anti-mouse GFP (1:500, Aves LABS), rat anti-mouse Gr-1 (1:100, Invitrogen), rat anti-mouse Ly-6G (1A8, 1:100, BD PharMingen), or Fusin (g-19) goat polyclonal (CXCR4, 1:30, Santa Cruz Biotechnology). Sections were rinsed and incubated with secondary antibody Fluorescein-labeled goat anti-chicken IgY (1:100, Aves LABS) for GFP; Cy3-conjugated goat anti-rat IgG (1:250, Jackson ImmunoResearch Laboratories) or Alexa Fluor 488 goat anti-rat (1:100, Invitrogen) for F4/80, CD11b, Gr-1, and Ly-6G; or Cy3-conjugated mouse anti-goat (1:250, Jackson ImmunoResearch Laboratories) for CXCR4. Slides were coverslipped and observed by fluorescence microscopy.
Culture of bone marrow-derived macrophages.
Bone marrow-derived macrophages (BMDMs) were prepared as previously described (Longbrake et al., 2007). Bone marrow cells were isolated as described above. The final preparation from each mouse was then cultured in one 10 cm non-tissue culture-treated Petri dish in culture medium (DMEM, 10% FBS, 1% penicillin/streptomycin, 1% GlutaMAX-1, 20% L-cell conditioned medium). After incubating for 24 h, adherent cells were discarded, and the nonadherent cells were replated in a cell culture dish and cultured in the same culture medium. At day 4, the medium was replaced with fresh culture medium, and cultures were maintained for 6–10 d. At the end of the culture period, at least 95% of cells were immunopositive for F4/80 and CD11b.
L-cell conditioned medium was obtained from culture supernatants of the cell line L929. Briefly, cells were incubated for 5 d, and the medium was collected, centrifuged, filtered through a 0.22 μm filter, and stored at −20°C.
Gelatin zymography.
Conditioned media from cultured BMDMs were collected and subjected to gelatin zymography. Cells were exposed to lipopolysaccharide (LPS) (0.1 μg/ml, Sigma), IL-1β (10 ng/ml, eBioscience), SDF-1α (100 ng/ml, R&D Systems), or TNFα (10 ng/ml, Invitrogen) for 18 h. Supernatants were concentrated using a Microcon filter (50K Membrane, Millipore), and equal amounts of protein (5 μg) were loaded on a 10% zymogram gel. After electrophoresis, the gel was incubated with renaturing buffer (Bio-Rad Laboratories) at room temperature for 30 min to restore the gelatinolytic activity of the proteins, then incubated with developing buffer (Bio-Rad Laboratories) at 37°C for 48 h. The gel was stained with Coomassie Blue and destained until clear bands became evident.
Migration assays.
Migration was quantified using Transwell culture chambers (24 well, 8 μm pore size, Corning Life Science). BMDMs (1 × 105) suspended in DMEM containing 0.4% FBS were loaded into the upper wells and allowed to migrate for 16 h. LPS (0.1 μg/ml) was added to the upper wells, and SDF-1α (100 ng/ml) was added to the lower wells. In some experiments, cells were preincubated with synthetic MMP inhibitors (Hsu et al., 2008). These included GM6001 (US Biologicals; Ki for human MMP-1: 0.4 nm; MMP-3: 26 nm; MMP-2: 0.5 nm; MMP-8: 0.1 nm; MMP-9: 0.2 nm), MMP-9 inhibitor I (Calbiochem; MMP-9 IC50: 5 nm; MMP-1 IC50: 1.05 mm; MMP-13 IC50: 113 nm), and the CXCR4 inhibitor AMD3100 (Sigma) for 1 h. Cells were then fixed with 4% formaldehyde and washed with PBS. To remove nonmigrating cells, the surface of the upper wells was gently scraped using cotton swabs. Cells migrating to the lower surface of the membrane were stained with DAPI, and photographed in five random, non-overlapping areas using 10× objective. The number of cells was then assessed by counting nuclei using MetaMorph software. All experiments were performed in triplicate.
Transendothelial migration.
Rat brain capillary endothelial cell line 4 (rBCEC4) (Blasig et al., 2001) cells were grown on type I collagen-coated plates in DMEM/F12 containing 10% FBS, 100 μg/ml heparin (Sigma), and 10 μg/ml endothelial cell growth factor (Roche). For trans-endothelial migration, rBCECs (5 × 104 cells) were seeded on type I collagen (10 μg/ml)-coated Transwell culture inserts (8 μm pore size) and grown to confluence for 3 d. To determine the confluency of the rBCEC monolayer, high-molecular-weight FITC-dextran was added to the upper chamber. Confluence was defined as the absence of fluorescence in the lower chamber. Before assays, monolayers were stimulated with TNFα (200 U/ml) and IFNγ (200 U/ml, AbD Serotec) for 24 h. After washing the monolayer with PBS, fluorescently labeled BMDMs were suspended in DMEM containing 0.4% FBS and added to the upper chamber. Quantum dots (Qtracker 625 cell labeling kit, Invitrogen) were used for labeling BMDMs. SDF-1α (100 ng/ml) was added to the lower chamber as a chemoattractant. Then, cultured macrophages were allowed to transmigrate for 16–18 h. Migrated fluorescently labeled macrophages were photographed in five random, non-overlapping areas using a 10× objective. Quantification of fluorescence was performed using the basic densitometric thresholding features of MetaMorph software, which quantifies pixel areas based on color thresholds. High and low thresholds were selected based on minimizing background and optimizing detection of desired structures. Once these parameters were validated across several plates, they then became the standard that was applied to the entire analysis. All experiments were performed in triplicate.
In vivo GM6001 studies.
Spinal cord-injured WT mice were treated with either GM6001 (100 mg/kg in 4% carboxy methylcellulose, i.p.), a general inhibitor of MMPs (Noble et al., 2002), or vehicle (4% carboxy methylcellulose, i.p.) from 4 d after injury and twice daily thereafter for 3 d. Mice were killed at 7 d after injury. A 2 cm spinal cord length of cord, centered over the site of injury, was sectioned longitudinally (20 μm in thickness) using cryostat. To determine the distribution of macrophages in the injured cord, two sections from the midline and showing the central canal were stained with F4–80 antibody for each animal, and then visualized with Cy-3-conjugated secondary antibody. All images were captured on a fluorescence microscope (4× objective). The maximal axial distribution of F4/80+ cells was determined using MetaMorph software (Molecular Devices) and was defined as the total length of cord showing an F4/80+ signal.
Proportional area measurements.
Proportional area was computed as the fraction of the area surrounding the lesion core with F4/80 staining divided by the total cross-sectional area of the cord at 7 d after injury (Popovich and Hickey, 2001). First to define the lesion epicenter, for each animal, a section, 20 μm in thickness, was selected every 200 μm and stained with cresyl violet. Spared white matter was quantified in each of these sections. The section showing the least spared white matter was defined as the lesion epicenter. Proportional area measurements were then conducted in two sections (40 and 60 μm caudal to the epicenter) stained with F4/80. Images from the two sections were digitized at low magnification that allowed the entire lesion area to be visualized within a single image frame. MetaMorph was then used to quantify F4/80+ cell areas based on color threshold. Upper and lower thresholds were defined using a similar logic to that described for in vitro assessment of transendothelial migrations of BMDMs. Thresholding was determined in two sections, prepared from two WT and two KO spinal cord-injured animals. The ratio of the area of F4/80 to the total scan area was reported as the proportional area (PA) for each section.
Adoptive transfer studies.
Bone marrow cells were isolated from c-fms-EGFP mice as described above. SB-3CT (25 mg/kg body weight), designed as a highly selective, mechanism-based inhibitor to MMP-2 and MMP-9 (Brown et al., 2000) (Ki was 14 ± 1 nm for human MMP-2 and 600 ± 200 nm for MMP-9), was administered intraperitoneally immediately after injury followed by a second dose immediately before bone marrow cell transplantation (Gu et al., 2005). The timing of administration of the CXCR4 antagonist AMD3100 [5 mg/kg of body weight, s.c., bolus (Nervi et al., 2009)] and vehicle (10% DMSO) was as described for SB-3CT. Following the second dose of drug or vehicle, each animal was immediately administered 200 μl of PBS containing 5 × 106 bone marrow cells in the tail vein in two consecutive doses at 4 and 6 h after injury. Mice were killed at 24, 48, or 72 h after transfer. For quantification of infiltrated GFP+ cells, longitudinal sections, 20 μm in thickness and centered over the epicenter, were prepared. For each animal, two sections were immunostained with anti-GFP antibody every 300 μm, and a total of eight sections were evaluated. Within each section, four separate images were taken using a 20× objective, two images on either side of the midline at lesion epicenter and another two images immediately rostral and caudal to the lesion epicenter and on either side of the midline. Infiltrated GFP+ cells were expressed as the number of cells per square millimeter.
Statistical analysis.
All tests were performed using GraphPad Prism version 4.0 (GraphPad Software). Data obtained from in vitro studies of cultured BMDMs and the in vivo drug treatment study were analyzed using ANOVA, followed by Bonferroni's post hoc test for multiple comparisons between genotypes or treatments. Two-tailed t tests were used to compare in vivo and in vitro data where two groups were specified. All data are presented as means ± SEM. A statistically significant difference was defined at p ≤ 0.05.
Results
SDF-1 and CXCR4 are increased in the injured cord
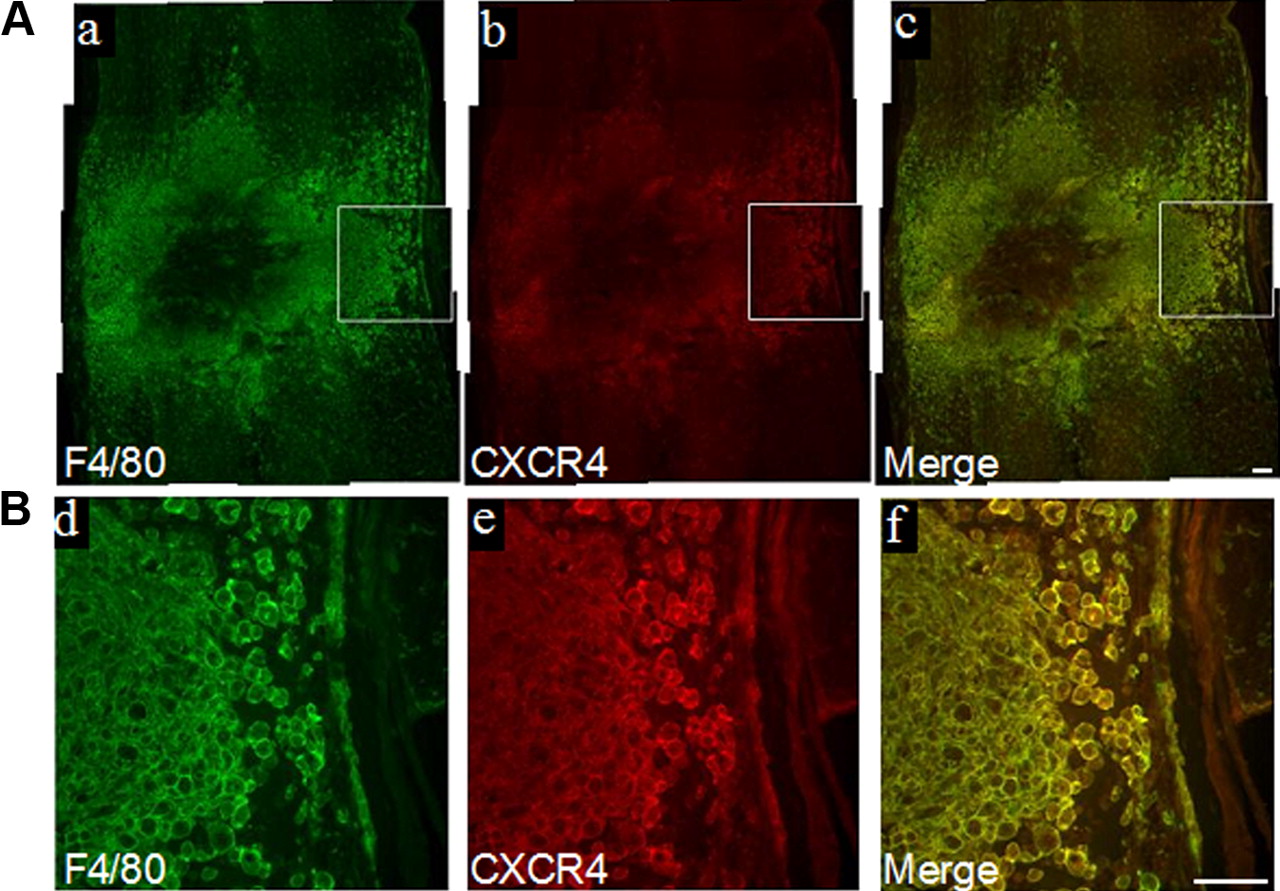
To confirm our hypothesis that the SDF-1 chemokine mediates the recruitment of macrophages, we first asked whether SDF-1 and its receptor CXCR4 are upregulated in the injured cord by SYBR Green real-time PCR analysis (RT2qPCR). Levels of both SDF-1 and CXCR4 were assayed from epicenter segment RNA samples collected at 3 and 7 d after injury (n = 5 per time point). Values were normalized to the level of three housekeeping genes and expressed relative to normalized values of uninjured controls (n = 5). Expression of SDF-1 and CXCR4 was upregulated by 2- and 9.71-fold, respectively, in injured cord at 3 d (Student's t test, p < 0.001) and 2.12- and 8.35-fold, respectively, at 7 d (Student's t test, p < 0.01) after injury compared with uninjured controls. Immunostaining for CXCR4 at 7 d after injury revealed prominent staining at the lesion epicenter, the region of maximal damage. CXCR4 colocalized with F4/80+ macrophages (Fig. 1A,B) that clustered around the central most damaged region.

SDF-1 and CXCR4 are upregulated in the injured cord. Double immunostaining at 7 d after injury reveals abundant F4/80+ cells that coimmunoexpress CXCR4 (A). Enclosed areas in a–c are shown at higher magnification in d–f (B). Scale bars, 100 μm (a–f).
Macrophages display gelatinolytic activity in the injured cord
We next confirmed by in situ gelatin zymography that macrophages express gelatinolytic activity in the injured spinal cord. An area of the epicenter that was rich in F4/80+ macrophages was chosen to compare MMP-9 WT and KO animals. While there was overt gelatinolytic activity surrounding WT macrophages at the lesion epicenter at 7 d after injury (Fig. 2A,B), a similar pattern of prominent activity was not noted in the MMP-9 KO macrophages (Fig. 2C,D). Gelatinolytic activity in WT animals colocalized with F4/80+ macrophages (Fig. 2E–G).

Macrophages express gelatinolytic activity in the injured spinal cord. In situ zymography reveals prominent gelatinase activity within the epicenter that is localized to F4/80+ macrophages in MMP-9 WT mice at 7 d after injury (A, B). Gelatinase activity appeared markedly reduced in the injured cord of the MMP-9 KO and there is no colocalization in F4/80+ macrophages (C, D). Higher magnification of the epicenter reveals localization of gelatinase activity in F4/80+ macrophages in WT mice (E–G). Scale bars: 50 μm (A–D), 100 μm (E–G).
Macrophages, derived from infiltrating bone marrow-derived cells, are a source of MMP-9 in the injured spinal cord
To evaluate the infiltration of blood-borne monocytes into the injured spinal cord, bone marrow stem cells expressing GFP were transplanted into irradiated mice (GFP chimeric mice). Animals were then subjected to a spinal cord contusion injury. At 7 d after injury, GFP+ cells (Fig. 3A), confirmed by immunolabeling with anti-GFP (Fig. 3F), were abundant at the lesion epicenter. Most of the GFP+ cells assumed a rounded macrophage-like phenotype (20–30 μm in diameter) (Fig. 3B) and expressed MMP-9 (Fig. 3C–E) and the macrophage marker F4/80 (Fig. 3F–H). That recruited GFP+ cells express MMP-9 raises the possibility that this protease may be involved in their trafficking across blood–spinal cord barrier after SCI.

Infiltration of bone marrow-derived GFP+ macrophage-like cells into the injured spinal cord. Irradiated WT mice, transplanted with GFP-expressing bone marrow cells (GFP chimeric mice), were subjected to SCI. GFP+ cells were visualized by fluorescence microscopy in longitudinal sections of the spinal cord, centered over the site of injury. At 7 d after injury, the infiltration of GFP+ cells was prominent in the injured cord (A). The majority of infiltrated GFP+ cells exhibited a macrophage-like phenotype (enclosed box in A). Higher magnification shows these cells are round (20–30 μm in diameter) with no processes (B). These GFP+ macrophage-like cells expressed MMP-9 (C–E). Double immunolabeling reveals colocalization of GFP (green in F), with F4/80 (red in G). Scale bars: 250 μm (A, B), 100 μm (C–H).
In vitro studies confirm MMP-9-dependent migration of BMDMs
To determine the dependency of migration on MMP-9, we then conducted in vitro migration assays. Three populations of macrophages are commonly studied in vitro, including resident peritoneal, thioglycolate-elicited peritoneal macrophages and BMDMs. Cytokine mRNA profiles, morphology, and functional characteristics of phagocytosis of these macrophages were compared (Longbrake et al., 2007). Since BMDMs exhibit phenotypic, functional, and inductive characteristics of macrophages elicited by SCI, we used them interchangeably in vitro to model the functional consequences of the infiltrating macrophages elicited by SCI. Supernatants, prepared from cultured BMDMs activated with LPS, IL-1β, SDF-1α, or TNFα, showed prominent expression of MMP-9 by gelatin zymography (Fig. 4A), whereas no MMP-9 expression was detected in cultured macrophages of MMP-9 KO mice (data not shown).

MMP-9-expressing BMDMs show enhanced cell motility. Zymography of supernatants from BMDMs stimulated with inflammatory cytokines for 18 h. While MMP-9 expression is prominent, there appears to be no change in MMP-2 (A). In vitro migration assays show greater cell motility in MMP-9-expressing WT BMDMs compared to MMP-9 KO BMDMs. Synthetic MMP inhibitors GM6001 and MMP-9 inhibitor I attenuate migration of WT BMDMs, but have no effect on KO BMDMs (B). ***p < 0.001.
WT BMDMs, activated with LPS, showed more prominent migration than KO BMDMs (Fig. 4B). Similarly, when WT BMDMs expressing MMP-9 were exposed to the synthetic MMP inhibitors GM6001 and MMP-9 inhibitor I, migration was reduced to basal levels, defined as that observed in the absence of stimulation by LPS (Fig. 4B).
SDF-1α mediates MMP-9-dependent transmigration of BMDMs across an endothelial monolayer
We further determined whether SDF-1 serves as a chemoattractant for BMDMs. BMDMs expressed CXCR4 (Fig. 5A), the receptor for SDF-1. We then added SDF-1α to the lower chamber of Transwells and plated cells on uncoated inserts. SDF-1α increased migration of cultured macrophages compared with the control group, and this effect was abrogated by the specific CXCR4 antagonist AMD3100 (Fig. 5B). SDF-1α-induced migration through uncoated membrane was not dependent on MMP-9 as evidenced by similar migration of WT and MMP-9 KO BMDMs. MMP-9 inhibitor did not, however, impair migration of WT BMDMs through uncoated filters (Fig. 5C).

SDF-1α-induced transendothelial migration of BMDMs is enhanced in the presence of MMP-9. Cultured BMDMs when stimulated with LPS, immunoexpress CXCR4 (A). There was a twofold increase in the migration of BMDMs across uncoated Transwells in the presence of SDF-1 in the bottom well. This migration, however, was effectively reduced in the presence of the CXCR4 inhibitor AMD3100 (B). SDF-1α induced a similar magnitude of migration in MMP-9 KO and WT BMDMs. The migration across uncoated Transwells was not affected by MMP-9 inhibitors (C). Transendothelial migration of WT and MMP-9 KO BMDMs was first studied across activated (TNF-α and IFN-γ) and unactivated endothelial cells with growth medium (10% FCS) in the bottom chamber. Transmigration of MMP-9 KO and WT BMDMs across unactivated endothelial cells is relatively modest. While transmigration of WT BMDMs across activated endothelial cells is markedly increased, no differences in transmigration are seen in MMP-9 KO BMDMs (D). When SDF-1 α was added to the bottom chamber as a chemoattractant, MMP-9 WT BMDMs showed greater migratory ability compared to MMP-9 KO BMDMs. No differences in transmigration are seen in MMP-9 KO BMDMs with or without SDF-1 α as the chemoattractant in lower chamber (E). AMD3100, a CXCR4 inhibitor (F), and an MMP-9 inhibitor (G) completely blocked SDF-1α-induced transendothelial migration. Results are expressed as percentages of transmigrated cells that are normalized to WT control. **p < 0.01, ***p < 0.001. Scale bar, 100 μm (A).
We next investigated the role of SDF-1α in the transendothelial migration of BMDMs, using a rat brain capillary endothelial cell line (rBCEC) (Blasig et al., 2001). A number of surface molecules (PECAM-1, ICAM-1, and VCAM-1) expressed by brain endothelium, either constitutively or upon treatment with the inflammatory cytokines TNF-α and IFN-γ, are involved in migration of activated leukocytes across the blood–brain barrier under inflammatory conditions (Weksler et al., 2005). We pretreated rBCEC monolayers grown on Transwells with TNF-α and IFN-γ for 24 h. The integrity of the monolayer was monitored using high-molecular-weight FITC-dextran. A limited amount of dextran (<10%) passed through the rBCEC monolayer (data not shown). When migration was evaluated with 10% FCS serving as the chemoattractant in the bottom chamber, migration of MMP-9 WT BMDMs across rBCEC monolayers increased upon pretreatment with TNF-α and IFN-γ (Fig. 5D), but little with MMP-9 KO BMDMs. The chemokine SDF-1α (Fig. 5E–G) increased trans-endothelial migration of BMDMs expressing MMP-9 by 2.6-fold compared to that in MMP-9 KO BMDMs, and no increase was seen in MMP-9 KO BMDMs (Fig. 5E). Both MMP-9 inhibitor I and the CXCR4 antagonist AMD3100 effectively blocked SDF-1α-induced trans-endothelial migration (Fig. 5F,G). These findings suggest that SDF-1α-induced trans-endothelial migration is MMP-9 dependent.
Macrophages are reduced in MMP-deficient mice after SCI
Next, the axial distribution of F4/80+ macrophages was evaluated in spinal cord-injured mice that were treated with the broad-spectrum MMP inhibitor GM6001 or vehicle from 4 to 6 d after injury. This window of treatment corresponds to the peak period of infiltration of macrophages into the injured cord (Sroga et al., 2003). At 7 d after injury, F4/80+ cells were primarily distributed at and away from the site of impact in the vehicle-treated group, whereas these cells were mostly clustered near the site of impact in the inhibitor-treated group (Fig. 6A). Treatment with GM6001 significantly reduced axial distribution of F4/80+ macrophages by 30% relative to vehicle-treated mice (N = 6/group, Student's t test, p < 0.05).

F4/80+ macrophages are reduced in MMP-deficient mice after SCI. Treatment with the broad-spectrum MMP inhibitor GM6001 from 4 to 6 d after SCI, when macrophage infiltration was most prominent, resulted in reduced axial distribution of F4/80+ macrophages relative to vehicle-treated mice at 7 d after injury (A). The proportional area of F4/80+ macrophages was reduced at the epicenter at 7 d after injury in MMP-9 KO relative to WT mice (B). Scale bars: 200 μm (A), 200 μm (B).
The density of F4/80+ macrophages, based upon measures of proportional area, was compared in MMP-9 KO and WT mice at 7 d after injury (Fig. 6B). Proportional area was computed as the fraction of the area surrounding the lesion core with F4/80 staining divided by the total cross-sectional area of the cord (Popovich and Hickey, 2001). While the total cross-sectional area was similar between the two groups (77,800 ± 2436 and 82,010 ± 2969 pixels), there was a significant reduction of 36% in the proportional area of F4/80 staining in the MMP-9 KO relative to the WT (N = 5/group, Student's t test, p < 0.01), indicating that MMP-9 deficiency markedly reduced recruitment of macrophages into the injured cord.
Adoptive transfer reveals MMP-9 as a cofactor in SDF-1 signaling migration of myeloid cells into the injured cord, and infiltrated myeloid cells assume a CD11b+ and F4/80+ phenotype
To determine whether MMPs specifically cooperate with SDF-1 to facilitate the recruitment of blood-borne monocytes into the injured spinal cord, mice were adoptively transferred with bone marrow myeloid cells freshly isolated from c-fms-EGFP mice (Sasmono et al., 2003), based upon studies in a model of retinal inflammation, which showed that freshly isolated bone marrow-derived CD11b+ monocytic cells circulate freely and traffic efficiently to the site of inflammation (Xu et al., 2005). Thus, we adoptively transferred bone marrow cells at 4 and 6 h after injury, and killed mice at 24 h after transfer.
It is noteworthy that GFP+ myeloid cells accumulated in the meninges in the SB-3CT group (Fig. 7A,B), suggesting that gelatinases may be used by these cells to infiltrate the injured cord via invasion of the parenchymal basement membrane or glia limitans.

Adoptive transfer confirms MMP-dependent migration of myeloid cells into the injured cord. Interestingly, there are very few GFP+ cells in the meninges in the vehicle-treated group (A), whereas there was an abundance of these cells in the meninges in the SB-3CT-treated group (B). At 24 h after adoptive transfer, most of the infiltrated GFP+ cells were CD11b+Gr1+ cells, which are characteristic of immature myeloid cells (C, D). However, by 48 h after transfer, many of the infiltrated GFP+ cells were Ly6G− (E). At 72 h after transfer, the cell bodies of the infiltrated GFP+ cells assumed a larger phenotype relative to those seen at 24 h after transfer and were F4/80+ (F). The presence of infiltrated GFP+ cells within the injured site was confirmed by immunostaining with anti-GFP antibody 24 h after transfer (G–J). Treatment with the MMP inhibitor SB-3CT or CXCR4 inhibitor AMD3100 reduced the number of adoptively transferred bone marrow-derived myeloid cells that were recruited to the injured cord to 70%, and further reduced to 55% when treated with SB-3CT and AMD3100 together (N = 5/group) (K). *p < 0.05, **p < 0.01. Scale bars: 50 μm (A, B, G–J), 100 μm (C–F).
At 24 h after transfer, infiltrated GFP+ cells were CD11b+Gr-1+, which is characteristic of immature myeloid cells (Fig. 7C,D), but F4/80− (data not shown). Notably, by 48 h after transfer, many of the infiltrated GFP+ cells were Ly-6G− (detected with mAb 1A8) (Fig. 7E). By 72 h after transfer, infiltrated GFP+ cells appeared larger, relative to earlier time points, and were F4/80+ (Fig. 7F), indicating that these infiltrating, adoptively transferred immature bone marrow myeloid cells continued to differentiate into mature macrophages within the lesion site.
To determine whether SDF-1 signals MMP-9-dependent migration, adoptive transferred animals were treated with (1) SB-3CT, a selective mechanism-based inhibitor of MMP-2 and MMP-9, (2) the CXCR4 antagonist AMD3100, (3) both SB-3CT and AMD3100, or (4) vehicle. There were 28–30% fewer GFP+ myeloid cells at the lesion site in the SB-3CT- or AMD3100-treated group (Fig. 7H,I), relative to the control group (Fig. 7G,K). When treated with both drugs, there was a 45% reduction in the number of infiltrated GFP+ myeloid-positive cells (Fig. 7J,K). These findings suggest that SDF-1 and MMP-9 enhance migration by independent mechanisms.
Discussion
Here we provide the first evidence for a synergistic partnership between MMP-9 and SDF-1/CXCR4 in mobilizing blood-borne monocytes to the injured spinal cord. Infiltrating monocytes expressed MMP-9 and the SDF-1 receptor CXCR4. SDF-1-induced transmigration of BMDMs expressing MMP-9 across cultured, brain-derived endothelial cells was blocked by inhibitors of MMP-9 and CXCR4. Reduction of macrophages in the injured cords of MMP-9-deficient mice and adoptive transfer of myeloid cells established their dependency on MMPs to transmigrate into the injured cord. Finally, adoptive transfer of myeloid cells, in spinal cord-injured mice treated with inhibitors to MMPs, CXCR4, or a combination of both, revealed synergism between SDF-1 and MMPs, in supporting the trafficking of blood-borne monocytes into the injured cord.
We propose a general model of regulatory cross-talk between injury signals originating from the injured cord and blood-borne monocytes that respond by infiltrating the damaged tissue. SDF-1 is a likely signal for the infiltration of monocytes. This cytokine, upregulated in ischemic brain (Ceradini et al., 2004; Hill et al., 2004; Krumbholz et al., 2006), increases the adhesion, migration, and homing of circulating CXCR4-positive neural progenitor cells (Ceradini et al., 2004; Imitola et al., 2004; Robin et al., 2006; Schönemeier et al., 2008) and monocytes (Hill et al., 2004) to the injured brain. SDF-1 may likewise serve as a key chemoattractant in regulation of homing and transmigration of blood-borne monocytes into the injured spinal cord.
While SDF-1 is a likely chemoattractant for monocytes, MMP-9 and cytokines such as TNF-α may facilitate their transmigration into the injured cord. TNF-α is rapidly upregulated within hours after SCI (Pan et al., 2002; Pineau and Lacroix, 2007). TNF-α upregulates MMP-9 expression and secretion in both a human monocytic cell line (MonoMac-6) and peripheral blood monocytes (Vaday et al., 2000). Moreover, in the presence of soluble or bound TNF-α, the MMP inhibitor GM6001 reduces chemotaxis through gels comprised of ECM substrates (Vaday et al., 2000). Thus, monocyte trafficking into the injured cord is likely dependent on an interplay between cytokines, chemokines (SDF-1), and proteolytic enzymes such as MMP-9.
Our study suggests that SDF-1, expressed in the injured cord, is a chemoattractant for infiltration of blood-borne monocytes. SDF-1 may also be important in mobilization of stem cells from bone marrow (BM) to peripheral blood. Plasma levels of SDF-1 increase in response to tissue damage in murine models of vascular injury and limb ischemia (Jin et al., 2006; Schober, 2008). In the case of limb ischemia, activated circulating platelets are one source of SDF-1 in plasma (Jin et al., 2006). Within minutes after vessel injury, platelets adhere to the exposed subendothelium, and these adherent/activated platelets release SDF-1. Within the ensuing hours and days after endothelial disruption, apoptotic vascular smooth muscle cells appear to account for the long-term release of SDF-1 (Massberg et al., 2006). An increase in circulating SDF-1 affects the gradient of this cytokine between blood and BM. With higher concentrations in blood, stem cells are preferentially mobilized from BM to blood.
In our adoptive transfer experiments, there was a reduction (28–30%) in the numbers of infiltrated GFP+ myeloid cells in the SB-3CT- and AMD3100-treated groups, relative to the control group, whereas there was a reduction of 45% in the group treated with both SB-3CT and AMD3100. These findings suggest that both SDF-1 and MMP-9 support transmigration. Importantly, the combination of SB-3CT and AMD3100 results in an additive effect on the transmigration capacity of monocytes, suggesting independent mechanisms of action. The latter finding is perhaps not surprising. While SDF-1 signals migration of cells toward regions containing higher concentrations of SDF-1, MMP-9 released by transmigrating monocytes likely degrades the endothelial basement membrane and the adjacent ECM, thus providing access to the injured spinal cord (Rosenberg, 2002).
Here we found MMP-9-related differences in migration of BMDMs in response to SDF-1 across bare Transwells relative to their transmigration across Transwells, coated with an endothelial cell layer. While deficiency in MMP-9 had no effect on migration, transmigration was significantly reduced in MMP-9 KO cells or WT cells, treated with an MMP-9 inhibitor. Such findings emphasize the distinction between migration and transmigration with the latter requiring additional steps to cross an endothelial interface. The dependency of transmigration on MMP-9 is consistent with its role in degradation of components of the endothelial junctional complexes and the adjacent basement membrane. The blood–brain/spinal cord barrier-associated protein zonula occludens-1, occludin, collagens, and laminins, comprising the endothelial basement membrane, are substrates for MMPs, including MMP-9 (Asahi et al., 2001; Lohmann et al., 2004; Caron et al., 2005; Navaratna et al., 2007; Yong et al., 2007; Buhler et al., 2009).
Other factors may also support the trafficking of monocytes into the injured cord. In both MMP-9-deficient mice and our adoptive transfer models treated with SB-3CT, the reduction in influx of F4/80+ macrophages or GFP+ myeloid cells was by ∼30%. This suggests the involvement of other factors in transmigration of monocytes, including other MMPs. One likely candidate is MMP-12. Deficiency of this protease results in reduced microglia/macrophage density in the injured spinal cord (Wells et al., 2003). In vitro, the requirement of MMP-14 (MT1-MMP) during human monocyte migration and endothelial transmigration has also been demonstrated. MT1-MMP is upregulated by monocytes following their attachment to fibronectin and to TNFα-activated endothelial monolayer, and monocyte transmigration across TNFα-activated endothelium is inhibited by anti-MT1-MMP mAb (Matías-Román et al., 2005). Monocyte stimulated with LPS and proinflammatory cytokines leads to induction of several other MMPs, including MMP-1, -2, and -3, which also play a central role in ECM degradation (Webster and Crowe, 2006). Beyond the effects of MMPs, there are a number of factors that can disrupt the barrier, including proinflammatory cytokines and chemokines (i.e., IL-1, TNF-α, IL-6, GM-CSF, MCP-1), thus allowing the trafficking of immune cells into the CNS (Stamatovic et al., 2005).
Few studies address the mechanisms underlying trafficking of leukocytes across the glia limitans. We found that GFP+ cells accumulated in the meninges in the SB-3CT-treated group, suggesting that MMP activity is required for these cells to cross the glia limitans into the adjacent parenchyma. SDF-1 is expressed in the meninges after SCI (Tysseling et al., 2011); thus, this cytokine may play a key role in signaling the recruitment of GFP+ cells. The glia limitans is composed of astrocytic foot processes and a distinct parenchymal basement membrane (Owens et al., 2008). Dystroglycan, a transmembrane receptor that anchors astrocytic endfeet to the basement membrane via high-affinity interactions with laminins 1 and 2, perlecan, and agrin, is a specific substrate of MMP-2 and MMP-9 (Agrawal et al., 2006). In recent studies involving experimental autoimmune encephalomyelitis, mice treated with a function-blocking antibody to an extracellular matrix metalloproteinase inducer (EMMPRIN) showed reduced clinical severity accompanied by decreased parenchymal invasion of leukocytes. Moreover, reduced invasion of leukocytes was associated with diminished MMP proteolytic activity at the level of the glia limitans, thus suggesting disruption of this barrier to parenchymal invasion (Agrawal et al., 2011). Our findings suggest that monocytes infiltrate the injured cord by several routes, a traditional intraparenchymal vascular route and an alternative pathway, beginning at the meningeal interface with infiltration via the Virchow Robin space and the adjacent glia limitans.
We found reduced numbers of GFP+ myeloid cells in the injured spinal cord at 24 h after their adoptive transfer in mice treated with either SB-3CT or AMD3100. While this early period is best known for neutrophil trafficking into the injured cord (Kigerl et al., 2006), there is growing evidence that monocytes likewise infiltrate early after injury. Circulating monocytes are highly mobile and rapidly recruited into inflamed tissues, simultaneously with neutrophils (Henderson et al., 2003; Gordon and Taylor, 2005). For example, monocytes are rapidly recruited to the peritoneal cavity after exposure to sterile thioglycolate, reaching values similar to those of neutrophils by 24 h. Similarly, infiltrated monocytes in the injured cord (CD45high CD11bhigh GR-1neg-int) increase by 12 h and remain high for at least 72 h (Saiwai et al., 2010), a time period that corresponds to the infiltration of neutrophils (Stirling and Yong, 2008; Saiwai et al., 2010).
In conclusion, we show that SDF-1/CXCR4 ligation and MMP-9 support the trafficking of blood-borne monocytes into the injured cord. Defining the signaling pathways that govern the migration of monocytes into the injured cord is an essential step toward understanding their contributions to injury and reparative processes and long-term neurologic recovery.
Footnotes
This work was supported by NINDS Grant R01 NS039278.
- Correspondence should be addressed to Haoqian Zhang, 513 Parnassus Avenue, Room HSE 722, San Francisco, CA 94143-0110. Haoqian.Zhang{at}ucsf.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}