

Abstract
Deposition of amyloid β peptides (Aβs) in extracellular amyloid plaques within the human brain is a hallmark of Alzheimer's disease (AD). Aβ derives from proteolytic processing of the amyloid precursor protein (APP) by β- and γ-secretases. The initial cleavage by β-secretase results in shedding of the APP ectodomain and generation of APP C-terminal fragments (APP-CTFs), which can then be further processed within the transmembrane domain by γ-secretase, resulting in release of Aβ. Here, we demonstrate that accumulation of sphingolipids (SLs), as occurs in lysosomal lipid storage disorders (LSDs), decreases the lysosome-dependent degradation of APP-CTFs and stimulates γ-secretase activity. Together, this results in increased generation of both intracellular and secreted Aβ. Notably, primary fibroblasts from patients with different SL storage diseases show strong accumulation of potentially amyloidogenic APP-CTFs. By using biochemical, cell biological, and genetic approaches, we demonstrate that SL accumulation affects autophagic flux and impairs the clearance of APP-CTFs. Thus, accumulation of SLs might not only underlie the pathogenesis of LSDs, but also trigger increased generation of Aβ and contribute to neurodegeneration in sporadic AD.
Introduction
Alzheimer's disease (AD) is characterized by extracellular deposits of amyloid β-peptide (Aβ) plaques and intraneuronal aggregates of hyperphosphorylated tau protein in the brain (Selkoe, 2001). Aβ derives from proteolytic processing of the β-amyloid precursor protein (APP). The initial cleavage of APP by the β-secretase/β-site APP-cleaving enzyme (BACE1) results in the secretion of the soluble ectodomain and the generation of a C-terminal fragment (CTF-β), which remains inserted in cellular membranes. CTF-β represents a substrate for γ-secretase that cleaves within the transmembrane domain, resulting in the production of Aβ (Walter et al., 2001). Mutations in the genes for presenilins that represent the catalytic subunits of γ-secretase or in the APP gene itself cause familial forms of early onset AD and commonly lead to increased generation and/or aggregation of Aβ. However, such mutations are very rare and account for only 1–5% of all AD cases (Tanzi and Bertram, 2001). Factors that could contribute to increased Aβ production in the pathogenesis of the much more common late-onset form of AD are largely unknown (St George-Hyslop, 2000).
Recently, it has been shown that Aβ could be generated during macroautophagy (herein referred to as autophagy) (Yu et al., 2005). Autophagy is a fundamental homeostatic process in eukaryotic cells that allows recycling of biomolecules. At the time of starvation, induction of autophagy provides amino acids and other components for the synthesis of essential macromolecules allowing cell survival (Levine and Kroemer, 2008). The autophagic process could be divided into distinct steps: induction of autophagosome formation, subsequent docking and fusion with lysosomes (maturation), and final breakdown of autophagic bodies (Klionsky and Emr, 2000). Basal autophagic activity is crucial for homeostatic turnover of cytoplasmic constituents in most tissues (Shintani and Klionsky, 2004). Neurons have a relatively high basal autophagic activity (Boland et al., 2008), and its inhibition causes neurodegeneration in mice (Cuervo et al., 2005; Hara et al., 2006; Komatsu et al., 2006). A number of aggregation-prone pathogenic proteins, including huntingtin, α-synuclein, and tau, appear to be degraded via autophagic pathways (Li et al., 2008; McCray and Taylor, 2008; Ravikumar et al., 2008; Wang et al., 2009). Thus, the regulation of autophagy could play an important role in the pathogenesis of neurodegenerative diseases (Eskelinen and Saftig, 2009).
Impairment of autophagy has also been associated with certain lysosomal storage disorders (LSDs) (Settembre et al., 2008). Several LSDs that are caused by impaired degradation of membrane lipids share pathological similarities with AD, such as extensive neurodegeneration, presence of intracellular tau protein aggregates, and neuroinflammation (Auer et al., 1995; Jeyakumar et al., 2003; Rojo et al., 2008). Moreover, the lipid composition is significantly altered in AD brain (Han, 2005). There is compelling evidence to support a role of membrane lipids, including cholesterol and sphingolipids (SLs) and glycosphingolipids (GSLs), in the proteolytic processing of APP (Simons and Ehehalt, 2002). However, the detailed mechanisms by which the accumulation of membrane lipids in endosomes and lysosomes could affect APP processing in these compartments are not well understood.
Materials and Methods
cDNA constructs, antibodies, and reagents.
The constructs encoding C99-EGFP, LC3-GFP, and LC3-RFP have been described earlier (Kabeya et al., 2000; Kaether et al., 2006). Anti-APP-CT (140), APP-ectodomain (5313), and anti-Aβ (2964) (Tamboli et al., 2005), anti-APP-CT (C1/6.1) (Mathews et al., 2002), and anti-cathepsin CT (generous gift from Dr. S. Höning, Cologne, Germany) antibodies have been described before. Antibodies against LC3 and 82E1 (Cell Signaling Technology), P70S6 kinase and phospho-P70S6 kinase (pP70S6K) (Abcam), lamp-2 (Iowa Hybridoma Bank), rab-7, beclin-1, and Ser 70 phospho-Bcl-2 (Santa Cruz Biotechnology), Bcl-2 (BD Transduction), and p62 (Sigma) were obtained from the respective companies listed. GSLs (Calbiochem), N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT), leupeptin, E64d, and vinblastine (Sigma), 3-methyladenine, trehalose, BH3I-1″, and rapamycin (Axxora), and EBSS and cholera toxin-FITC (Invitrogen) were purchased from the respective sources listed.
Cell culture, transfection, and immunocytochemistry.
Embryonic fibroblasts of wild-type (WT) and lamp-1/lamp-2 double knock-out (lamp-KO) mice were described previously (Eskelinen et al., 2004). Mouse melanoma B16, GM95, and embryonic fibroblasts of control wild-type and atg5-knock-out (atg5-KO) mouse were obtained from the Riken Cell Bank. Additional cell lines were obtained from ATCC. Cells were cultured in DMEM supplemented with 10% fetal calf serum. Transfection of cells with recombinant cDNA constructs was performed with Lipofectamine (Invitrogen) according to the manufacturer's instructions. Clones were selected with 100 μg/ml zeocine (Invitrogen). For immunocytochemistry, cells grown on polylysine-coated glass coverslips were fixed in 4% paraformaldehyde before processing for immunofluorescence as described previously (Tamboli et al., 2008).
Electron microscopy.
Cells were fixed in 3% glutaraldehyde for 3 h followed by washing with 0.1 m HEPES buffer. Cells were later progressively dehydrated in a graded series of ethanol (30–100%) and embedded in Epon. Ultrathin sections (50–60 nm) were cut using an Ultracut microtome and placed on copper grids for analysis. Grids were stained and contrasted with uranyl acetate and lead citrate, followed by examination with Zeiss TEM900 electron microscope.
In vitro γ-secretase assay.
Cells were lysed in hypotonic buffer (10 mm Tris, pH 7.6, 1 mm EDTA, and 1 mm EGTA) on ice for 10 min. The cell suspension was then passed through a needle 15 times. The cell debris and nuclear fraction were pelleted by centrifugation, and the supernatant was further centrifuged at 13,200 rpm for 1 h to obtain a membrane pellet. The membrane pellet was resuspended in citrate buffer containing protease inhibitors and incubated at 37°C for 2 h. After 2 h, the tubes were centrifuged at 13,200 rpm for 1 h. Supernatant was analyzed by immunoblotting for generation of APP intracellular domain (AICD), whereas APP-CTFs were detected in pellets.
Metabolic labeling, immunoprecipitation, and Western immunoblotting.
Metabolic labeling of APP and APP-CTFs was performed as described before (Tamboli et al., 2005). Briefly, cells were starved at 37°C in methionine-free, serum-free medium for 60 min and then labeled with [35S]methionine/[35S]cysteine (ICN) at 37°C for 10 min. Cells were then washed with PBS and chased in medium supplemented with 10% FCS and excess amounts of unlabeled methionine for the indicated periods of time. Cells were lysed in STEN buffer (50 mm Tris, pH 7.6, 150 mm NaCl, and 2 mm EDTA) supplemented with 1% NP-40/1% Triton X-100/2% BSA on ice for 10 min. Lysates were clarified by centrifugation for 20 min at 13,200 rpm and immunoprecipitated for 3 h at 4°C. Cellular APP was immunoprecipitated with APP ectodomain antibody before immunoprecipitation by APP-CTF antibody. After separation by SDS-PAGE, proteins were transferred to nitrocellulose membrane (Schleicher & Schuell) and analyzed by autoradiography or phosphoimaging. Alternatively, proteins were detected by immunoblotting using enhanced chemiluminescence reagent ECL (GE Healthcare).
Aβ measurements by ELISA.
Secreted Aβ peptides in conditioned medium were quantified by a sandwich immunoassay using the Meso Scale Discovery Sector Imager 2400 as described previously (Page et al., 2008). Briefly, streptavidin-coated 96-well multiarray plates were incubated with biotinylated 2D8 capture antibody. This was followed by addition of media samples and Aβ peptide standards (Bachem). Ruthenylated C-terminal-specific anti-Aβ38 (Meso Scale Discovery), anti-Aβ40, or anti-Aβ42 antibodies were used as detection antibodies. For detection, Meso Scale Discovery Read buffer was added, and the light emission at 620 nm after electrochemical stimulation was measured using the Meso Scale Discovery Sector Imager 2400 reader. The corresponding concentrations of Aβ peptides were calculated using the Meso Scale Discovery Workbench software.
Isolation and analysis of cellular sphingolipids.
Cells were homogenized in 1 ml of water, and aliquots for protein determination were saved. Lipids were extracted for 24 h at 37°C in chloroform/methanol/water (2:5:1, v/v/v). After evaporation in a stream of nitrogen, phospholipids were degraded by mild alkaline hydrolysis with methanolic NaOH (100 mm) for 2 h at 37°C. The lipid extracts were desalted by reversed-phase chromatography on LiChroprep RP18, applied to TLC plates (Merck), and developed in chloroform/methanol/ 0.22% aqueous CaCl2 55:45:10 (v/v/v). For quantitative analytical TLC determination, increasing amounts of standard lipids (GM1, GD1b) were applied. After development, plates were air dried, sprayed with 8% (w/v) H3PO4 containing 10% (w/v) copper (II) sulfate pentahydrate, and charred at 180°C for 10 min, and lipids were quantified by photo densitometry (Shimadzu) at λ = 595 nm.
Data analysis and statistics.
ECL signals were measured and analyzed using an ECL imager (ChemiDoc XRS, Bio-Rad) and the Quantity One software package (Bio-Rad). Statistical analysis was done using SD and Student's t test. Significance values are indicated by asterisks as follows: *p < 0.05; **p < 0.01; ***p < 0.001. Note that expression of all analyzed proteins was normalized to cellular actin levels.
Results
Accumulation of SLs stabilizes APP-CTFs
To selectively increase the levels of GSLs in cellular membranes, human neuroglioma H4 cells were incubated with a mixture of purified bovine brain GSLs. As compared to endogenous levels, we observed an approximately twofold increase in the cellular levels of individual GSLs (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Exogenous GSLs appeared to accumulate at the cell surface as well as within intracellular vesicular compartments (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). The addition of exogenous GSLs to human H4 cells led to marked accumulation of APP-CTFs that represent substrates for γ-secretase, and to a lesser extent of full-length APP (Fig. 1A). Similar effects of GSLs on APP-CTFs were also observed in several other cell types, including human neuroblastoma SHSY-5Y (Fig. 1B), human embryonic kidney (HEK293), and HeLa cells (supplemental Fig. S2A,B, available at www.jneurosci.org as supplemental material).

SLs regulate cellular APP-CTF levels. A, B, SHSY-5Y (A) and H4 (B) cells were incubated without (control) or with (GSLs) 50 ng/μl purified bovine brain gangliosides for 48 h, and cellular APP (APP-fl), APP-CTFs, and actin were detected by Western immunoblotting. C, H4 cells were incubated with 50 ng/μl of individual GSL or total bovine brain GSLs (mix) for 48 h as indicated. APP-CTFs were analyzed in cellular membranes by Western immunoblotting. D, Primary fibroblasts from indicated sphingolipid storage disease patients and healthy control were obtained and expanded by culturing them further. Cellular APP and APP-CTFs were detected in isolated membranes of these cells. Numbers below the blots indicate the ratio of APP CTFs to full-length APP as determined by quantitative ECL imaging. E, APP-CTFs were immunoprecipitated from mouse melanoma control (B16) and GSL-deficient (GM95) cells using an APP-CT antibody and detected by Western immunoblotting. Full-length APP (APP-fl) expression in cellular membranes isolated from B16 and GM95 cells was analyzed by Western blotting. Expression of APP-CTFs was analyzed by ECL imaging (B16—100 ± 16.58%, GM95—21.83 ± 8.22%, p ≥ 0.018). Actin was detected as protein loading control. F, H4 cells, stably overexpressing C99-EGFP, were incubated with 50 ng/μl GSLs. Cellular C99-EGFP, full-length APP, and actin were detected by Western immunoblotting (* and ** represent C99-EGFP fragments further processed by β- and α-secretase, β′-CTF-EGFP and α-CTF-EGFP, respectively). C99-EGFP levels were quantified by ECL imaging (control—100 ± 1.55%, +GSLs—115.24 ± 2.1%, p ≥ 0.008).
Accumulation of APP-CTFs was also observed upon application of the major individual brain GSLs GM1, GD1a, GD1b, and GT1b as well as with sphingomyelin (Fig. 1C; supplemental Fig. S2C, available at www.jneurosci.org as supplemental material). Phosphatidylserine or phosphatidic acid did not increase CTF levels, suggesting that the metabolism of APP-CTFs is selectively affected by SLs. To prove that SL storage indeed affects APP metabolism, we analyzed APP-CTFs and full-length APP in human primary fibroblasts obtained from patients with distinct LSDs, i.e., Niemann–Pick A and B (NPA and NPB), Tay–Sachs, and Sandhoff's diseases (Kolter and Sandhoff, 2006). These cells showed prominent accumulation of APP-CTFs (Fig. 1D). However, fibroblasts of a patient with Farber's disease, which selectively accumulate ceramide due to ceramidase deficiency, showed no accumulation of APP-CTFs. Notably, mouse melanoma GM95 cells that completely lack GSLs due to a genetic defect in glucosylceramide synthase (Ichikawa et al., 1994) had strongly reduced levels of APP-CTFs compared to control cells (Fig. 1E). We also confirmed the effect of GSLs in cells stably overexpressing a GFP-tagged variant of APP-CTF-β, C99-EGFP, which mimics the APP-CTF generated after β-secretase cleavage of APP. Addition of GSLs slightly but significantly increased the levels of C99-EGFP (Fig. 1F). The much weaker effect of SLs on C99-EGFP as compared to the effects on endogenous APP-CTFs might be due to already very high levels of C99-EGFP upon overexpression. We also checked the effect of GSLs on cells overexpressing Swedish APP, which is preferentially cleaved by β-secretase to generate predominantly β-CTFs. APP-CTFs generated from Swedish APP also accumulated upon GSL treatment (supplemental Fig. S2D, available at www.jneurosci.org as supplemental material). Together, these data demonstrate a role of SL accumulation in the metabolism of APP and/or APP-CTFs.
To analyze the metabolism of APP and APP-CTFs in more detail, pulse chase experiments were performed. Cellular proteins in control and GSL-treated cells were labeled with [35S]-methionine, and turnover of labeled APP and APP-CTFs was analyzed. APP undergoes maturation by N′ and O′-glycosylation in control and GSL-treated cells as indicated by the appearance of a slower-migrating form during the chase (Fig. 2A) (Walter et al., 1997, Tamboli et al., 2005). In the pulse-chase experiments, APP-CTFs are generated from processing of its radio-labeled precursor APP and thus appear only after 1 h chase, coinciding with APP maturation. This is followed by steady increase in CTF signal for up to 4 h due to efficient processing of labeled APP during this time and relatively slower clearance of APP-CTFs. The generation of APP-CTFs appeared after 1 h of chase and was similar in treated and control cell (Fig. 2A). While CTF-α was the predominant species present in H4 cells, CTF-β and CTF-β′ (CTF generated by alternative β-secretase cleavage at Glu-11 site) were also detected (Fluhrer et al., 2002; Lee et al., 2003).

Stabilization of APP-CTFs by GSLs. A, After culturing in the absence or presence of 50 ng/μl GSLs for 48 h, H4 cells were labeled with [35S]-methionine for 10 min and chased for the indicated time periods. GSLs were excluded from both pulse and chase media of cells. APP was immunoprecipitated from cell lysates using APP ectodomain antibody, followed by immunoprecipitation with APP-CT antibody to precipitate APP-CTFs. Full-length APP and APP-CTFs were detected by phosphoimaging. CTFs generated by β-secretase cleavage (β CTF) and α-secretase cleavage (α CTF) are indicated by arrowheads. The CTFs generated by alternative β-secretase cleavage are indicated by an asterisk. B–E, H4 cells cultured with and without 50 ng/μl GSLs for 48 h were labeled with [35S]-methionine for 10 min and chased for the indicated time periods. APP and APP-CTFs were immunoprecipitated and detected as above (B). Time-dependent metabolism of β APP-CTFs (C), α CTFs (D), and total CTFs (E) was analyzed by ECL imaging quantification. F, H4 cells were grown with and without 50 ng/μl GSLs for 48 h; cells were then labeled with [35S]-methionine for 10 min and chased for 16 h. Pulse chase as well as detection of cellular APP and APP-CTFs was performed as described above. G, Quantification of APP-CTF expression in control and GSL-treated cells after 2 and 16 h chase. H, The rate of APP-CTF degradation in control and GSL-treated cells. Almost complete degradation of APP-CTFs is observed in control cells (99.03%) after 16 h, whereas only 33.5% of CTFs were degraded in GSL-enriched cells when compared to amounts present at 2 h.
Since the levels of radiolabeled APP-CTFs increased within 4 h of chase in control as well as in GSL-treated cells, we prolonged chase periods in the next experiments. Degradation of CTFs was observed after 6–8 h in HEK293 cells (supplemental Fig. S2E, available at www.jneurosci.org as supplemental material) and was evident after 10–12 h chase period in H4 cells (Fig. 2B). GSLs appeared to increase the stability of APP-CTFs. Importantly, CTF-α and amyloidogenic CTF-β both are stabilized by GSLs (Fig. 2C–E). After 16 h, almost all APP-CTFs radiolabeled after the pulse were degraded in control cells (Fig. 2F). In contrast, APP-CTFs in GSL enriched cells were much more stable, indicating attenuated degradation (Fig. 2F,G). When compared with initial levels of APP-CTFs generated after 2 h, 99.03% of APP-CTFs were degraded in control cells, whereas only 33.5% of APP-CTFs were degraded in GSL-treated cells after 16 h (Fig. 2H). After maturation, full-length APP was efficiently turned over in both control and GSL-treated cells (Fig. 2F,G). Thus, accumulated GSLs selectively impair the clearance of APP-CTFs, which ultimately results in strong accumulation of APP-CTFs. There was also a trend toward increased APP levels in the presence of GSLs; however, rate of APP degradation within 16 h was similar in control and GSL-treated cells, indicating selective effect of GSLs on APP-CTF degradation (Fig. 2H).
Sphingolipid-mediated induction of autophagy causes accumulation of APP-CTFs in autophagic vacuoles and subsequent processing via γ-secretase
Since deficiency of γ-secretase could also lead to accumulation of APP-CTFs, we tested whether SLs affect the activity of γ-secretase and thereby stabilize APP-CTFs. In vitro γ-secretase assays were performed with purified cellular membranes in the absence or presence of exogenous GSLs. Consistent with previous results (Osenkowski et al., 2008), the addition of GSLs to isolated membranes rather increased γ-secretase activity as indicated by increased production of AICD (Fig. 3A, top two panels). γ-Secretase activity was also increased in isolated membranes from cells that have been pretreated with GSLs as compared to those from control cells (Fig. 3A, bottom two panels). Moreover, GSL treatment increased the generation of both extracellular and intracellular Aβ from endogenous APP in human SH-SY5Y cells (Fig. 3B). GSLs also significantly increased the secretion of the Aβ variants Aβ38, Aβ40, and Aβ42 in cells stably overexpressing APP C99-EGFP (Fig. 3C). Additionally, GSL treatment of cells pretreated with γ-secretase inhibitor DAPT further increased the levels of APP-CTFs (supplemental Fig. S3, available at www.jneurosci.org as supplemental material), confirming that accumulation of CTFs by GSLs is not mediated by inhibition of γ-secretase.

Induction of autophagy by GSLs increases extracellular Aβ. A, Membranes were isolated from SHSY-5Y cells and subjected to in vitro γ-secretase assay in the absence or presence of exogenous GSLs (top two panels). Alternatively, γ-secretase activity was analyzed in isolated membranes from SHSY-5Y cells that were preincubated in the presence or absence of GSLs for 48 h (bottom two panels). APP-CTFs and AICD were detected in the pellet and supernatant, respectively, by Western immunoblotting using antibody 140 directed against the cytoplasmic domain of APP. B, Human SH-SY5Y cells were treated with GSLs for 48 h and endogenously generated Aβ was immunoprecipitated from conditioned media (secreted) as well as from RIPA buffer cell lysates (intracellular) and estimated by Western immunoblotting followed by ECL imaging. C, H4 cells, stably overexpressing C99-EGFP, were treated with 50 ng/μl GSLs for 48 h, and secreted Aβ38, Aβ40, and Aβ42 were measured by ELISA. D, H4 cells were incubated in the absence (control) or presence (+GSLs) with 50 ng/μl GSLs for 48 h. Endogenous LC3 expression was analyzed by immunocytochemistry. Scale bar, 25 μm. E, H4 cells were treated with GSLs for 48 h, and Bcl-2, phospho-Bcl-2 (pBcl-2; Ser 70), and beclin-1 (bec-1) were detected in cell lysates by immunoblotting, followed by quantification using ECL imaging. F, H4 cells were treated with 10 μg/ml rapamycin (+rapa), 100 mm trehalose (+tre), or 20 μm BH3 mimetic BH3I-1 (+BH3) for 24 h. Cellular APP-CTFs, APP, and actin were detected by immunoblotting. APP-CTF levels in percentage were quantified using ECL imaging.
Previous studies implicated lysosomal and autophagic pathways in the metabolism of APP-CTFs (Haass et al., 1992; Yu et al., 2005; Nixon, 2007). Accordingly, cell treatment with leupeptin, a reversible inhibitor of lysosomal serine and thiol proteases that also affects autophagic clearance, led to strong accumulation of APP-CTFs (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). To check whether GSLs could affect the autophagic process, we analyzed the localization of LC3 in control and GSL-treated cells by immunostaining. Control cells showed weak cytosolic staining, whereas GSLs caused an accumulation of LC3 in autophagic vesicles in nearly 20% cells (Fig. 3D). Increase in autophagic vesicles in GSL-treated cells could be due to either increased induction or reduced clearance of autophagic vesicles. LSDs have been previously linked with enhanced autophagy via beclin-1 (Pacheco et al., 2007). To test whether GSLs also affect induction of autophagy via this pathway, we analyzed the expression of beclin-1 and its binding partner bcl-2. There was a trend toward increased beclin-1 expression upon GSL application. GSLs also reduced the total Bcl-2 levels and increased phosphorylated Bcl-2. Because phosphorylation of Bcl-2 inhibits the interaction with beclin-1, these data are consistent with an increased induction of autophagy by free beclin-1 (Wei et al., 2008) (Fig. 3E). In addition, induction of autophagy by BH-3 mimetic, which disrupts interaction between Bcl-2 and beclin-1, also led to accumulation of APP-CTFs (Ghiotto et al., 2010). Further, induction of autophagy by rapamycin (Laplante and Sabatini, 2009) and trehalose (Sarkar and Rubinsztein, 2008) caused the accumulation of APP-CTFs (Fig. 3F). Together, these data suggest that increased induction of autophagy might contribute to the GSL-dependent accumulation of APP-CTFs. This led us to analyze GSL-dependent localization of APP-CTFs in autophagic compartments. To differentiate between full-length APP and APP-CTFs, we costained H4 cells with antibodies against APP C terminus (APP-CT) and APP N terminus (APP-NT). While a staining of compartments with both antibodies could indicate the presence of full-length APP, a selective detection with APP-CT might represent localization of APP-CTFs. In control cells, a strong costaining in juxtanuclear structures indicates the presence of full-length APP in Golgi compartments (Fig. 4A; supplemental Fig. S5, available at www.jneurosci.org as supplemental material). Vesicles selectively stained with APP-CT antibodies were relatively rare. Notably, the treatment with GSLs strongly increased the number of APP-CTF-containing cytosolic vesicles, while the juxtanuclear localization of full-length APP was not strongly altered (Fig. 4B; supplemental Fig. S5, available at www.jneurosci.org as supplemental material). To test whether these APP-CTF-containing vesicles involve autophagic compartments, we transfected the cells with LC3-GFP and performed costains with APP-NT and APP-CT antibodies in the presence and absence of GSLs. We found few LC3-GFP-containing vesicles stained with APP-NT antibody in control and GSL-treated cells. Costaining between APP-NT and LC3-GFP did not change significantly depending on GSLs. Importantly, GSLs increased the colocalization of LC3-GFP with APP-CT, thus indicating accumulation of APP-CTFs in autophagic vesicles in the presence of GSLs (Fig. 4C).

Localization of APP-CTFs within autophagic vacuoles. A, Control and 50 ng/μl GSL-treated (48 h) H4 cells were fixed, permeabilized, and stained with polyclonal APP-ectodomain antibody 5313, monoclonal APP-CT antibody C1/6.1, and respective secondary fluorescent antibodies. Staining was analyzed by microscopy. B, C, H4 cells were transiently transfected with LC3-GFP and treated with GSLs for 48 h. Control and GSL-treated cells were later stained with APP ectodomain antibody 5313 (B) and APP-CT antibody 5818 (C). Analysis of LC3-GFP-expressing and APP/APP-CTF-containing vesicles was performed by microscopy. Scale bar, 25 μm.
Accumulation of GSLs impairs autophagic clearance
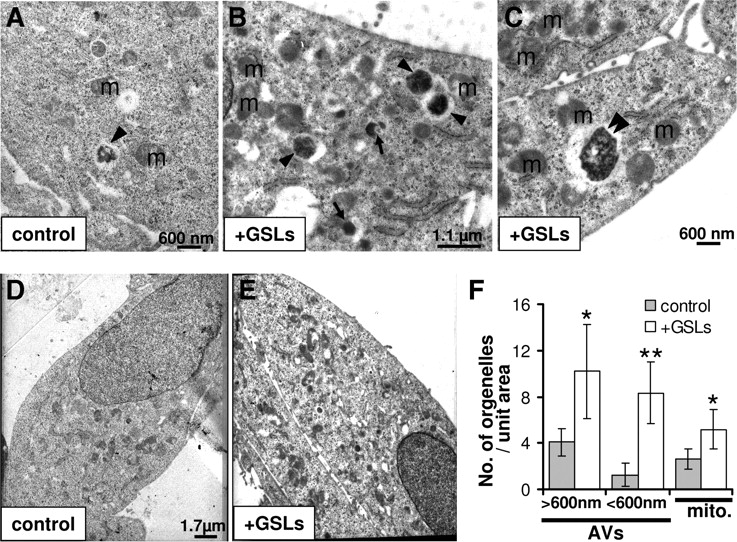
To gain further insights into GSL-dependent regulation of autophagy, we performed ultrastructural analysis using electron microscopy (Fig. 5). In contrast to controls (Fig. 5A,D), GSL-treated cells (Fig. 5B,C,E) contained several electron-dense (arrowheads and arrows) as well as clear and occasionally multinucleated (double arrowhead) autophagolysosomes with 300–2000 nm diameter (Fig. 5A–C). Some of these vacuoles appeared to be either in a process of fusion or in close proximity with each other. Vacuoles with diameter >600 nm (arrows) might represent autophagosomes/endosomes recently fused with lysosomes, whereas ones with diameter <600 nm might be residual bodies with minimal or no degrading activity (arrows). Such a pattern with the presence of electron-dense autophagic vacuoles (AVs) has been previously observed upon inhibition of autophagosome proteolysis or inhibition of autophagosome–lysosome fusion/maturation (Boland et al., 2008). Additionally, the number of mitochondria increased significantly in GSL-treated cells, and a lot of these mitochondria were damaged and swollen, indicating impaired turnover via mitophagy (Fig. 5) (Chen and Chan, 2009).

GSLs impair autophagic flux. A–E, Electron micrographs of control (A, D) and 48 h, 50 ng/μl GSL-treated (B, C, E) H4 cells. Electron-dense AVs and lysosomal compartments with diameter >600 nm are indicated with arrows and with diameter <600 nm are indicated with arrowheads. Double arrowhead indicates multinucleated AV. Mitochondria are indicated as “m.” F, Number of different size AVs (electron dense and clear) and total number of mitochondria (mito.) per unit area were quantified using seven different overview micrographs (similar to D and E) from control as well as GSL-treated cells.
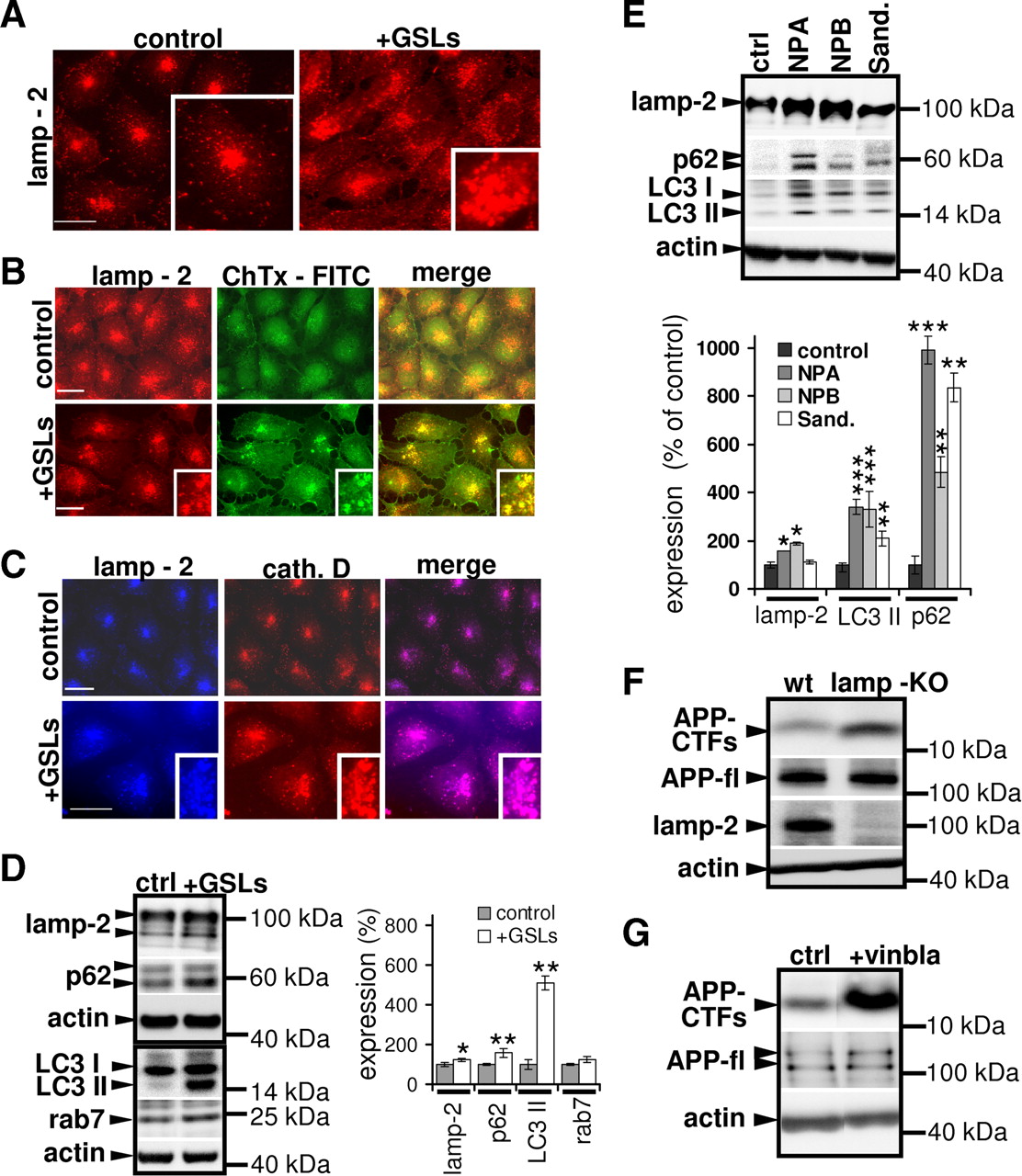
To further investigate the GSL-dependent changes in lysosomal function and morphology, we analyzed the subcellular distribution of lamp-2. Control as well as GSL-treated H4 cells showed predominantly punctuate distribution of lamp-2, indicating localization of lamp-2 in cytoplasmic vesicles/lysosomes. These vesicles tended to concentrate in the juxtanuclear region (Fig. 6A). Notably, treatment with GSLs led to increased number of cells with dispersed lamp-2 stain and appearance of lamp-2-positive larger cytosolic vesicles as compared to control cells. Quantitative analysis revealed that 31.71 ± 10.17% of GSL-treated cells, but only 8.63 ± 5.7% of control cells (p < 0.01) showed such enlarged compartments. These enlarged lamp-2-positive vesicles also contained GM1, one of the major components of the GSLs, as well as lysosomal enzyme cathepsin D (Fig. 6A–C). The presence of enlarged lysosomes upon accumulation of GSLs was also confirmed by direct staining with LysoTracker dye (supplemental Fig. S6A, available at www.jneurosci.org as supplemental material). While lamp-2 protein levels were modestly increased, levels of p62, an autophagic substrate protein (Jaakkola and Pursiheimo, 2009), and LC3 II were increased significantly by GSL treatment (Fig. 6D). Interestingly, LC3 II/I ratio (control—0.98 ± 0.21, +GSLs—4.61 ± 0.60, p ≥ 0.01) and LC3 I (control—100 ± 9.5%, +GSLs—124.29 ± 18.55%, p ≥ 0.049) and LC3 II (control—100 ± 29.63%, +GSLs—547.68 ± 50.90%, p ≥ 0.01) levels increased upon GSL application. Thus, the increased LC3 II/I ratio might not only be due to increased conversion of LC3 I to LC3 II, but also involves strong accumulation of LC3 II by decreased consumption during the autophagic process. This further supports impaired clearance of autophagy by GSLs. Also, levels of rab-7, another protein associated with AVs, were modestly increased upon GSL application. Moreover, increase in total lamp-2, p62, and LC3 II levels were detected in fibroblasts obtained from patients with different sphingolipid storage diseases (Fig. 6E). Notably, processing of lysosomal protease cathepsin D was not significantly affected by GSLs, as evidenced by unaltered levels of 31 kDa mature cathepsin D fragment (supplemental Fig. S6B, available at www.jneurosci.org as supplemental material). Together these data indicate that GSLs could promote induction of autophagy as well as impair efficient clearance/maturation during this process. Both effects might contribute to accumulation of APP-CTFs in AVs.

Impaired autophagic clearance contributes to accumulation of APP-CTFs. A–C, Control and 72 h GSL-treated (+GSLs) cells were fixed, permeabilized, and stained using monoclonal antibody against lamp-2 (A), antibody against lamp-2 and cholera toxin-FITC (10 μg/ml), which binds to GM1 (B), or antibodies against lamp-2 and cathepsin-D (cath. D) (C) followed by respective secondary Alexa antibodies against primary antibodies. Distribution of lamp-2 and its colocalization with GM1 or cathepsin D was analyzed by microscopy. Presence of lamp-2-positive aggregates was estimated by evaluating staining from control and GSL-treated cells, 100 each (control: 8.63 ± 5.7%, +GSLs: 31.71 ± 10.17%, p ≥ 0.01). Scale bar, 25 μm. D, H4 cells were treated with GSLs for 72 h, and indicated proteins were detected with immunoblotting; ECL imaging was used for quantification. E, Primary fibroblasts from NPA/NPB and Sandhoff's disease (Sand.) patients and a healthy control were analyzed for expression of cellular lamp-2, p62, LC3 II, and actin by Western immunoblotting, followed by ECL imaging quantification. F, Lysates were prepared from wild-type (MEF-WT) and lamp-KO mouse embryonic fibroblasts. APP-CTFs, full-length APP (APP-fl), lamp-2, and actin were detected by Western immunoblotting as indicated. APP-CTF levels were quantified using ECL imaging (WT—100 ± 9.76%, lamp-KO—258.37 ± 67.47%, p ≥ 0.05). G, H4 cells were treated with 10 μm vinblastine (+vinbla.) for 6 h, and APP-CTFs, APP, and actin were detected in cellular lysates by Western immunoblotting. APP-CTF levels were quantified using ECL imager (control: 100 ± 10.32%, + vinblastine: 225.92 ± 10.42%, p ≥ 0.007).
To prove that impaired autophagic clearance affects the metabolism of APP-CTFs, we analyzed their levels in embryonic fibroblasts from WT and lamp-KO mice. Lamp proteins have been shown to be crucial for fusion of autophagosomes with lysosomes (Saftig et al., 2008); therefore, lamp-1 and 2 deficient mouse embryonic fibroblasts (lamp-KO) represent a genetic model to study the impact of impaired autophagic maturation. We observed strong accumulation of APP-CTFs in lamp-KO cells (Fig. 6F), suggesting an important role of lamp-2-dependent autophagic clearance in APP-CTF metabolism. In addition, pharmacological inhibition of autophagosome maturation by vinblastine (Kovács et al., 1982), also caused strong accumulation of endogenous APP-CTFs (Fig. 6G) as well as of overexpressed C99-EGFP in H4 cells (supplemental Fig. S7A, available at www.jneurosci.org as supplemental material). However, extracellular Aβ levels did not change significantly upon vinblastine treatment, most likely due to inhibition of release of excess Aβ generated within AVs, as vinblastine is known to inhibit microtubule-dependent vesicle transport (supplemental Fig. S7B, available at www.jneurosci.org as supplemental material). Accordingly, intracellular Aβ levels were strongly increased in vinblastine-treated cells (supplemental Fig. S7C, available at www.jneurosci.org as supplemental material).
GSLs do not inhibit the fusion of autophagosomes with endosomes and lysosomes
Starvation of cells by culturing them in a medium devoid of amino acids and growth factors (EBSS) is known to promote lysosomal function and induce efficient clearance of aggregated proteins by lysosome-dependent autophagy (Williams et al., 2006). Therefore, next, we tested whether GSL-dependent electron-dense structures containing undigested organelles and other cellular material (Fig. 5) can be cleared upon incubation in EBSS. Incubation of control cells for 6 h gave rise to relatively clear, big vacuoles sometimes with scattered amorphous material (indicated by asterisk). These AVs represent autolysosomes with efficient proteolytic ability (Fig. 7Aa). Induction of autophagy using EBSS in H4 cells treated with GSLs further increased the number of such clear autolysosomes; however, electron-dense vacuoles, likely AVs with undigested organelles (Fig. 7Ab–Ad), were still present. These results further support an impaired autophagic maturation by increased GSLs, which apparently cannot be overcome by additional induction of autophagy by starvation.

Fusion of autophagosomal compartments with lysosomes and endosomes in the presence of GSLs. A, Electron micrographs of H4 cells. Cells were incubated in the presence (b–d) or absence (a) of GSLs (50 ng/μl) for 48 h followed by starvation in EBSS medium ± GSLs. AVs and lysosomal compartments are indicated by asterisks and fusion events by arrows. B, C, H4 cells were transiently transfected with LC3-RFP or empty vector and incubated with 50 ng/μl GSLs for 48 h. Cells were later fixed and stained using antibody against lamp-2 (B) and EEA-1 (C). The colocalization of lamp-2 and EEA-1 with LC3-RFP was analyzed by immunocytochemistry. Scale bar, 25 μm.
Interestingly, AVs in the process of fusion were markedly increased upon EBSS treatment in the presence of GSLs (Fig. 7Ab). Increased number of AVs in the process of fusion might be either due to increased fusion of autophagosomes with endosomes/lysosomes or due to inefficient completion of fusion event in the presence of excess GSLs. To get additional insight into fusion processes, we analyzed the effect of GSLs on vesicular colocalization of LC3-RFP with lamp-2 or the endosomal marker EEA1. We observed colocalization of weakly stained, peripheral lamp-2-positive vesicles with LC3, whereas strongly labeled juxtanuclear condensed lamp-2 vesicles mostly lacked LC3 in control as well as GSL-treated cells (Fig. 7B, top and middle panels). However, we observed presence of LC3-RFP in some of the lamp-2-containing enlarged vesicles in GSL-treated cells (Fig. 7B, bottom). Thus, fusion between lamp-2-positive lysosomes and LC3-containing autophagosomes seems not to be strongly altered by GSLs. On the other hand, colocalization between EEA1 and LC3-RFP was significantly increased in the presence of GSLs, indicating increased fusion of endosomes with autophagosomes (Razi et al., 2009) (Fig. 7C). Together these data rule out an inhibition of fusion as a major reason for impaired autophagic flux in SL enriched cells. Note that for above studies, we specifically used LC3-RFP and not LC3-GFP, as GFP fluorescence is known to be unstable in acidic environment.
GSLs impair starvation-induced clearance of APP-CTFs
In contrast to treatment with GSLs, cells incubated with EBSS did not show presence of electron-dense AVs using EM analysis. Rather, the detection of translucent vesicles indicates proteolytically efficient lysosomal compartments in these cells (Figs. 5, 7). Moreover, induction of autophagy with EBSS also did not cause appearance of lamp-2-positive enlarged vesicles; instead, lamp-2-containing vesicles appeared to be more condensed in the juxtanuclear region (supplemental Fig. S8A, available at www.jneurosci.org as supplemental material). Thus, unlike the stimulation of autophagy by GSLs, EBSS seems to involve efficient turnover of AVs. Accordingly, induction of autophagy by EBSS led to consumption of p62 as well as rab-7 and lamp-2, proteins known to be associated with AVs, suggesting efficient autophagic clearance (Jäger et al., 2004; Saftig et al., 2008). Induction of autophagy upon incubation with EBSS led to increased levels of total LC3 as well as LC3 II after 6 h. APP-CTF levels decreased within 1 and 6 h, whereas levels of APP dropped slightly with time in control cells upon incubation with EBSS (Fig. 8). These data strongly indicate that autophagy could promote the clearance of potentially amyloidogenic APP-CTFs. This is further supported by the significant decrease of APP C99-GFP upon induction of autophagy by EBSS (supplemental Fig. S8B, available at www.jneurosci.org as supplemental material). Importantly, EBSS also reduced the generation of Aβ38, Aβ40, and Aβ42 (supplemental Fig. S8C, available at www.jneurosci.org as supplemental material).

GSLs impair starvation-induced degradation of APP-CTFs. H4 cells were incubated in EBSS for the indicated time periods, and the different proteins [APP-CTFs; full-length APP (APP-fl)] in cell lysates were detected by Western immunoblotting. EBSS (con), Incubation in EBSS in the absence of GSLs. EBSS+GSLs (+p), Incubation in EBSS in the presence of GSLs after pretreatment of cells with GSLs (50 ng/μl) for 48 h. EBSS + GSLs (−p), Incubation in EBSS in the presence of GSLs without pretreatment. Quantification was done by ECL imaging, and trend lines were plotted to understand the time-dependent metabolism of individual protein. Note that the initial amount of each protein was wet to 100%.
Next to assess the impact of GSLs on lysosome-dependent degradation of APP-CTFs, we studied the metabolism of APP-CTFs upon starvation using EBSS in the presence of GSLs (Fig. 8). To differentiate between acute and prolonged effect of GSLs, we additionally pretreated one set of cells with GSLs for 48 h before EBSS incubation (Fig. 8). In control cells, starvation led to rapid degradation of APP-CTFs, while LC3 II transiently increased, indicating induction of autophagy. Levels of p62, rab-7, and lamp-2, three AV-associated proteins, also decreased during starvation, demonstrating efficient autophagic flux. The presence of GSLs during starvation decreased the clearance of these AVs. LC3 II levels increased steadily in the cells where GSLs were added to cells only during incubation with EBSS, whereas LC3 II levels did not change significantly in the cells that were pretreated with GSLs for 48 h. Note that the incubation of cells with GSLs for 48 h itself increased LC3 II levels, as indicated in Figure 6D. Thus, already higher LC3 II levels do not increase further upon induction of autophagy with EBSS. Overall transient increase and subsequent decrease upon autophagy induction indicates efficient turnover of LC3 II in control cells, whereas LC3 II turnover seems to be impaired in GSL-treated cells. More importantly, in contrast to efficient clearance of APP-CTFs during starvation in control cells, GSL almost completely inhibited APP-CTF degradation. Rather, we observed an increase in APP-CTFs after 1 and 6 h. Because the increased levels of CTFs in the presence of GSLs cannot be attributed solely to inhibition of their degradation, GSLs might affect the generation of APP-CTFs by increasing the expression and/or processing of APP under nutritional stress. Accordingly, total APP levels transiently increased upon EBSS treatment in GSL-treated cells (Fig. 8). The combined data strongly indicate that GSLs inhibit the starvation-induced degradation of APP-CTFs in lysosomal and autophagic compartments.
Discussion
Abundance of autophagosomes and late autophagic vacuoles in dystrophic neurites is characteristic for AD brains as well as lipid storage diseases such as NPC (Nixon, 2007; Pacheco and Lieberman, 2008). Recent studies indicate that impaired clearance of autophagic vacuoles can contribute to this pathology (Boland et al., 2008). Other studies have also linked lysosomal lipid accumulation to autophagic dysfunction (Boland et al., 2008). However, factors that initiate changes in the autophagic pathway and thereby might trigger AD pathogenesis are largely unknown. The present data indicate that accumulation of SLs could induce key cytopathological changes characteristic for AD, i.e., (1) alteration of the autophagic/lysosomal system, (2) increased generation of Aβ, and (3) accumulation of APP-CTFs. Thus, our data indicate a close functional relation between lipid accumulation in vesicular compartments and the metabolism of APP, in particular in the clearance of potentially amyloidogenic APP-CTFs involving autophagic processes.
Our data indicate that the accumulation of SLs plays a dual role in autophagy. While promoting the induction of autophagy, SLs also impair the efficient turnover of AVs, resulting in their accumulation. To confirm the induction of autophagy by SLs, cells were treated with E64d, a cysteine protease inhibitor that impairs LC3 II degradation, in the presence or absence of GSLs (Clement et al., 2009). If GSLs only inhibit autophagic flux, the effect of both additions on LC3 II levels would be equal to that of E64d alone, whereas it would be additive if induction of autophagy and LC3 expression is increased. We observed synergistic effects of the combined treatment with GSLs and E64d on LC3 II levels, thereby supporting a dual role of GSLs in the autophagic process, i.e., promoting the induction of initial AV formation and impairment of autophagic clearance (supplemental Fig. S9, available at www.jneurosci.org as supplemental material). The induction of autophagy is regulated by several signaling cascades involving mTOR, AMPK, class I PI3K/Akt, p70S6K, and class III PI3K (PI3KC3) (Laplante and Sabatini, 2009). Another important regulatory protein in autophagy induction is beclin-1. When bound to Bcl-2, beclin-1 is inactive. Starvation induces the phosphorylation of Bcl-2 by JNK1 at Thr69, Ser70, and Ser87, resulting in the dissociation from beclin-1, thereby allowing formation of autophagosomes (Wei et al., 2008). Previous studies reported upregulation of beclin-1-dependent autophagy in models of NPC that are associated with impaired trafficking and accumulation of cholesterol (Pacheco et al., 2007). Our data demonstrate that GSLs increased the phosphorylation of Bcl-2, which would lead to dissociation from beclin-1 and induction of autophagy. Ceramide has been shown to also promote the phosphorylation of Bcl-2 (Pattingre et al., 2009). Notably, the accumulation of GSLs also decreased the cellular levels of total Bcl-2, which might decrease complex formation of BCL-2, and beclin-1, which might further promote induction of autophagy (Levine et al., 2008).
Importantly, the accumulation of APP-CTFs upon lipid storage was not caused by inhibition of γ-secretase. Rather, SLs stimulated γ-secretase activity in vitro, which is consistent with previous data (Osenkowski et al., 2008). Accordingly, we found increased production of intracellular and secreted variants of Aβ. Lysosomes and AVs could contribute to both generation and degradation of APP-CTFs (Haass et al., 1992; Nixon, 2007). Here, we show that cell starvation led to rapid degradation of APP-CTFs. Notably, the starvation-induced clearance of APP-CTFs was strongly inhibited by GSLs. The metabolism of known autophagic substrates p62, LC3 II, and AV-associated proteins such as lamp-2 and rab-7 was also significantly impaired. The inhibition of cellular clearance activity by GSLs was further demonstrated at the ultrastructural level by the accumulation of electron-dense and undigested material in AVs. Thus, although autophagy induction could contribute to increased CTF levels, efficient autophagic flux would not induce its accumulation (as indicated by starvation-induced clearance of CTFs). Therefore, both processes—induction of autophagy and simultaneous inhibition of AV turnover—might together contribute to accumulation of APP-CTFs in sphingolipid storage conditions.
The GSL-dependent inhibition of APP-CTF clearance might involve incomplete maturation of AVs. However, the colocalization of LC3-RFP and lamp-2 in GSL-treated cells indicates efficient fusion of autophagosomes and lysosomes. Recent studies also suggested fusion of autophagosomes to endosomes as an obligatory step in autophagy maturation (Razi et al., 2009), but the increased colocalization between EEA1 and LC3 in GSL-treated cells is rather indicative of increased fusion between autophagosomes and endosomes. The fusion of endosomes and lysosomes that contain major fractions of APP-CTFs with autophagosomes could lead to efficient sorting of APP-CTFs to AVs. Indeed, we showed localization of APP-CTFs in LC3-positive compartments upon GSL accumulation. Thus, APP-CTFs might be mainly produced in endosomal/lysosomal compartments involving α- and β-secretase activities, and then sorted to AVs upon fusion. Although we observed efficient maturation of cathepsin D, effect of GSLs on other lysosomal hydrolases cannot be ruled out. The impaired clearance of APP-CTFs and other material destined for degradation might also involve changes in the membrane lipid composition in lysosomal and autophagic compartments, and/or sterical hindrance by accumulated lipids. It will be interesting to further dissect how accumulated GSLs impair the capacity of lysosomal and autophagic clearance.
The importance of basal autophagy in neuronal metabolism has been recently demonstrated in mice with conditional deletion of autophagy-related genes (Komatsu et al., 2006). These mice developed strong neurodegeneration associated with accumulation of ubiquitin-positive material partly resembling cytopathological phenotypes of AD. Our data indicate that basal autophagy could also contribute to the metabolism of APP-CTFs. Because of the close relation of lysosomal degradation and autophagy, it is difficult to selectively target one process over the other. Vinblastine, which impairs fusion of AVs with lysosomes and subsequent degradation of AVs, as well as leupeptin, which inhibits lysosomal hydrolases and thereby also prevents degradation of autophagic cargo, both caused accumulation of APP-CTFs. APP-CTFs also accumulated in lamp-KO cells. In addition to their role in autophagy, lamp proteins are essential for normal lysosomal function and activity. Thus, APP-CTF accumulation in lamp KO cells or upon incubation with leupeptin could also result from inhibited degradation in lysosomes. Treatment of H4 cells with 3-methyladenine (3-MA), a class III PI3K inhibitor that more selectively inhibits the initiation of autophagy, also increased APP-CTF levels (supplemental Fig. S10A, available at www.jneurosci.org as supplemental material) (Seglen and Gordon, 1982). Upon inhibition of autophagic induction by 3-MA, APP-CTFs might be shunted to non-degrading compartments such as endosomes. The ratio of APP-CTFs to full-length APP was also significantly increased in mouse embryonic fibroblasts of atg5-KO mice that have selective defect in autophagy (Kuma et al., 2004) (supplemental Fig. S10B, available at www.jneurosci.org as supplemental material). Together, these findings strongly support an involvement of basal autophagy in APP-CTF metabolism.
Our findings are consistent with recent studies indicating autophagic impairment in lysosomal storage disorders such as NPC, GM1-gangliosidosis, and mucopolysaccharidoses (Liao et al., 2007; Pacheco and Lieberman, 2008; Settembre et al., 2008; Takamura et al., 2008). Thus, the autophagic–lysosomal dysfunction could initiate a pathogenic cascade by impaired recycling of metabolites and accumulation of cellular waste products ultimately leading to neurodegeneration. The accumulation of APP-CTFs might not only be induced by accumulation of SLs, but might be a more general feature of LSDs. Interestingly, autophagic activity could also be impaired by the pharmacological inhibition of GSL biosynthesis, while supplementation with gangliosides partially rescued this phenotype (Wei et al., 2009). Together, the present data indicate that the lipid composition of intracellular vesicular compartments plays crucial roles in the regulation of lysosomal and autophagic activity.
Accumulating evidence suggests that certain membrane lipids might increase during aging and AD (Ariga et al., 2008; Yamamoto et al., 2008). Our data provide strong evidence for an increased production of Aβ upon accumulation of GSLs. Thus, age-dependent changes in the membrane-lipid composition of endosomal and lysosomal compartments could underlie the accumulation of APP-CTFs and Aβ observed in sporadic AD. Moreover, GSL storage could also impair the autophagic flux and induce accumulation of AVs, a characteristic cytopathological feature in AD brains (Nixon, 2005). The processing of APP-CTFs by γ-secretase within these compartments might contribute to increased generation of intracellular and secreted Aβ. Interestingly, membrane lipids could also stimulate BACE1 activity (Kalvodova et al., 2005). As BACE1 together with γ-secretase was detected in lysosomes and AVs (Yu et al., 2005), these compartments might indeed be a major location of Aβ production under pathological conditions.
The accumulation of APP-CTF in the brain is a characteristic feature of AD (Chang and Suh, 2005). Importantly, transgenic overexpression of APP-CTFs in mice is highly detrimental and induces AD-like pathological features (Chang and Suh, 2005), including impairment of calcium homeostasis (Kim et al., 2000) and long-term potentiation (Nalbantoglu et al., 1997). Moreover, CTFs also trigger strong inflammation and astrocytosis (Chong, 1997; Bach et al., 2001) and eventually neurodegeneration (Yankner, 1996). Recent studies also showed that βCTFs are responsible for inducing endosomal pathology characteristic of AD (Jiang et al., 2010).
Notably, mouse and cell models of NPC disease caused by impaired trafficking and metabolism of cholesterol also show strong accumulation of APP-CTFs. Further, NPC in humans is associated with intraneuronal aggregates of tau (Jin et al., 2004). Thus, the age-dependent accumulation of membrane lipids could induce both major neuropathologic events in AD, neurofibrillary tangles and amyloid plaques. Because the relation between these two pathologies is not understood (Burns and Duff, 2002), it is intriguing to speculate that accumulation of lipids is an early event in the pathogenesis of AD and could drive both pathologies at initial stages. The present data also indicate that stimulation of autophagy could be a useful strategy in AD therapies and/or prevention targeted toward Aβ reduction, provided autophagic clearance is not impaired; otherwise, such approaches might be rather countereffective.
Footnotes
-
This work was supported by the Bundesministerium für Bildung und Forschung (01GI0708), the Deutsche Forschungsgemeinschaft (SFB645, KFO177), and the Mizutani Foundation to J.W., and the BONFOR program to I.Y.T. and J.W. We thank Drs. C. Haass and S. Höning for providing the C99-EGFP construct and cathepsin D antibody, respectively. We also thank Drs. E. H. Koo and H. P. Hauri for antibodies against APP and giantin, respectively. We also thank Drs. T. Yoshimori and N. Mizushima for providing LC3-GFP construct.
- Correspondence should be addressed to Dr. Jochen Walter, Molecular Cell Biology, Department of Neurology, University Hospital Bonn, Sigmund-Freud-Strasse 25, 53127 Bonn, Germany. Jochen.Walter{at}ukb.uni-bonn.de





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}