

Abstract
The NMDAR plays a unique and vital role in subcellular signaling. Calcium influx initiates signaling cascades important for both synaptic plasticity and survival; however, overactivation of the receptor leads to toxicity and cell death. This dichotomy is partially explained by the subcellular location of the receptor. NMDARs located at the synapse stimulate cell survival pathways, while extrasynaptic receptors signal for cell death. Thus far, this interplay between synaptic and extrasynaptic NMDARs has been studied exclusively in cortical (CTX) and hippocampal neurons. It was unknown whether other cell types, such as GABAergic medium-sized spiny projection neurons of the striatum (MSNs), which bear the brunt of neurodegeneration in Huntington's disease, follow the same pattern. Here we report synaptic versus extrasynaptic NMDAR signaling in striatal MSNs and resultant activation of cAMP response element binding protein (CREB), in rat primary corticostriatal cocultures. Similarly to CTX, we found in striatal MSNs that synaptic NMDARs activate CREB, whereas extrasynaptic NMDARs dominantly oppose CREB activation. However, MSNs are much less susceptible to NMDA-mediated toxicity than CTX cells and show differences in subcellular GluN2B distribution. Blocking NMDARs with memantine (30 μm) or GluN2B-containing receptors with ifenprodil (3 μm) prevents CREB shutoff effectively in CTX and MSNs, and also rescues both neuronal types from NMDA-mediated toxicity. This work may provide cell and NMDAR subtype-specific targets for treatment of diseases with putative NMDAR involvement, including neurodegenerative disorders and ischemia.
Introduction
NMDARs play a critical role in neuronal transmission. Calcium influx through NMDARs initiates signaling for synaptic plasticity, gene transcription, and survival (Dingledine et al., 1999; Hardingham et al., 2002; Hardingham and Bading, 2003, 2010; Léveillé et al., 2008, 2010). In contrast, excessive Ca2+ entry through NMDARs causes glutamate excitotoxicity (Choi, 1987; Tymianski et al., 1993; Arundine et al., 2003; Léveillé et al., 2010).
This dichotomous signaling has several potential explanations: stimulation intensity, specific protein–protein interactions, subunit composition, or subcellular location of the receptor (Papadia et al., 2005; Köhr, 2006; Hardingham and Bading, 2010). Although all may contribute to the downstream consequences of NMDAR activation, the role of NMDAR subcellular location is perhaps the most comprehensively investigated. In this model, synaptically localized NMDARs signal cell survival, while those located extrasynaptically signal cell death (Hardingham and Bading, 2010). The receptor's subcellular location also affects its protein–protein interactions, influencing downstream NMDAR-mediated Ca2+ signaling (Lau and Zukin, 2007).
The role of NMDAR subunit composition is less clear. GluN2B-containing NMDARs are reported to mediate distinct plasticity signaling, such as that required to induce LTD (Liu et al., 2004; Massey et al., 2004), although this is disputed (Morishita et al., 2007). GluN2B-containing receptors may preferentially signal cell death (Li et al., 2006; Liu et al., 2007; Tu et al., 2010); although other studies indicate that both GluN2B- and GluN2A-containing receptors can activate both cell death and survival (von Engelhardt et al., 2007; Martel et al., 2009). One possible unifying explanation is that GluN2B-containing receptors may reside preferentially, although not exclusively, at extrasynaptic sites (Tovar and Westbrook, 1999; Barria and Malinow, 2002; Thomas et al., 2006; Petralia et al., 2010).
Regardless of subunit composition, synaptic NMDARs activate cellular antioxidant defenses (Papadia et al., 2005), initiate signaling through extracellular signal-related kinase (ERK) (Ivanov et al., 2006), affect histone acetylation (Wittmann et al., 2009), suppress death pathways (Léveillé et al., 2010), and phosphorylate cAMP response element binding protein (CREB) (Hardingham et al., 2002; Léveillé et al., 2008), a transcription factor that regulates gene expression important for survival and plasticity (Lonze and Ginty, 2002). In contrast, extrasynaptic NMDARs shut off cell survival pathways (Hardingham et al., 2002), induce mitochondrial dysfunction (Gouix et al., 2009), and activate pro-death molecules (Léveillé et al., 2010). Thus far, disparate synaptic/extrasynaptic NMDAR signaling has been investigated in cortical (CTX) and hippocampal neurons; it is unknown whether nonglutamatergic subcortical projection neurons, such as GABAergic medium-sized spiny projection neurons of the striatum (MSNs; >90% of striatal cells), follow the same pattern. The striatum receives glutamatergic input from the cortex and thalamus, and is critical for executive function and movement. Striatal dysfunction is central to neurodegenerative diseases such as Parkinson's (Kreitzer and Malenka, 2007) and Huntington's disease (Milnerwood and Raymond, 2010). Evidence suggests that signaling pathways are different in MSNs compared with CTX cells (Arundine et al., 2003; Fan et al., 2010); therefore, we used corticostriatal cocultured neurons to investigate synaptic and extrasynaptic NMDAR signaling in MSNs versus CTX cells.
Materials and Methods
Transfection and plating.
Cultures were prepared as in Fan et al. (2010); briefly, pregnant Wistar rats were halothane anesthetized and decapitated at gestational day 17–18. Embryos were extracted and the brains were removed and placed on ice in HBSS (Invitrogen); cells were isolated by papain digestion and plated in 24-well plates at a concentration of 300,000 cells/ml. For transfections, 3–5 million cells were suspended in 100 μl of electroporation buffer (Mirus Bio) with 1–5 μg of endonuclease-free DNA, placed in a cuvette (0.2 μm; Biorad) and electroporated (AMAXA nucleofector I: program 03 for cortex and 05 striatum). Solution was removed from the cuvette and resuspended in D minimum essential medium (Invitrogen) plus 10% FBS (DMEM+), and plated as described above. After 2–4 h DMEM+ was replaced with 500 μl of plating medium (PM, 2% B27, Invitrogen; penicillin/streptomycin; 2 mm α-glutamine; and neurobasal medium, Invitrogen). At DIV 4, 500 μl of PM was added, with subsequent half media changes every 3–5 d. DNA plasmids included yellow fluorescent protein (YFP) on a CAG promoter (a gift from S. Kaech and G. Banker, Oregon Health Sciences University, Portland, with permission from J. Miyazaki for the CAG promoter) (Niwa et al., 1991; Kaech and Banker, 2006), luciferase on a cAMP-response element (CRE) promoter (Stratagene), control luciferase on a minimal non-CRE-dependent promoter (Stratagene), and YFP-GluN2B. Enhanced YFP followed by the linker LVPRGSRSR was inserted by site-directed mutagenesis and PCR into rat GluN2B (Kim et al., 2005, a gift from M. Sheng, Genentech) between amino acids 2 and 4 of the mature N terminus in a modified pLentiLox 3.7 vector with a synapsin promoter, and the sequence of the coding region was confirmed.
For corticostriatal cocultures, either CTX or striatal MSNs were YFP nucleofected to differentiate between cell types and then plated with nontransfected cells, either MSNs or CTX, respectively, at a ratio of 1:1.
Microfluidic chambers.
Coverslips (24 × 40 mm) were washed in 50% HCl for 2 h, sonicated for 30 min, and washed once in 100% EtOH, then twice in 70% EtOH. Microfluidic isolation chambers were prepared as in Park et al. (2006). Both chambers and coverslips were irradiated for >1 h under ultraviolet light. Coverslips were placed in culture plates, and chambers pressed down onto the coverslips. Borate-buffered PDL was placed in the chamber wells and incubated for 24–72 h, then washed three times with sterile dH2O for 10 min at 37°C. Chambers were filled with PM for at least 30 min. Cells were plated at a density of 4–5 million cells/ml. The media was removed from the wells, and 10 μl of culture was added to the top wells, and 5 μl to the bottom wells. After 45 min at 37°C, 150 μl of PM was added to the top and bottom wells. Cells were maintained at 37°C, 5% CO2, and half the media was changed every 3–5 d.
Treatments.
To stimulate synaptic NMDARs, neurons were treated with a synaptic stimulation mixture containing the following: bicuculline (50 μm; Tocris Bioscience), 4-aminopyridine (4-AP, 2.5 mm; Tocris Bioscience), nifedipine (5 μm; Tocris Bioscience), glycine (10 μm; Sigma), and strychnine (2 μm; Tocris Bioscience) mixed in conditioned media as in Hardingham et al. (2002). Control medium also contained nifedipine, glycine, and strychnine at the same concentrations. To selectively stimulate extrasynaptic NMDARs, cells were first treated with synaptic stimulation mixture for 10 min at 37°C, 5% CO2, followed by synaptic stimulation mixture plus MK-801 (10 μm; Tocris Bioscience) for 5 min to irreversibly block synaptic NMDARs. Cells were rinsed three times with media to remove MK-801, and then treated with NMDA (15 or 30 μm; Sigma) for 15 min. In chamber experiments, neurons were treated for 30 min with synaptic stimulation mixture or control media.
For toxicity assays, neurons were pretreated for 1 h with either conditioned media alone or else containing memantine (10 or 30 μm; Tocris Bioscience) or ifenprodil (3 μm; Tocris Bioscience), then treated with the following concentrations of NMDA (in μm:) 15, 30, 60, 100, and 500 in conditioned media ± memantine or ifenprodil for 10 min. Drugs were removed and the cells were incubated in conditioned media for 6 h, then fixed in 4% PFA with 4% sucrose.
Immunocytochemistry.
Coverslips were fixed in 4% PFA + 4% sucrose for 10 min and rinsed three times with PBS. Next, coverslips were washed, permeabilized (5 min, PBS plus 0.03% Triton X-100; Sigma, PBST) and blocked (30 min, 10% NGS in PBS). Primary antibodies were incubated overnight with shaking at 4°C in PBST plus 2% NGS, subsequently incubated at room temperature (RT) for 1 h, washed three times with PBST, and incubated for 1.5 h at RT with secondary antibodies (Alexa 488- and Alexa 568-conjugated α-mouse and α-rabbit, Invitrogen; anti-guinea pig AMCA, The Jackson Laboratory).
For live staining, cells were incubated for 10 min at 37°C with α-green fluorescent protein (GFP; cross-reactive with eYFP) in conditioned media, rinsed two times with media, fixed in PFA-sucrose for 10 min, rinsed three times with PBS, and incubated with secondary antibody for 1.5 h. For labeling with PSD-95 and VGLUT1, cells were subsequently permeabilized with methanol for 5 min at −20°C, rinsed three times with PBS, washed for 5 min with PBST, then treated with primary antibodies overnight, washed three times with PBST, and incubated with secondary antibodies as above. For nuclear staining, cells were treated with 5 μm Hoechst 33342 (Invitrogen) for 10 min then washed three times with PBST. Coverslips were slide mounted with Fluoromount (SouthernBiotech).
Antibodies included: α-phosphorylated CRE-response element binding protein, Serine-133 (pCREBSer133, mouse, Millipore 05–667, 1:750), α-GFP(chicken, AbCam ab13970, 1:1000), α-glutamic acid decarboxylase 65 (GAD-65; mouse, Millipore Bioscience Research Reagents MAB 351R, 1:1000), PSD-95 (mouse, Thermo Scientific MA 1–045, 1:1000), VGLUT1 (guinea pig, Millipore Bioscience Research Reagents, AB 5905, 1:4000).
Microscopy and image analysis.
Images were acquired using a Zeiss Axiovert 200 M fluorescence microscope. To assess nuclear pCREBSer133, 0.4 μm z-stacks (three images centered on the nucleus) taken at 63× magnification (NA: 1.4) were flattened using the extended focus projection function (Axiovision 4.6). All exposure times were constrained within experiments and analysis was conducted on unprocessed (raw) images. Nuclear and cytoplasmic (background) pCREBSer133 fluorescence was quantified in ImageJ (National Institutes of Health) as mean intensity within a region of interest covering the nucleus and an average of three small circles within the perinuclear cytoplasm. The nuclear pCREBSer133 intensity was measured, and the ratio of nuclear fluorescence to background cytoplasmic signal was calculated for each cell. Since the values obtained by raw intensity were similar to those obtained by within-cell normalization to cytoplasmic background fluorescence, normalized values were used. For analysis of toxicity, the proportion of apoptotic eYFP-transfected neurons in each condition was scored based on dendritic and nuclear morphology as a percentage of 100 cells per condition (Fan et al., 2010).
For morphological analysis, cells transfected with YFP were imaged using a 40× objective (NA: 1.3, 0.400 μm stacks). Dendrites, soma, and spines were manually reconstructed and quantified in IGOR using custom software routines (Milnerwood et al., 2010).
For colocalization analysis, cells transfected with YFP-GluN2B were stained with antibodies raised against GFP, VGLUT1, and PSD-95. Images (63×, 8–15 z-stacks of 0.4 μm) were acquired, and extended focus projections were created from 4 to 5 images containing all of the visible dendritic surface staining and the glass-attached somatic membrane. For cluster detection and localization images were manually thresholded with the experimenter blind to condition, and analysis of colocalization was conducted in small regions of interest around one primary dendrite and two secondary dendrites. After thresholding, the VGLUT1 clusters were dilated one point in ImageJ to increase the probability of counting as colocalized VGLUT1 and YFP-GluN2B clusters that were opposed but not overlapping. Colocalization was calculated using an ImageJ colocalization plugin (http://rsb.info.nih.gov/ij/plugins/colocalization.html) as in Tapia et al. (2011).
Luciferase assay.
Striatal cells were transfected on the day of plating with luciferase CRE-reporter or negative control plasmids containing luciferase on a minimal-non-CRE promoter (Luciferase Assay Kit; Stratagene) and plated with CTX cells. At DIV 20–21, cells were treated for 5 h with either forskolin (10 μm; Tocris Bioscience); synaptic stimulation mixture; or conditioned media containing nifedipine, glycine, and strychnine. Cells were rinsed twice with PBS and washed with lysis buffer (Luciferase Assay Kit; Stratagene) with shaking for 15 min. Cells were scraped, vortexed, and spun at 12,000 g, 4°C, 2 min. The supernatant was removed, placed on ice, and immediately assayed for luciferase activity using a luminometer (TD-20/20; Turner Designs) and luciferase assay buffer (Luciferase Assay Kit; Stratagene).
Electrophysiology.
Whole-cell patch-clamp recordings were performed on cortical or striatal neurons identified by YFP fluorescence in coculture at DIV 20–23. Electrophysiology experiments were conducted under voltage-clamp at a holding potential of −70 mV, as described previously (Fernandes et al., 2007). Briefly, neurons were superfused at room temperature with extracellular solution containing the following: 167 mm NaCl, 2.4 mm KCl, 10 μm MgCl2, 10 mm glucose, 10 mm HEPES, 2 mm CaCl2, pH 7.2. Tetrodotoxin (TTX; 0.3 μm), glycine (10 μm), and picrotoxin (PTX; 100 μm) were added before use to control and agonist solutions. Rapid switching between solutions was achieved using a computer-controlled solenoid system attached to a theta tube. In the control condition, 1 mm NMDA was applied for 3 s and repeated five times at intervals of 60 s. Synaptic bursting (induced by network disinhibition with 100 μm PTX and 10 μm 4-AP during transient wash out of TTX) was used to effect an irreversible, stable, and selective blockade of synaptic NMDARs with the transient addition of MK-801 (10 μm) before reapplying TTX, and recording extrasynaptic NMDAR-mediated whole-cell currents. Ifenprodil (3 μm) treatment of isolated NMDAR currents was by bath application for 5 min, followed by NMDA agonist application in the presence of ifenprodil for eight trials. Intracellular recording solution contained the following (in mm): 130 CsMe, 5 CsCl, 4 NaCl, 1 MgCl, 10 HEPES, 5 EGTA, 5 lidocaine, 0.5 GTP, 10 Na-phosphocreatine, 5 MgATP, titrated to pH 7.2 using CsOH. Data were acquired using an Axopatch 200B amplifier and analyzed using pCLAMP 10.2 software (Molecular Devices). Whole-cell current density was calculated as peak NMDA current/cell capacitance.
Data analysis.
Data are presented as mean ± SEM and statistical analyses by appropriate ANOVA and t tests were conducted in Prism 4 software (GraphPad) as detailed in the text.
Results
Coculture characterization
As all excitatory glutamatergic connections onto striatal MSNs derive from neurons outside the striatum, MSNs in striatal monoculture receive little, if any, glutamatergic input (Shehadeh et al., 2006). In the absence of glutamate transmission, MSNs do not develop their characteristic spiny morphology (Segal et al., 2003). To study synaptic and extrasynaptic NMDAR-mediated transmission onto GABAergic subcortical projection neurons of the striatum (MSNs or striatal cells) in vitro, we reproduced the corticostriatal pathway by mixing cortical and striatal cells on the day of plating. To determine the relative abundance of GABAergic cells, cultures were stained for GAD-65, an enzyme required for production of the major MSN neurotransmitter GABA: neurons with perinuclear GAD-65 were classified as inhibitory. Equal mixing of CTX and MSN cells reduced the percentage of GABAergic neurones typically found in striatal monoculture from ∼80 to ∼50% (Fig. 1A,B). The reduced proportion of inhibitory cells in cocultures suggests that striatal neurons receive much more excitatory glutamatergic input due to the inclusion of cortical cells. This glutamatergic transmission allows investigation of synaptic signaling through NMDARs in cultured MSNs.

Coculture characterization. A, Proportion of inhibitory cells. Cortical monocultures (left; CTX), corticostriatal cocultures (middle), and striatal monocultures (right; MSN) labeled with α-GAD-65 and Hoechst. Arrows indicate cells with perinuclear GAD-65. B, Coculture composition. Corticostriatal cocultures contain fewer inhibitory neurons than striatal monocultures. Differences in composition analyzed by one-way ANOVA, F(2,11) = 89.51, p < 0.0001, n = 4 cultures, Bonferroni's post-test **p < 0.01. C, Baseline nuclear pCREBSer133. Right, Without stimulation, cells were fixed at DIV 21 and stained for pCREBSer133 (inset). Left, Within sister cultures, average nuclear fluorescence was normalized to CTX monoculture. One-way ANOVA, F(2,6) = 28.2, p < 0.001, n = 3 cultures, 15–17 cells/culture. MSNs in monoculture were significantly different from CTX monoculture, #p < 0.05, one-sample t test. CTX co, Corticostriatal coculture with labeled cortical neurons; MSN co, coritcostriatal coculture with labeled striatal neurons; CTX mono, cortical monocultures; MSN mono, striatal monocultures.
Calcium influx through synaptic NMDARs regulates CREB signaling in cortical and hippocampal neurons by increasing CREB phosphorylation at sites including Serine 133 (pCREBSer133). The phosphorylation state of CREB is analogous to CREB activity levels, since nuclear pCREBSer133 regulates transcriptional activity (Hardingham et al., 2002). Therefore, we measured levels of nuclear pCREBSer133 in mature (DIV 21) MSNs in coculture and mono-culture (Fig. 1C). Mono-cultured MSNs have only 66% of the level of nuclear pCREBSer133 observed in mono-cultured CTX cells. Providing striatal MSNs with glutamatergic input in coculture increases nuclear pCREBSer133 to levels similar to CTX cells (91%). The data demonstrate that increasing glutamatergic transmission onto striatal MSNs increases basal CREB activation.
It has been shown that striatal neurons only develop large numbers of spines when in coculture with glutamatergic cells, and that this is dependent on synaptic transmission (Segal et al., 2003). We quantified the morphology of striatal neurons in mono-culture and coculture (Fig. 2). Striatal MSNs in coculture exhibited more complex dendritic architecture (Fig. 2A,B) and more dendritic spine-like protrusions (0.3 vs 0.12 protrusions/1 μm on average; Fig. 2A,C). In contrast, the morphology of CTX cells was unaltered by coculture with striatal cells (data not shown).

Coculture morphology. A, Representative MSNs from sister cultures grown in monoculture (left) and coculture (right). Striatal neurons were transfected with YFP and amplified post fixation with cross-reactive α-GFP. Cocultured MSNs show a more complex dendritic architecture and greater number of dendritic protrusions. B, Quantification of dendritic arborization. Analysis of sholl lengths (left) and sholl crossings (right). Lengths were analyzed by two-way ANOVA. The main effect of culture condition was significant, F(1,240) = 46.81, p < 0.0001. The main effect of distance from the soma was significant, F(9,240) = 37.87, p < 0.0001. The interaction of culture condition and distance from the soma was significant, F(9,240) = 37.87. Crossings were analyzed by two-way ANOVA. There was a significant main effect of culture condition, F(1,216) = 50.39, p < 0.0001. The main effect of distance from the soma was significant, F(8,216) = 33.96. The interaction was significant, F(8,216) = 3.027. For both sholl lengths and crossings Bonferroni's post-test was used **p < 0.01, ***p < 0.001. C, Quantification of dendritic protrusions. The density of protrusions (left) and interval between protrusions (right) were measured and analyzed by two-way ANOVA. The main effect of culture condition was significant, F(1,92) = 17.83, p < 0.0001, Bonferroni's post-test *p < 0.05. Cumulative probability of the interprotrusion interval was analyzed by two-way ANOVA. The main effect of culture condition was significant, F(1, 3358) = 668.9, p < 0.0001. The main effect of dendrite length was significant, F(145, 3358) = 92.38, p < 0.0001. The interaction was significant, F(145, 3358) = 6.818, p < 0.0001. Bonferroni's post-test, *p < 0.05, ***p < 0.001. For B,C, n = 13–15 cells from each of 3 separate cultures.
Synaptic versus extrasynaptic NMDA receptor activation
To determine the effect of synaptic versus extrasynaptic NMDAR activation, we used medium density cocultures of a 1:1 ratio of CTX and striatal neurons (MSNs). To selectively stimulate synaptic NMDA receptors on both MSNs and CTX, cells were treated with a synaptic stimulation mixture containing bicuculline (50 μm) to block GABAA receptors and 4-AP (2.5 mm) to block A-type K+ channels (Hardingham et al., 2002), resulting in disinhibition, exaggerated membrane depolarization, action potential generation, and glutamate release at the synapse. All conditions included the NMDAR coagonist glycine (10 μm), nifedipine (5 μm) to block L-type Ca2+ channels and ensure that Ca2+ influx is through NMDA receptors, and strychnine (2 μm) to block glycine receptors. Treatment with synaptic stimulation mixture increased nuclear pCREBSer133; subsequently treatment with bath NMDA decreased nuclear pCREBSer133 (Fig. 3Ai,Aii, representative examples). Analysis of raw nuclear pCREBSer133 intensity (Fig. 3Aii, left) provided very similar results to nuclear pCREBSer133 when normalized to background cytoplasmic fluorescence values within each cell (Fig. 3Aii, right). Therefore, in subsequent experiments, we chose to use the within-cell nuclear/cytoplasmic pCREBSer133 ratio to reduce variability and control for between and within culture staining efficacy.

Stimulation of synaptic versus extrasynaptic NMDA receptors. A, Synaptic stimulation activates pCREBSer133. For synaptic stimulation, cells were treated with synaptic stimulation mixture that contained the following (in μm): 50 bicuculline, 5 nifedipine, 10 glycine, and 2 strychnine with 2.5 mM 4-AP. Cultures were treated with media or synaptic stimulation mixture (syn) for 15 min, followed by media or NMDA to stimulate all NMDARs for 15 min. For stimulation of extrasynaptic NMDARs (syn-block-NMDA), neurons were treated with synaptic stimulation mixture for 10 min, then stimulation mixture plus MK-801 (10 μm) for 5 min to block synaptic NMDARs, rinsed 3 times with media, and treated with NMDA for 15 min to stimulate extrasynaptic receptors. After treatment, all conditions were placed in conditioned media for 30 min. Representative examples (Ai) showing the localization of pCREBSer133 in neurons grown in 24-well culture dishes at DIV 21, and treated with media (left), synaptic stimulation mixture (middle), and synaptic stimulation mixture followed by bath NMDA to stimulate all NMDARs (syn-NMDA; right). pCREBSer133 staining is diffuse throughout the cytoplasm without treatment (left, inset) and dramatically increased in the nucleus after synaptic stimulation (middle, inset); nuclear pCREBSer133 is reduced back to baseline levels when synaptic stimulation is followed by bath NMDA (right, inset). Scale bar, 50 μm. Synaptic stimulation increases raw nuclear pCREBSer133 intensity and normalized nuclear pCREBSer133 in both CTX and MSN. Since the results obtained by raw intensity values (Aii, left) and normalized nuclear pCREBSer133 levels (Aii, right) were similar, for subsequent analysis normalized nuclear intensity values were used. Nuclear pCREBSer133 intensity analyzed by one-way ANOVA, F(7,567) = 14.22, p < 0.0001. Bonferroni's post-test ***p < 0.001, n = 59–79 cells (10–15 cells from each of 5 cultures). Normalized pCREBSer133 analyzed by one-way ANOVA, F(7,567) = 30.74, p < 0.0001. Bonferroni's post-test ***p < 0.001); ns, nonsignificant. To show that the increase in pCREB led to CRE-mediated gene transcription (Aiii), MSNs were transfected with a luciferase reporter on a CRE promoter (pCRE Luc) or a restriction enzyme site on a luciferase promoter as a negative control (pLuc MCS) on the day of plating and grown in coculture with CTX. Neurons were treated with forskolin (10 μm), synaptic mixture, or subjected to a media change for 5 h. All values are normalized to media condition. Analyzed by one-way ANOVA, F(3,12) = 24.3, p < 00001. Group means compared with media condition using a one sample t test, *p < 0.05; n = 4 cultures. B, Transient MK-801 exposure during synaptic stimulation produces a stable block of synaptic NMDARs in cocultured MSNs, as evidenced by block of slow NMDAR-mediated components of synaptic bursts. Representative traces of stimulated, and MK-801 blocked, bursting (Bi). This synaptic NMDAR block reduces whole-cell currents by ∼50% (Bii; n = 5 from 3 separate cultures), and remaining extrasynaptic NMDAR currents are stable after washout of MK-801. C, Synaptic activation in microfluidic isolation chambers. Cultures were grown in microfluidic chambers (Ci,Cii) with CTX on one side (Ci, left), and MSN on the other (Ci, right). The two compartments are fluidically separate, but axons can grow through micro-channels between them (Ci, dotted lines). Representative examples of neurons treated with synaptic stimulation mixture (Cii). When only the CTX cells were treated with synaptic stimulation mixture, nuclear pCREB increased in both CTX and MSNs (Cii,Ciii). Blocking only MSN NMDARs with AP5 prevented this increase in MSN but not CTX (Ciii). Analyzed by Kruskal-Wallis ANOVA, p < 0.0001, Dunn's multiple-comparison post-test, *p < 0.05,**p < 0.01,***p < 0.001, n = 40–78 cells (10–15 cells from each of 4 separate cultures).
Baseline nuclear/cytoplasmic pCREBSer133 showed a small nonsignificant reduction in MSNs compared with CTX (Fig. 3Aii, right; similar to Fig. 1C). Synaptic stimulation of NMDARs in these cocultures increased nuclear pCREBSer133 in both CTX and MSNs (Fig. 3Aii). Normalized nuclear pCREBSer133 in CTX increased from 1.1 (±0.03) to 2.0 (±0.1), while in MSNs increased from 1.0 (±0.05) to 1.7 (±0.07). When synaptic stimulation was followed by bath application of NMDA, signaling to CREB was shut off in both CTX and MSNs; nuclear pCREBSer133 levels in CTX were 1.3 (±0.05), while MSN levels were 1.2 (±0.06). In agreement with a previously reported dominant effect of extrasynaptic over synaptic signaling in glutamatergic cells (Hardingham et al., 2002), the effect of bath NMDA was similar to that of selectively stimulating extrasynaptic NMDARs, which drove the nuclear pCREB Ser133 down to 1.1 (±0.05) in CTX and 1.0 (±0.03) in MSNs. Therefore, in subsequent treatments, we used bath NMDA to shut off CREB after synaptic stimulation. To ensure that the MK-801 block of synaptic NMDARs was stable, and that MK-801 washed out before subsequent NMDA application, we conducted the synaptic block protocol while performing whole-cell patch-clamp recordings on cocultured MSNs. The slow NMDA-receptor-mediated components of network excitatory synaptic bursts were rapidly eliminated by MK-801 (Fig. 3Bi), and whole-cell NMDA currents were stably reduced by ∼50% with the remaining 50% representing nonsynaptic NMDARs (Fig. 3Bii).
An increase in nuclear pCREBSer133 would be expected to trigger enhanced transcription of a variety of genes. To test whether the increase in nuclear pCREBSer133 following synaptic stimulation effectively activated gene transcription in MSNs, we transfected striatal neurons with a luciferase reporter driven by CRE. Indeed, we found that synaptic stimulation for 5 h led to a threefold increase in luciferase expression compared with incubation with media alone (Fig. 3Aiii). Forskolin, which directly activates PKA signaling to CREB, was used as a positive control.
Microfluidic isolation culture platforms (Park et al., 2006) allow the growth of corticostriatal cocultures in which CTX and MSNs are plated in somatodendritic and fluidic isolation; connectivity is maintained between compartments via small channels through which axons can grow, providing the MSNs with glutamatergic input from CTX (Fig. 3Ci). This system permits straightforward cell identification and discrete drug treatment, providing a convenient and clear method to prove that a downstream effect is caused by direct glutamate release from CTX cells onto MSNs.
Only the CTX cells were treated with synaptic stimulation mixture (Fig. 3Cii). Therefore, any effect on MSNs was likely caused by synaptic glutamate transmission and not direct depolarization. Indeed, after CTX treatment with synaptic mixture, nuclear pCREBSer133 increased significantly in both CTX and MSNs (Fig. 3Cii). In CTX, nuclear pCREBSer133 increased from 2.0 (±0.02) to 3.7 (±0.02); in MSNs, pCREBSer133 increased from 2.4 (±0.03) to 3.0 (±0.02). Application of the NMDAR antagonist D-AP5 (100 μm) to MSNs prevented this increase in MSNs only, indicating that the increase in nuclear pCREBSer133 is NMDAR-mediated. The CTX cells were unaffected, demonstrating that the chambers are indeed fluidically separate. In fact, the nuclear pCREBSer133 in CTX cells increased slightly (Fig. 3Cii, left; not significant) compared with the synaptic mixture alone.
Blocking extrasynaptic NMDARs prevents pCREBSer133 shutoff
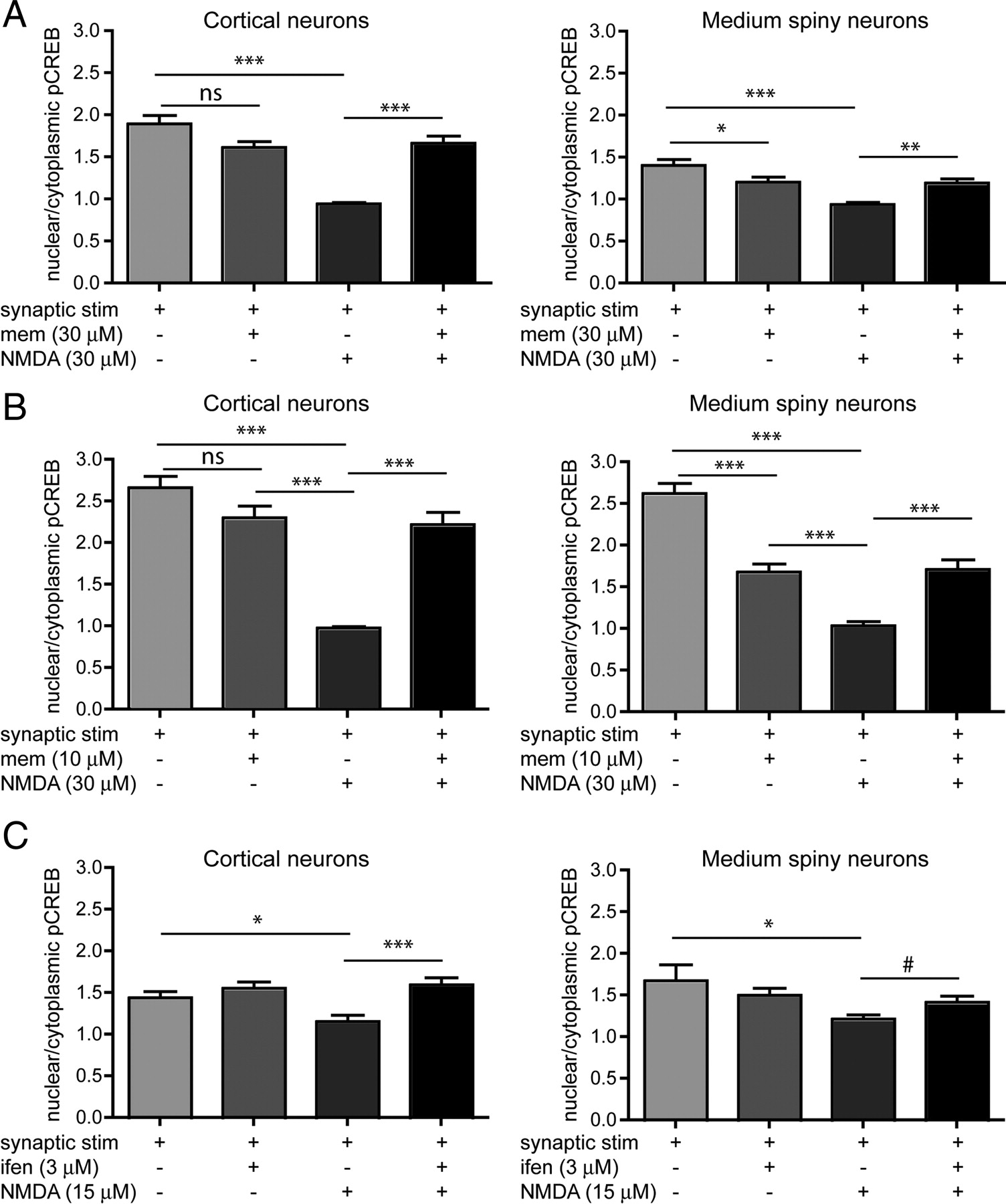
Memantine, an open-channel NMDAR blocker (uncompetitive antagonist) with a fast off-rate, has been shown to preferentially block extrasynaptic NMDARs at low concentrations (Chen and Lipton, 2006; Xia et al., 2010); at 1 μm memantine was ∼2-fold more potent at extrasynaptic sites, blocking 50% of extrasynaptic NMDA-induced current and only 25% of synaptic NMDA-mediated EPSCs (Xia et al., 2010). Elsewhere, 1 μm memantine has been shown to have no effect on synaptic burst-induced calcium influx, but to effectively block ∼50% of extrasynaptic NMDAR current and associated cell death in cortical cells (Léveillé et al., 2008). In our hands 1 μm memantine did not rescue from bath NMDA-induced CREB shutoff (data not shown) in MSNs or CTX cells. Therefore, we tested whether 10 μm or 30 μm memantine could protect from CREB shutoff by bath NMDA (30 μm). Previous experiments were performed using 15 μm NMDA; however, for these memantine experiments, we increased NMDA to 30 μm to provide a stronger shutoff and clearer signal, since our results with memantine at 1 μm differed from published reports.
Neurons were preincubated with memantine or subjected to a media change 15 min before synaptic stimulation. Addition of 30 μm memantine to the bathing medium during incubation with the synaptic mixture resulted in a small reduction in pCREBSer133 activation, which was significant for MSNs (1.4 ± 0.07 to 1.2 ± 0.06) but not CTX cells (1.9 ± 0.10 to 1.6 ± 0.07; Fig. 4A). Addition of 10 μm memantine to the medium during synaptic mixture incubation had a similar effect, with a significant reduction in MSNs (2.6 ± 0.1 to 1.6 ± 0.10) but not CTX (2.6 ± 0.10 to 2.26 ± 0.14; Fig. 4B). However, both concentrations of memantine were effective in preventing the bath NMDA-induced shutoff of pCREBSer133 in both CTX and MSNs (Fig. 4A,B). After treatment with 30 μm NMDA, the nuclear pCREBSer133 in MSNs was 0.9 (±0.02); whereas with 30 μm memantine present it was 1.2 (±0.05). In CTX, NMDA brought pCREBSer133 down to 0.9 (±0.1), whereas with 30 μm memantine present pCREBSer133 remained at 1.66 (±0.08). Together with data showing that bath application of NMDA is equivalent to selective activation of extrasynaptic NMDARs in reducing nuclear pCREBSer133 (Fig. 3C), these data suggest that the shutoff of CREB is mediated predominantly by extrasynaptic NMDARs in both CTX and MSNs in coculture.

Preventing CREB shutoff. A–C, Neurons were preincubated with inhibitors [memantine (mem) or ifenprodil (ifen)] or conditioned medium for 15 min, then treated with synaptic stimulation mixture ± inhibitor for 15 min, then treated with NMDA ± inhibitor or medium ± inhibitor, and finally incubated in conditioned medium ± inhibitor for 30 min. A, Blocking extrasynaptic NMDARs with 30 μm memantine rescues from CREB shutoff, but affects synaptic activation of CREB in MSNs. Analyzed by one-way ANOVA, cortical neurons F(3,218) = 29.25, p < 0.0001, MSNs F(3,193) = 12.55, p < 0.0001. n = 45–57 cells (10–15 cells from each of 3 separate cultures). B, Blocking extrasynaptic receptors with 10 μm memantine rescues from CREB shutoff, but also affects synaptic stimulation of CREB in MSNs. Analyzed by one-way ANOVA, cortical neurons F(3,188) = 36.07, p < 0.0001, MSNs F(3,185) = 43.31, p < 0.0001. n = 41–50 cells (10–15 cells from each of 3 separate cultures). C, Blocking GluN1/GluN2B heterodimers rescues from CREB shutoff. Ifenprodil rescues CTX from CREB shutoff more effectively than MSNs. Analyzed by one-way ANOVA. For cortical neurons, F(3,258) = 6.724, p < 0.001, for MSN, F(3,242) = 2.672, p < 0.05. n = 57–64 cells (from 4 separate cultures). Difference between MSN NMDA and NMDA + ifenprodil calculated by unpaired t test with Welch's correction, #p < 0.05. Differences between all other groups analyzed by Bonferroni's post-test, for A, B, and C, *p < 0.05, **p < 0.01, ***p < 0.001; ns, nonsignificant.
Blocking GluN2B-containing receptors prevents pCREBSer133 shutoff
Ifenprodil, a selective inhibitor of GluN2B heterodimers (Williams, 1993), prevented, at least partially, the bath NMDA-induced shutoff of CREB (Fig. 4C). In MSNs, the nuclear pCREBSer133 decreased from 1.5 (±0.07) after synaptic stimulation in the presence of ifenprodil, to 1.2 (±0.05) after NMDA treatment; with the addition of ifenprodil during NMDA exposure, the ratio was maintained at 1.4 (±0.08). In CTX, the pCREBSer133 decreased from 1.5 (±0.07) after synaptic stimulation plus ifenprodil to 1.2 (±0.07) after bath NMDA; with ifenprodil present during NMDA stimulation, the ratio remained at 1.6 (±0.08). However, the rescue of NMDA-induced CREB shutoff was more robust in CTX than MSNs (Fig. 4C), since in MSNs it was only significant by direct t test, not by ANOVA Bonferroni's post-test. The data suggest that GluN2B-containing receptors may be more involved in extrasynaptic NMDAR-mediated CREB shutoff in CTX cells than in MSNs. It is also possible that the distribution of GluN2B-containing receptors is different in CTX versus MSNs.
Localization of GluN2B-containing receptors
Since both ifenprodil (which blocks GluN2B-containing NMDAR heterodimers) and memantine (which preferentially blocks extrasynaptic NMDARs) prevent NMDA-induced CREB shutoff, we wanted to determine the proportion of GluN2B located at the synapse and at extrasynaptic sites in corticostriatal cocultures. Due to a lack of reliable N-terminal epitope antibodies for GluN2B subunits, CTX and MSNs were transfected with YFP-GluN2B and grown in microfluidic chambers. At DIV 21 live cultures were incubated with α-GFP antibodies to label GluN2B-containing surface receptors then fixed, permeabilized, and stained with α-VGLUT1 to mark presynaptic glutamatergic terminals, and α-PSD-95 to mark the postsynaptic density (Fig. 5A). While live cell incubation with anti-GFP antibodies may cause artificial aggregation of diffuse YFP-NR2B, it allows amplification of the extrasynaptic receptor signal, which was otherwise too low to assess. Excitatory synapses were defined by colocalization of VGLUT1 and PSD-95.

Localization of GluN2B subunits. MSN (striatal neurons in corticostriatal coculture) and CTX (cortical neurons in corticostriatal coculture) were transfected with a YFP-tagged GluN2B subunit and grown in chambers for 21days in vitro. A, Representative examples of CTX (top) and MSNs (middle, bottom) transfected with a YFP-GluN2B subunit, live stained with α-GFP, fixed, and stained with α-PSD-95 as a postsynaptic marker and α-VGLUT1 as a presynaptic marker. B, Dendritic puncta density. CTX cells express more GluN2B-YFP clusters on secondary dendrites than MSNs. CTX and MSNs show similar distributions of VGLUT1 on secondary dendrites. MSNs show an increased density of PSD-95 on secondary dendrites compared with CTX. Significant by one-way ANOVA, F(5,120) = 18.46, p < 0.0001. Bonferroni's post-test, *p < 0.05,**p < 0.01. C, Dendritic puncta colocalization. The percentage colocalization between markers on dendrites was similar between CTX and MSN except for VGLUT1 colocalization with PSD-95, one-way ANOVA, F(5,120) = 8.00, p < 0.0001, Bonferroni's post-test, *p < 0.05. D, Somatic puncta density and colocalization. MSNs expressed more GluN2B on the soma than cortical cells. A greater percentage of VGLUT1 was associated with GluN2B in MSNs compared with cortical cells. One-way ANOVA, F(5,120) = 16.52, p < 0.0001, Bonferroni's post-test, *p < 0.05, **p < 0.01. B–D, n = 4–10 cells per culture from each of 3 separate cultures. E, Representative NMDAR currents from cocultured cortical neuron (left) or MSN (right), evoked by application of 1 mm NMDA (solid line) in control conditions, followed in the same neuron by NMDA application with treatment of 8 –12 min of 3 μm ifenprodil (Ifen). Scale bar, 1nA and 1s. F, Mean peak current densities in both cortical neurons (CTX; 38.00 ± 4.32, n = 8 from 3 cultures) and MSN (46.86 ± 4.25, n = 8 from 3 cultures) are reduced by ifenprodil (ifen) treatment (20.10 ± 2.87 in CTX, 23.97 ± 2.69 in MSNs, *p < 0.05 by one-way ANOVA, followed by Bonferroni's post-test).
CTX cells exhibited significantly more GluN2B clusters on secondary dendrites than MSNs (42.8 ± 4.1 clusters per 100 μm2 in CTX, 29.6 ± 2.5 clusters per 100 μm2 in MSNs), although the densities of VGLUT1 clusters were similar (12.4 ± 1.7 clusters per 100 μm2 in CTX, 14.0 ± 1.5 clusters per 100 μm2 in MSNs). Interestingly, the density of PSD-95 clusters was lower in CTX than in MSNs (16.3 ± 2.1 clusters per 100 μm2 in CTX, 25.4 ± 2.9 clusters per 100 μm2 in MSNs; Fig. 5A,B), suggesting that MSNs may use PSD-95 at excitatory synapses more generally than do pyramidal cells, which rely upon other MAGUKS (such as PSD-93) in subpopulations of excitatory synapses (Elias et al., 2006). The percentage of VGLUT1 colocalized with PSD-95 was significantly higher in MSNs (40.7% ± 5.9 in CTX, 49.2% ± 7.2 in MSNs), but the percentage of VGLUT1 associated with YFP-GluN2B was not significantly different between the two cell types (32.5% ± 5.1 in CTX, 23.4% ± 4.8 in MSNs; Fig. 5C).
A major difference in GluN2B expression between MSNs and CTX was on the soma. MSNs expressed significantly more GluN2B clusters on the soma than CTX cells (30 ± 3 per 100 μm2 in CTX, 45 ± 2 per 100 μm2 in MSNs). In addition, a much greater percentage of the VGLUT1 clusters located on the soma were associated with GluN2B in MSNs than CTX (Fig. 5D; MSN, 29% ± 5; CTX, 8% ± 5).
To compare the proportion of NMDAR-mediated current carried by GluN2B heterodimers, whole-cell patch-clamp recording was performed. Despite the increased density of surface GluN2B punctae on dendrites of CTX and on the soma of MSNs, the ifenprodil-sensitive proportion of whole-cell NMDA-evoked current density was not significantly different for CTX and MSNs (Fig. 5E,F). Thus, despite differences in GluN2B localization, the overall density of functional surface GluN2B heterodimers was similar between these two neuronal types.
NMDA-induced toxicity and rescue
Both CTX and MSNs exhibit an age-dependent increase in NMDA-induced cell death between DIV 14 and 21 (Fig. 6A); however, at both time points striatal MSNs are markedly (and significantly) less susceptible to NMDA-induced toxicity than CTX cells. For example, at DIV 21, following 60 μm NMDA treatment, striatal MSNs show an average of 64% apoptotic nuclei, while CTX shows an average of 94% apoptotic nuclei (Fig. 6A,Bi,C). Regardless of the difference in sensitivity to NMDA toxicity, both CTX and MSN cells were rescued completely from NMDA-induced apoptosis by either memantine or ifenprodil (Fig. 6Bii,C).

NMDA-induced toxicity. A, MSNs and CTX display different sensitivities to NMDA-induced toxicity. Neurons at DIV 14 (left) and DIV 21 (right) were treated with NMDA for 10 min, then incubated in conditioned medium for 6 h, after which cells were fixed, stained with anti-GFP and Hoecsht, and assessed for cell death according to dendritic and nuclear morphology. Striatal MSNs are less susceptible to cell death than cortical neurons. Analyzed by two-way ANOVA. The interaction between NMDA concentration and cell type was significant: DIV 14, F(5,36) = 24.01, p < 0.0001; DIV 21, F(5,24) = 13.52, p < 0.0001, Bonferroni's post-test, *p < 0.05, **p < 0.01, ***p < 0.001, n = 4 cultures each genotype for DIV 14 and DIV 21. B, Representative examples of striatal MSNs after treatment. YFP-transfected striatal neurons grown in corticostriatal coculture were pretreated with conditioned medium ± inhibitors for1 h, then treated with 30 μm NMDA ± inhibitors for 10 min. After 6 h in conditioned medium, cells were fixed and cell death was assessed according to dendritic and nuclear morphology. After NMDA treatment, cells appeared damaged, with blebbed or missing dendrites (Bi, bottom) compared with controls (Bi, top). Both memantine (Bii, top) and ifenprodil (Bii, bottom) rescued dendritic morphology, and transfected neurons showed healthy, round nuclei compared with NMDA treatment (Bi, bottom). C, Rescue from NMDA-induced toxicity in CTX (left) and MSNs (right). Neurons were pretreated for 1 h± inhibitors, then 30 μm NMDA for CTX, 60 μm NMDA for MSNs, for 10 min ± inhibitors, then placed in conditioned medium for 6 h. Memantine (mem) rescues from NMDA-induced toxicity at both 10 and 30 μm, at DIV 14 and DIV 21. Ifenprodil (ifen) similarly rescues from NMDA-induced toxicity at 3 μm. For CTX, significant by one-way ANOVA, F(7,16) = 346.5, p < 0.0001, n = 3 cultures. For MSNs, significant by one-way ANOVA, F(7,16) = 70.41, p < 0.0001, n = 3 cultures. Differences between groups analyzed by Bonferroni's post- test,***p < 0.001.
Discussion
We demonstrate, for the first time, that striatal MSNs show increased and decreased nuclear pCREBSer133 after stimulation of synaptic and extrasynaptic NMDARs, respectively. However, we found differences in NMDAR signaling and GluN2B subcellular distribution between striatal MSNs and CTX. Notably, MSNs in coculture were less susceptible to NMDA-induced toxicity. Furthermore, CTX cells exhibited a higher density of GluN2B-containing clusters on dendrites, but a similar density of VGLUT1 clusters and synapses, compared with MSNs; suggesting that more (extrasynaptic) dendritic GluN2B-containing NMDARs in CTX cells are associated with increased cell death. Striatal MSNs expressed somatic GluN2B-containing NMDAR clusters, often associated with VGLUT1, suggesting somatic synapses not observed in CTX.
Synaptic NMDAR signaling to CREB
CREB is a key regulator of signal transduction; synaptic Ca2+ transmission increases CREB phosphorylation, leading to plasticity, pro-survival, and anti-apoptotic gene transcription (Greer and Greenberg, 2008; Hardingham and Bading, 2010; Milnerwood and Raymond, 2010). Providing MSNs with glutamatergic input in coculture with CTX cells increased basal nuclear pCREBSer133 to levels similar to CTX cells, consistent with positive modulation of pCREBSer133 by excitatory synaptic transmission.
Excitatory synaptic signaling positively modulates neuronal survival (Hardingham et al., 2002; Ivanov et al., 2006; Léveillé et al., 2010), and we found that coculture of MSNs prolonged MSN viability (data not shown) to >4 weeks compared with an average of <2 weeks for mono-culture. Moreover, excitatory input increased dendritic complexity and protrusion density such that cocultured MSNs resembled those in vivo (Milnerwood et al., 2010). This observation complements a seminal report (Segal et al., 2003), with the modest advance that we determined the striatal origin of MSNs by YFP transfecting them before plating.
Extrasynaptic NMDAR-mediated CREB shutoff
Importantly, we confirmed the antagonistic relationship between synaptic and extrasynaptic NMDAR survival signaling in subcortical GABAergic projection neurons. This was previously shown in predominantly glutamatergic cultures (Hardingham et al., 2002; Léveillé et al., 2010). Moreover, our data support the dominance of extrasynaptic over synaptic NMDAR activation in opposing survival signaling.
Previous studies in cortical and hippocampal cultures suggest that low memantine concentrations preferentially block extrasynaptic NMDAR activity, whereas higher concentrations also block synaptic transmission (Chen and Lipton, 2006; Léveillé et al., 2008; Xia et al., 2010). Treatment with low concentrations of memantine protects against NMDA-induced toxicity in vitro (Léveillé et al., 2008; Papadia et al., 2008; Gouix et al., 2009; Bordji et al., 2010), as well as neurotoxicity and learning deficits in chemical lesion models (Misztal et al., 1996; Lee et al., 2006) and neurodegenerative disease models (Okamoto et al., 2009; Martinez-Coria et al., 2010; Milnerwood et al., 2010). However, higher doses of memantine produce deleterious effects (Chen et al., 1998; Chen and Lipton, 2006; Okamoto et al., 2009).
As previously, memantine was neuroprotective at higher concentrations (Léveillé et al., 2008; Papadia et al., 2008, Okamoto et al., 2009), in this case against extrasynaptic CREB-off signaling in CTX and MSN cells at 10 and 30 μm. However, these concentrations significantly disrupted pCREBSer133 signaling produced by enhanced synaptic transmission in MSNs (10 μm reducing pCREBSer133 levels ∼40%), consistent with a report of 10 μm memantine blocking 40% of synaptic NMDAR-mediated currents in hippocampal autapses (Xia et al., 2010). This block was not significant in CTX cells, demonstrating a cell-specific variation in memantine's ability to block synaptic signaling in vitro.
Although memantine partially disrupted synaptic CREB signaling at 10 and 30 μm, both concentrations prevented extrasynaptic NMDAR-mediated reductions in pCREBSer133 levels. At 1 μm, memantine did not perturb synaptic signaling, but extrasynaptic CREB-off signaling was unaffected (data not shown). This suggests that some synaptic blockade can be tolerated therapeutically, if reducing dominant cytotoxic extrasynaptic signaling enhances the net effect of pro-survival signaling from remaining synaptic transmission. Indeed, 10 and 30 μm memantine completely prevented NMDAR toxicity in both striatal and CTX cells. However, chronic treatment with 30 mg/kg (high dose) of memantine in vivo increased striatal neuronal degeneration in a Huntington's disease mouse model (Okamoto et al., 2009); thus, further investigation of memantine dose effects, especially in different neuronal types and brain regions, is warranted.
Role of GluN2B-containing receptors
In contrast to memantine, the GluN2B-subunit-selective antagonist ifenprodil did not interfere with synaptic pCREBSer133 activation at a concentration that prevented extrasynaptic CREB-off signaling. Similarly to memantine, ifenprodil rescued both CTX and striatal cells from NMDA-induced apoptosis.
One interpretation is that all GluN2B-containing receptors are located at extrasynaptic sites, as suggested by some studies (Kew et al., 1998; Tovar and Westbrook, 1999; Barria and Malinow, 2002). This seems unlikely based on colocalization of YFP-GluN2B with VGLUT1 and PSD-95 and a wealth of literature (Groc et al., 2006; Thomas et al., 2006; Harris and Pettit, 2007; Petralia et al., 2010). Synaptic GluN2B-containing receptors may be in triheteromeric complexes (GluN1/2A/2B), since 3 μm ifenprodil preferentially blocks GluN1/2B diheteromers (Williams, 1993; Hatton and Paoletti, 2005). Alternatively, if synaptic GluN1/2B diheteromers are blocked by ifenprodil as demonstrated in hippocampal cultures (Liu et al., 2007), they may not affect synaptic pCREBSer133 activation or survival signaling. Recent studies link GluN2B-containing NMDARs selectively to cell death signaling (Kim et al., 2005; Liu et al., 2007; Tu et al., 2010), and putatively pathophysiological extrasynaptic signaling is eliminated by ifenprodil in both Huntington's and Alzheimer's disease models (Milnerwood et al., 2006, 2010; Li et al., 2011). However, GluN2B-containing receptors can also signal for survival (von Engelhardt et al., 2007; Martel et al., 2009). Both subcellular location and subunit composition likely affect NMDAR signaling, but regardless of receptor composition, downstream signaling is probably also altered by site-specific protein interactions (Li et al., 2002, 2003; Yi et al., 2007).
We found that GluN2B subcellular localization differed in CTX and MSNs. Dendritic VGLUT1 and density of synapses were equivalent; however, GluN2B surface cluster density was higher in secondary dendrites of CTX cells compared with MSNs. The data suggest CTX dendrites have more GluN2B-containing receptors that are not associated with synapses, and therefore more extrasynaptic GluN2B receptors. This may explain larger pCREBSer133 reductions following extrasynaptic NMDAR activation and greater sensitivity to NMDA-induced toxicity in CTX cells.
Curiously, we also observed large, numerous somatic GluN2B clusters in MSNs compared with few in CTX cells. Furthermore, >50% of these MSN clusters were associated with VGLUT1, suggesting somatic excitatory synapses. If these do represent functional synapses, GluN2B-containing NMDAR calcium signaling would occur near cellular transcription machinery. However, this may be a developmental or cell culture-specific phenomenon; somatic excitatory synapses have not been described in adult striatum (Smith and Bolam, 1990), although they are found in the developing visual CTX (Bähr and Wolff, 1985). Although this observation may not be relevant beyond our 3-week-old culture system, it may contribute to differences observed between CTX and MSNs in vitro, such as differential NMDAR toxicity.
Differences in NMDA-induced toxicity between CTX and MSNs
Disparate downstream signaling may also explain differences in NMDAR-mediated apoptosis observed between these cell types. Inhibition of neuronal nitric oxide synthase or disruption of the GluN2B-PSD-95 interaction rescue cortical and hippocampal neurons, but not MSNs, from cell death (Aarts et al., 2002; Fan et al., 2010). Apoptotic p38 MAPK signaling depends on the GluN2B–PSD-95 interaction in hippocampal neurons (Soriano et al., 2008), whereas survival signaling through ERK1/2, another MAPK, is differentially regulated by synaptic/extrasynaptic NMDAR activation in CTX and hippocampal neurons (Ivanov et al., 2006; Léveillé et al., 2008). Future studies will be required to investigate distinct signaling pathways in CTX and MSNs in coculture.
Role of extrasynaptic NMDARs
Extrasynaptic NMDARs are activated by glutamate spillover during high-frequency synaptic release in the hippocampus (Lozovaya et al., 2004; Harris and Pettit, 2007; Tzingounis and Wadiche, 2007). In acute slices, striatal MSNs exhibit an ∼50% increase in NMDAR-mediated charge after blockade of glutamate transport (Milnerwood et al., 2010), indicating the presence of extrasynaptic receptors. MSNs have a low resting membrane potential, and high levels of excitatory input (∼1000 Hz) are required to drive upstate transitions and action potential firing (Wilson and Kawaguchi, 1996; Wolf et al., 2005), which might render glutamatergic inputs to MSNs predisposed to spillover. Recent work shows that brief glutamate uncaging drives depolarizing plateaus in distal dendrites of MSNs (Plotkin et al., 2011), which would result in sustained relief of Mg2+ block. Glutamate transmission at extrasynaptic NMDARs may then occur through other means, such as release from glia (Hamilton and Attwell, 2010), cholinergic interneurons (Higley et al., 2011), and dopaminergic terminals (Stuber et al., 2010).
Glutamate transporters maintain synaptic integrity by preventing spillover between synapses (Tzingounis and Wadiche, 2007); it is possible that extrasynaptic NMDAR signaling, at a local dendritic level, could similarly prevent the spread of signal transduction under physiological conditions. This putative system could be deleterious when overactivated, as in stroke, trauma, and Huntington's disease.
Conclusions
Striatal GABAergic MSNs show similarities as well as a few key differences in synaptic/extrasynaptic NMDAR signaling compared with cortical glutamatergic neurons in coculture. Extrasynaptic NMDAR activity has been implicated in neurodegenerative disorders and stroke (Hardingham and Bading, 2010; Gladding and Raymond, 2011). More detailed understanding of cell and location-specific NMDAR signaling may lead to therapeutic strategies for the treatment of neurological diseases.
Footnotes
This study was supported by operating grants from the Canadian Institutes of Health Research (CIHR; MOP-102517 to L.A.R. and MOP-69096 to A.M.C.). K.S. holds a CIHR Frederick Banting and Charles Best Canada Graduate Scholarship. A.M.C. holds a Canada Research Chair. A.J.M held CIHR, Huntington's Disease Society of America and Huntington Society of Canada Fellowships while elements of this work were conducted. We are grateful for technical support from Lily Zhang, Tulin Okbinoglu, Ashley George, and Kia Duthie.
- Correspondence should be addressed to Dr. Lynn A. Raymond, Department of Psychiatry, University of British Columbia, 4N3-2255 Wesbrook Mall, Vancouver, BC V6T 1Z3, Canada. lynnr{at}exchange.ubc.ca





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}