

Abstract
Alzheimer's disease (AD) is the major cause of dementia. During the development of AD, neurofibrillary tangles progress in a fixed pattern, starting in the transentorhinal cortex followed by the hippocampus and cortical areas. In contrast, the deposition of β-amyloid (Aβ) plaques, which are the other histological hallmark of AD, does not follow the same strict spatiotemporal pattern, and it correlates poorly with cognitive decline. Instead, soluble Aβ oligomers have received increasing attention as probable inducers of pathogenesis. In this study, we use microinjections into electrophysiologically defined primary hippocampal rat neurons to demonstrate the direct neuron-to-neuron transfer of soluble oligomeric Aβ. Additional studies conducted in a human donor–acceptor cell model show that this Aβ transfer depends on direct cellular connections. As the transferred oligomers accumulate, acceptor cells gradually show beading of tubulin, a sign of neurite damage, and gradual endosomal leakage, a sign of cytotoxicity. These observations support that intracellular Aβ oligomers play a role in neurodegeneration, and they explain the manner in which Aβ can drive disease progression, even if the extracellular plaque load is poorly correlated with the degree of cognitive decline. Understanding this phenomenon sheds light on the pathophysiological mechanism of AD progression. Additional elucidation will help uncover the detailed mechanisms responsible for the manner in which AD progresses via anatomical connections and will facilitate the development of new strategies for stopping the progression of this incapacitating disease.
Introduction
The progressive accumulation of specific protein aggregates along anatomical connections is a common hallmark of major non-infectious neurodegenerative diseases, such as Alzheimer's (AD), Huntington's and Parkinson's diseases (Braak and Braak, 1995; Braak et al., 2004). AD is characterized by progressive cognitive impairments and memory deficiencies (Blennow et al., 2006), with pathology that progresses from the entorhinal cortex to all of the hippocampal subregions and cortical areas via anatomical connections (Harris et al., 2010). The pathophysiological process responsible for this disease progression is not yet understood. Clinical data show that the distribution of tangles formed by the microtubule-associated protein tau is better correlated with cognitive decline than the plaques formed by β-amyloid (Aβ) accumulation (Braak and Braak, 1991). These observations are contradicted by cellular and animal models that indicate that Aβ accumulation (most likely the intracellular soluble oligomeric form) drives the disease (LaFerla et al., 2007; Gouras et al., 2010) and precedes tau-related neurotoxicity (Hardy and Selkoe, 2002). This finding suggests that the transneuronal transfer of tau (Clavaguera et al., 2009; Frost et al., 2009) is unlikely to explain the disease progression.
The neuronal dysfunction induced by Aβ can progress trans-synaptically from the neurons in the entorhinal cortex that selectively overexpress amyloid precursor protein (APP) to the terminal end of the dentate gyrus (Harris et al., 2010). The exogenous intracerebral injection of Aβ aggregates taken from brain extracts of AD patients induces cerebral β-amyloidogenesis that progresses from the injection site in APP transgenic mice (Kane et al., 2000; Meyer-Luehmann et al., 2006). Soluble Aβ aggregates appeared to be the most potent inducer, but the pathology associated with these aggregates failed to propagate in wild-type mice (Langer et al., 2011). Furthermore, Aβ reduces the number of spines and inhibits synaptic plasticity in neurons that neighbor neurons overexpressing APP (Wei et al., 2010). The present literature provides several lines of evidence suggesting that exogenous Aβ seeds are responsible for the induction of pathology, but thus far no study has addressed whether Aβ can drive the progression of pathology by means of direct transfer between neurons. To date, studies of transgenic animal models (Kane et al., 2000; Meyer-Luehmann et al., 2006; Eisele et al., 2009) have been unable to differentiate between direct neuron-to-neuron transmission of intracellular Aβ and the secondary induction of endogenous Aβ production. This distinction is crucial for both understanding the disease pathology and finding new target sites for treatment and intervention.
In the following study, we demonstrate direct evidence of the transmission of soluble oligomeric Aβ (oAβ) via connections between neuronal cells in two scenarios: (1) after microinjection in individual primary hippocampal rat neurons and (2) after donor and acceptor cell coculturing. This transmission causes disruption of tubulin structures and has cytotoxic effects. This effect strengthens the pathological role of intracellular Aβ and suggests that there is a correlation between aggregate propagation and neurodegeneration. The finding of direct cell-to-cell Aβ transfer sheds important light on the manner in which AD may be propagated through anatomical connections.
Materials and Methods
Preparation of soluble oAβ.
Freshly made oAβ1–42 were prepared and used immediately for each experiment. Lyophilized Aβ(1–42),5-TMR (AnaSpec) that had been resuspended in 1,1,1,3,3,3-hexafluoro-2-propanol were used to prepare oAβ1–42–TMR by diluting 5 mm Aβ(1–42),5-TMR in Me2SO to a concentration of 100 μm in HEPES buffer, pH 7.4. The solution was immediately vortexed and sonicated for 2 min and then incubated at 4°C for 24 h (Stine et al., 2003; Catalano et al., 2006; Rönicke et al., 2011). The scrambled Aβ1–42 (catalog #25382; AnaSpec) was labeled by incubating the peptide with TMR (6-carboxytetramethylrhodamine, succinimidyl-ester; Invitrogen) in HEPES buffer, pH 7.4, for 24 h in 4° C at a 1:2 ratio. After using a PD-10 column (GE Healthcare) to remove free dye, the scrambled Aβ1–42–TMR was processed in the same manner as the Aβ1–42–TMR.
Characterization of oligomers.
Samples of the prepared oligomer solutions (10 μl) of Aβ1–42–TMR and Aβ1–42 (which had concentrations of 30 and 10 μm, respectively) were placed on carbon-coated nickel grids. After 2 min, the solutions were removed from the grids via blotting with a filter paper, and the grids were washed with dH2O. Staining was conducted by a 20 s application of 10 μl of 2% uranyl acetate, after which the grids were blotted dry. The dry grids were viewed in a Jeol 1230 transmission electron microscope equipped with an ORIUS SC 1000 CCD camera. To characterize the oligomers of Aβ1–42–TMR and Aβ1–42, nonreducing SDS-PAGE and native-PAGE western blots using anti-Aβ (clone WO2; Millipore) were conducted. The presence of any intracellular fibrils or protofibrils were detected using Congo red staining; the oligomer-containing cells were fixed, rinsed with tap water, and then rinsed for 1 min with Mayer's hematoxylin solution. The cells were then placed in solution A [10 g of NaC1 in 1000 ml of 80% (v/v) ethanol] for 10 min and then transferred directly to solution B (2 g of Congo red and 10 g of NaC1 in 1000 ml of 80% ethanol), incubated for 20 min, and rinsed briefly (10 s) in two changes of ethanol. Bright green birefringences of the Congo red staining specific for amyloid fibrils or protofibrils were observed using crossed polarizers in polarization microscopy. The viability of the cells in the presence of Aβ1–42–TMR and Aβ1–42 oligomers was revealed using standard MTT (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) and XTT (sodium 3′-[1-(phenylaminocarbonyl)-3, 4-tetrazolium]-bis (4-methoxy-6-nitro) benzene sulfonic acid hydrate) assays.
Rat neuronal cultures.
Primary cultures of rat hippocampal neurons were prepared in the same manner described previously (Granseth et al., 2006). Hippocampi were dissected from P0–P1 Sprague Dawley rat pups of either sex, treated with papain, and plated on n = 0 glass coverslips. They were then incubated at 37°C in modified Eagle's minimum essential medium supplemented with 1% N2 and 10% horse serum in a humidified atmosphere with 5% CO2. Neural networks were allowed to develop for 1 week before the intracellular loading of oAβ1–42–TMR.
Neocortices from newborn Sprague Dawley rats of either sex were dissected to obtain cultures of rat primary cortical neurons. The neurons were either seeded in extracellular matrix (ECM) gels as described above or 35 mm Petri dishes and cultured for at least 10 d in Neurobasal A media supplemented with 0.5 mm l-glutamine, 1% penicillin/streptomycin, 2% B-27 supplement, and 5–10 ng/ml β-FGF. Half of the culture medium was replaced every 2–3 d.
Whole-cell patch-clamp recordings.
Samples of oAβ1–42 were loaded into selected neurons via diffusion from a patch-clamp pipette in whole-cell configuration. Hippocampal cultures were incubated in buffer containing 136 mm NaCl, 2.5 mm KCl, 10 mm HEPES, 2 mm CaCl2, 1.3 mm MgCl2, 10 mm d-glucose, and 100 U/ml penicillin/streptomycin. The pipette buffer constituents included the following (in mm): 140 KCl, 10 NaCl, 10 HEPES, 4 MgATP, 0.4 mm K-GTP, and 0.2 mm EGTA. Borosilicate glass microelectrodes with filament and tip resistances of ∼5 MΩ were tip loaded with buffer that did not contain any oligomers or dyes and then backfilled with buffer that contained 200 nm oAβ1–42–TMR and 50 μm Alexa Fluor-488 10,000 molecular weight dextran. In the experiments aborted as a result of a failure to achieve gigaseals, no dye loading occurred. This finding indicates that there was no significant leakage of oligomers before establishing the whole-cell configuration. The neural identity of each cell was confirmed by evoking the voltage-gated membrane currents responsible for generating action potentials with depolarizing voltage-clamp steps (Fig. 1F). These voltage steps were applied using an EPC9 patch-clamp amplifier (HEKA) controlled with the Pulse software package. Access resistance was <20 MΩ, and loading lasted 15 min and was confirmed by epifluorescence imaging while in the whole-cell setup. After 24 h of incubation, each culture was fixed with 4% paraformaldehyde and mounted using ProLong anti-fade (Invitrogen).

Transmission of oAβ1–42 through neurites of hippocampal neurons. A, Confocal image of a neuron injected with 200 nm oAβ1–42–TMR (white arrow) 24 h earlier. Note the asymmetric spread of fluorescence to glial cells with the injected cell at the periphery and interruptions by clear areas (hatched in white). Yellow arrow points to neurons to which oAβ1–42–TMR has transferred, and red arrow points to a neuron with no oAβ1–42–TMR. Scale bar, 250 μm. B, Neurons with oAβ1–42–TMR in green (visualized by subtracting glial fluorescence in C from original fluorescence in A) with DIC image. Some neurons contain oAβ1–42–TMR, but others do not. Arrows as in A. C, Distribution of oAβ1–42–TMR fluorescence in glial layer in blue (visualized by 50-μm-diameter Gaussian filtering of image in A) with DIC image. Note absence of neurons in areas in which fluorescence is interrupted. Arrows as in A. D, Average fluorescence intensity at soma of neurons (green) corrected for local glial fluorescence and background, plotted against distance from the injected neuron. Control cells plotted against an arbitrarily selected cell > 1 mm from the injected cell and average fluorescence intensity of glia (blue) corrected for background and plotted against distance from injected cell. Hatched lines are 3.3 SDs of control cell fluorescence. All analyses were made from the original, unfiltered image. au, Arbitrary units. E, Pseudocolor images of oAβ1–42–TMR fluorescence in glia (a) and dendrites (b) 24 h after injection and just after injection (c). The intensities have been individually normalized to reveal granular fluorescence. Arrows points to dendrites of injected neuron. Note that fluorescence at the time of injection is evenly distributed in the cytosol. Scale bar, 20 μm. F, DIC image during whole-cell patch clamp and membrane currents evoked by depolarizing voltage steps in the injected neuron. Scale bar, 50 μm.
Culture and differentiation of donor and acceptor cells.
All SH-SY5Y cells were pretreated with 10 μm retinoic acid (RA; Sigma-Aldrich) for 7 d before the experiment. The acceptor cells were prepared by seeding predifferentiated SH-SY5Y cells (ECACC; Sigma-Aldrich) in ECM gel (1:1; Sigma-Aldrich) in 10-mm glass-bottom Petri dishes (MatTek). The cells were differentiated for 10 d with brain-derived neurotrophic factor, neuregulin β1, nerve growth factor, and vitamin D3; this differentiation procedure has been described previously (Agholme et al., 2010). After 10 d, the cells were well differentiated; they had long, branched neurites, expressed several neurospecific markers, including synapse protein Sv2, and developed synapse-like structures, axonal vesicle transport, and mature splicing forms of tau. The acceptor cells were further prepared by staining differentiated cells with EGFP-tagged (green) endosomal (Rab5a), lysosomal (Lamp1), and tubulin proteins using organelle lights BacMam-1.0 or 2.0 (Invitrogen). The donor cells were prepared by incubating predifferentiated cells (10 μm RA for 7 d in 35-mm cell-culture Petri dishes) with 100–500 nm oAβ1–42–TMR for 3 h at 37°C in a 5% CO2 atmosphere (Hu et al., 2009). After extensive PBS washing (two washes of 10 min each at 37°C with 5% CO2) and trypsinization, the donor cells [40,000 cells per dish mixed (1:1) with prechilled ECM gel] were cocultured for 24 h with 10 μm RA on the top of the ECM gel-differentiated acceptor cells. Trypsination was conducted to remove the traces of oligomers that remained on the extracellular surfaces of the donor cells after extensive washing. Nondifferentiated acceptor and donor cells without neuronal connections were also cultured; in this scenario, the acceptor cells were treated with differentiating factors but not pretreated with RA, and the donor cells were cocultured in the absence of RA.
Receptor pharmacology.
The cocultured donor and acceptor cells were incubated for 24 h with 10 μm RA at 37°C in an atmosphere with 5% CO2, in the presence of one of the following: 20 μm tetrodotoxin (TTX), which is a voltage-gated sodium channel antagonist; a solution of 100 μm AP-5 and 100 μm (+)-MK-801 hydrogen maleate [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate], which serves as an NMDA receptor antagonist; 100 μm GYKI 52466 [4-(8-methyl-9H-1,3-dioxolo [4,5-h][2,3]benzodiazepin-5-yl)-benzenamine hydrochloride], which is an AMPA receptor antagonist; 100 nm α-bungarotoxin, which is an antagonist of the α7 subunit containing nicotinic acetylcholine receptors (nAChRs); 2 μg/ml anti-α-prion receptor protein (αPrP) mouse IgG (sc-71930; Santa Cruz Biotechnology); or without any antagonist as a control.
Immunocytochemistry.
Oligomers of Aβ, synaptic vesicles, and lysosomal membranes were immunostained with a 1:200 solution of rabbit anti-oAβ (A11) (Invitrogen), a 1:500 solution of mouse anti-Sv2 (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA), and a 1:100 solution of anti-LAMP2 (Southern Biotechnology). The secondary antibodies were Alexa Fluor 488-conjugated and Alexa Fluor 595-conjugated goat anti-mouse IgG (Invitrogen).
Image analysis.
An inverted LSM 700 Carl Zeiss confocal microscope was used to take images of the fixed cells (4% paraformaldehyde in PBS), the live cells, and time-lapse image sequences. The images were taken sequentially from the differential interference contrast (DIC) mode and red and green channels using 20×/0.8, 40×/1.30 oil-immersion, and 63×/1.40 oil-immersion plan-apochromatic DICII objectives; transmitted light was used for DIC, and 543 and 488 nm lasers were used for excitation in the red and green channels, respectively. An inverted Carl Zeiss Axiovert-200M Fluorescence Microscope was also used to visualize the live cells in the DIC mode and red (568 nm) and green (488 nm) excitation channels using a plan-apochromatic 63×/1.40 oil-immersion DIC-M27 lens. NIH Image J was used to analyze the colocalization area of the red oAβ1–42–TMR. The movements of oAβ1–42–TMR that had colocalized with lysosomes were followed using time-lapse video imaging with a confocal microscope. The acceptor cells incubated with 500 nm oligomers for 24 h were used to track oAβ1–42–TMR colocalized lysosomes. Lysosomes were stained with 200 nm Lysotracker green (DND-26; Invitrogen). In addition, oAβ1–42–FITC was used to study lysosomal colocalization. Velocity quantitation (PerkinElmer Life and Analytical Sciences) was used to track the movements of the oligomers and the vesicles.
Statistics.
Data were expressed as the mean ± SEM, and statistical comparisons were made using two-tailed unpaired Student's t tests with a significance level of p < 0.05 using the GraphPad Prism software package (GraphPad Software). Every batch of cell cultures has been treated as one independent experiment (n = 1).
Results
oAβ is transferred from neuron to neuron after injection into a single electrophysiologically defined primary neuron
To investigate whether Aβ can be transferred between neurons, we prepared cells from rat hippocampi for primary cell cultures. After 1 week in culture, the nerve cells formed a neuronal network on top of a layer of glial cells. Red fluorescent tetramethylrhodamine-tagged Aβ1–42, directly purchased from AnaSpec, was used to prepare oAβ1–42–TMR. Fluorescently labeled oligomers were then introduced to individual neurons by including 200 nm oAβ1–42–TMR in the intracellular buffer during whole-cell patch-clamp recording. The membrane conductances recorded while cells were in a patch-clamp configuration showed that all of the injected cells were neurons (Fig. 1F). Twenty-four hours after being injected with Aβ, the cultures were fixed and mounted on glass slides. The injected neuron could be located when the cultures were examined with confocal microscopy in 3 of 14 cultures. The fluorescently labeled oligomers were not confined to the injected neuron; they had spread to surrounding neurons and cells in the glial layer (Fig. 1A). This result shows that oAβ can be transferred between cells.
The spread of oAβ1–42–TMR from the injected neuron (indicated by the white arrow in Fig. 1C) was asymmetric in the sense that the injected cell was in the periphery of an approximately circular area with a diameter of 550 μm. To better visualize the oAβ1–42–TMR transfer to glia, the fluorescence image was Gaussian filtered (diameter of 50 μm) and superimposed on a DIC-enhanced image of the neuronal network (Fig. 1C). This type of filtering suppresses neuronal fluorescence because a typical neuron under these conditions has a soma diameter that is <20 μm, whereas the underlying glia cells have a diameter of ∼50 μm. The distribution of oligomers in the glial layer was also interrupted by clear areas (note the hatched area in Fig. 1). This pattern cannot be explained by the passive leakage of the oligomer from the injected neuron or the diffusional spread of labeled Aβ between cells in the glial syncytium.
Similarly, to better visualize the neurons that contained oAβ1–42–TMR (Fig. 1B), the filtered fluorescence image was subtracted from the original fluorescence image. The asymmetric spread of fluorescence was also apparent in the neuronal network. Neurons close to the injected cell might contain little or no oAβ1–42–TMR (cell indicated with red arrow in Fig. 1B, injected cell indicated with white arrow), whereas a more distant cell might contain a large amount of the oligomer (Fig. 1B, yellow arrow). When a neuron contained the oligomer, the glial cell underneath it also contained the oligomer (Fig. 1A–C). When the uptake of oAβ1–42–TMR was quantified in the original, unfiltered, fluorescence image by measuring the average intensity of the fluorescence at the nerve cell soma, it was significantly increased (>3.3 SEM) in the cells within a distance of 600 μm from the injected cell (Fig. 1D). This finding was also the case among glia. The colocalization of the oligomer between neurons and glia suggests that the molecule can be exchanged between these cell types. However, because it appears that the oAβ1–42–TMR does not spread between glia (see above), the expansion of the affected area is consistent with transfer between neurons in the neuronal network.
At the time of injection, oAβ1–42–TMR fluorescence was diffusely distributed in the cytosol and readily diffused to the distal regions of the dendrites (Fig. 1Ec). By the time that the oligomer had spread to nearby cells, the fluorescence in the injected neuron had become granular (Fig. 1Eb, white arrows point to the dendrites of the injected cell). The fluorescence inside neurons and glia that had taken up the oAβ1–42–TMR was also granular in nature (Fig. 1Ea,Eb). Therefore, the transfer of the oligomer appears to depend on either the formation of the oligomer aggregates inside the cytosol or its incorporation into endosomal compartments inside the cell.
The reason for the failure to locate the injected cell in 11 of 14 cultures might be because the injected cell detached from the network after the combined trauma of the whole-cell recording and oAβ1–42 cytotoxicity (Zhang et al., 2002). Moreover, no other cells that contained oAβ1–42–TMR were found among these cultures, which suggests that the nonspecific leakage of the oligomer from a damaged cell was not sufficient for labeling nearby cells.
Cell-to-cell transmission of Aβ oligomers can also be shown using a donor–acceptor coculture method with both rat neurons and human neuronal-like cells
To further examine the Aβ transmission, a donor–acceptor coculture method that used rat neocortex neurons in primary culture was used. Donor cells fed for 3 h with a 500 nm solution of oAβ1–42–TMR were reseeded in a three-dimensional ECM (3D-ECM) gel after extensive washing and trypsinization. The reseeded neocortical donor cells were morphologically healthy after 24 h in culture. The donor cells were reseeded on top of 3D-ECM-cultured neocortical acceptor cells transfected with a green EGFP-tagged endosomal marker, Rab5a. After 24 h of coculturing, the transmitted oAβ1–42–TMR was readily detected in many acceptor cells; it was mainly localized in the soma but was also observed in the neurites. oAβ1–42–TMR was often colocalized with endosomes (Fig. 2A).

Transmission of oAβ1–42–TMR between neuronal cells. Live-cell images of cocultured donor (containing oAβ1–42–TMR; red) and acceptor cells (transfected with EGFP-tagged lysosomal Lamp1 or endosomal Rab5a; green) using both rat cortical neurons (A) and human differentiated SH-SY5Ycells (neuronal-like) (B–D). A, Neocortical donor cells (red arrows) fed with a 500 nm oligomer solution demonstrate the transfer of oligomers to connected acceptor cells (yellow arrows) and frequent colocalization with the endosomes of the connected neurites (white arrowheads). Human partially differentiated SH-SY5Y donor cells fed with 500 nm (B), 250 nm (C), and 100 nm (D) oligomer solutions show transmission of oAβ1–42–TMR (red) from donor cells (red arrows) to most but not all highly differentiated SH-SY5Y, neuronal-like, acceptor cells (yellow arrows). Arrowheads indicate connections between donor and acceptor cells. The acceptor cell denoted with a white arrow in C has not internalized oAβ1–42–TMR. Superimposed images of the red and green channels on a DIC image and red and green confocal images. n = 6. Scale bar: A–D, 10 μm.
To confirm that the transfer of oAβ was also possible between human cells, we used human SH-SY5Y cells differentiated into mature neuron-like cells as described previously (Agholme et al., 2010) in a donor–acceptor coculture setting, such as the one described above. As seen in Figure 2, after 24 h, the transfer of oAβ from donor cells (oAβ1–42–TMR) to connected acceptor cells (mature, differentiated neuron-like cells that had been transfected with green EGFP-tagged lysosomal marker Lamp1 or endosomal marker Rab5a) occurred. The transfer occurred when the donor cells were preincubated for 3 h with oAβ1–42–TMR at all of the investigated concentrations (100, 250, and 500 nm; Fig. 2B–D). The oAβ1–42–TMR observed within the acceptor cells is unlikely to have originated from a nonspecific contamination by the oligomers after donor cell loading because trypsin treatment was performed to remove any extracellular oligomers (Frost et al., 2009). Furthermore, unlike protofibrils and fibrils, soluble oAβ is not resistant to trypsin digestion (Chauhan et al., 2005). Donor cell uptake was specific for oAβ1–42; no uptake of scrambled oAβ1–42–TMR was detected (Fig. 3A). The uptake of exogenous oAβ1–42–TMR after being added directly to the culture media of ECM gel-differentiated cells for 3 h was observed at all of the tested concentrations (10 to 500 nm). Labeled oligomers were only found intracellularly in the donor cells, and these cells appeared to be healthy and spread with numerous small neurites after trypsinization and reseeding in the ECM gel (Fig. 3B). Therefore, it is most probable that the oAβ1–42–TMR in acceptor cells originated exclusively from internalized oligomers in donor cells. Nevertheless, we investigated the possibility that nonspecific transmission from the minuscule amounts of oAβ1–42–TMR in the culture media occurred (Hu et al., 2009) by incubating the acceptor cells with pullout media from donor cell cultures at different time points (0.5, 5, and 24 h). No fluorescence was detected in the pullout media at the relevant excitation and emission wavelengths, and the fluorescence detected in acceptor cells was not different from the background fluorescence level (Fig. 3C). Thus, the possibility of an artifact attributable to an exogenous oligomer source in the current experiments is unlikely.

Transmission is specific, and oAβ1–42 requires neuritic connections. A, No uptake was detected in the ECM gel-differentiated SH-SY5Y cells that had been incubated with 250 nm scrambled oAβ1–42–TMR for 24 h (n = 3). B, After 24 h, donor cells fed with 500 nm oAβ1–42–TMR and reseeded on ECM gels appeared to be healthy and have small neurites. No fluorescence was present outside the cells (n = 3). C, Differentiated ECM-gel cells incubated for 24 h with pullout media that were taken 5 h after the reseeding of the donor cells showed no uptake (n = 3). D, Superimposition of red, green, and DIC images shows no oAβ1–42–TMR (red) transmission between undifferentiated acceptor (green; white arrows) and donor (red; red arrows) cells that were neighboring but unconnected. Superimposed image and channels provided separately (n = 2). Note the absence of labeled Aβ in unconnected acceptor cells in the red channel. Scale bars: 10 μm.
Effective transfer of oAβ is dependent on neuritic contacts between cells
To investigate whether the close apposition of cells is sufficient for Aβ transfer, donor and acceptor SH-SY5Y cells were cocultured without differentiating factors, resulting in undifferentiated cells that did not develop neurites. Under these conditions, no transfer of oAβ1–42–TMR took place, even when cells were in immediate proximity (Fig. 3D), which suggests that specific connections are necessary for this enhanced cell-to-cell transmission. In contrast, after 24 h under the conditions of normal differentiation, the short neurites of the donor cells made a number of connections with the highly branched neurites of the acceptor cells. Immunohistochemistry confirmed that the connected neurites contained the synaptic vesicle marker Sv2, which indicated that synaptic connections were present (Fig. 4A), although it remains to be shown whether these are functional. Under these conditions, red oAβ1–42–TMR was present in the neurites of both the donor (unstained) and acceptor (green) cells (Fig. 2) after 24 h of incubation. Additional evidence of the direct, neuritic transfer of oAβ1–42 is shown by time-lapse imaging in which red fluorescent granules can be seen to travel in neurites at a velocity of 0.061 ± 0.009 μm/s, similar to the measured velocity of late endosomes or lysosomes (Fig. 4D). However, the transmission of oAβ1–42–TMR was not universal; not all of the acceptor cells connected to donor cells received oligomers (Fig. 2C, white arrow). The neurites in 40.43 ± 2.45% of the acceptor cells were found to be in direct contact with the neurites of donor cells. Of the connected acceptor cells, 48.5 ± 4.3% contained oAβ1–42–TMR, whereas only 10.95 ± 2.25% of the acceptor cells that did not have demonstrated donor cell connections showed incorporated oligomers (Fig. 4B). However, it is possible that the presence of oligomers in unconnected cells has been overestimated because it is difficult to identify all of the potential connections in a 3D cell culture system. The differences in transfer to connected acceptor cells versus unconnected ones were further characterized by using the proportion of the cell body occupied by oAβ1–42–TMR fluorescence as a measurement of the amount of transferred oligomer. This measurement showed that the amount of transfer to connected cells (22.19 ± 4.6% of the total area) was significantly higher compared with that of unconnected acceptor cells (4.49 ± 3.41% of the total area). In control cells incubated with donor cell media, the area of fluorescence was only 1.4 ± 0.38% (Fig. 4C).

Quantification and characterization of transmission specificity. A, Sv2 immunostaining indicates the presence of synaptic connections between partially differentiated SH-SY5Y donor cells (yellow arrow) with short neurites and highly differentiated SH-SY5Y acceptor cells (white arrow); white arrowhead indicates synaptic vesicles in connected neurites (n = 2). B, Quantification of differentiated SH-SY5Y acceptor cells containing transferred oAβ1–42–TMR. Of all of the acceptor cells, 40.43 ± 2.45% were connected to donor cells, and 58.82 ± 2.49% were unconnected. Of the connected acceptor cells, 48.5 ± 4.3% were positive for fluorescent Aβ oligomers, whereas 10.95 ± 2.25% of the unconnected acceptor cells demonstrated fluorescence attributable to the presence of Aβ oligomers; data from 75 images of five cultures. C, The areas containing transmitted oligomers were significantly higher (22.19 ± 4.6%) within connected acceptor cells than either unconnected acceptor cells (4.49 ± 3.41%) or negative controls (1.4 ± 0.38%); data from 20 images of two sets of cultures. Images in B and C were taken from areas in which donor and acceptor cells were evenly distributed. **p < 0.01, ***p < 0.001. D, Time-lapse confocal image sequence of the cell-to-cell transmission of oligomers (red) from partially differentiated SH-SY5Y donor (fed with 500 nm Aβ) to highly differentiated SH-SY5Y acceptor (Rab5a) cells (green; n = 3). Transfer of discrete accumulations of oligomers could be followed; one such is indicated by white arrowheads. Scale bars: A, 10 μm; D, 5 μm.
Transmission of oAβ slowly and gradually causes cytotoxicity
In AD pathology, disease progression is accompanied by a spread of neurodegeneration. Therefore, we investigated whether oAβ transmission caused a decrease in cell viability. No significant toxicity was observed using an XTT assay, MTT assay, or nuclear staining of oAβ1–42–TMR-loaded donor and acceptor cells after 48 h in coculture (results not shown). Changes in subcellular morphology, such as the formation of beaded tubulin structures (Shah et al., 2009) and changes in the distribution of discrete endosomal protein Rab5a (Song et al., 2011), are early signs of cytotoxicity. Similarly, these measurements indicated that, after 2 d of incubation, cells were healthy; tubulin in the neurites of the acceptor cells was evenly dispersed (Fig. 5A), and the distribution of Rab5a in the acceptor cells was uniformly dotted (Fig. 5B). However, after ≥3 days of incubation (and increasing with both the incubation time and intracellular oligomer concentration), both donor and acceptor cells gradually lost their cellular shapes; the acceptor cells began to form tubulin beads, and the discrete endosomal (Rab5a) staining was disrupted. All of these morphological changes indicated cytotoxicity (Fig. 5). In contrast, the acceptor cells that had little or no oligomer content retained a uniform tubulin structure and discrete arrangement of endosomal structures. Even after 5–8 d of incubation, the acceptor cells that had not absorbed the transmitted oligomers displayed a more normal morphology, more intact tubulin, and more regularly spaced endosomal structures, which indicated that they were healthier than the cocultured donor and acceptor cells that contained intracellular oligomers (Fig. 5).
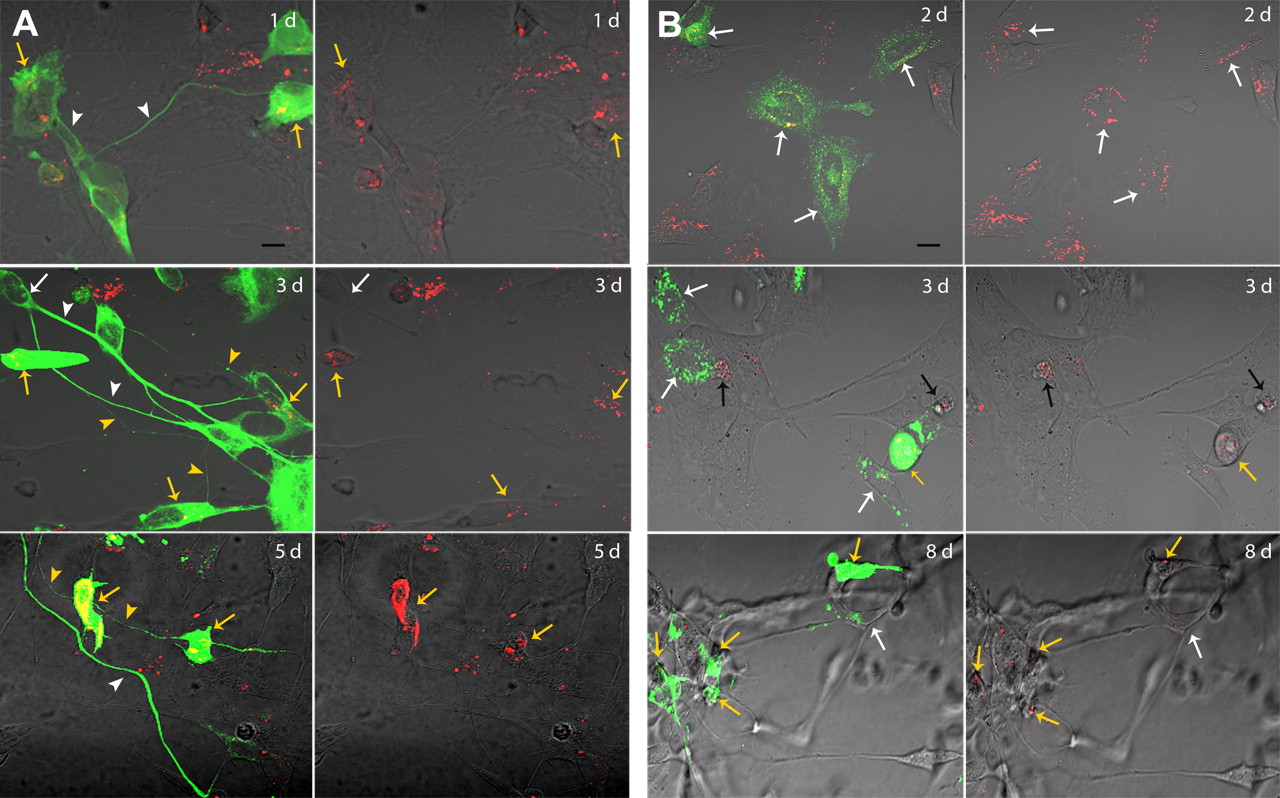
Transmission of oAβ results in slow cytotoxicity. A, oAβ internalized in donor and acceptor cells, prepared using differentiated human SH-SY5Y cells, eventually causes neurodegeneration, which is revealed by the presence of beaded tubulin structures. oAβ1–42–TMR-containing acceptor cells (yellow arrows) retained an evenly distributed tubulin structure (white arrowheads) in the neurites after 24 h of incubation. The beaded tubulin structure (yellow arrowheads) began to form in the neurites of most acceptor cells that contained oAβ1–42–TMR after 3 d of incubation. Neurites not connected directly with the oligomer-containing acceptor cells retained their tubulin structures (white arrowhead), even after 5 d (n = 4). B, Internalized oAβ1–42–TMR in both donor and acceptor cells has gradual subcellular toxic effects and eventually causes cell death. After 2 d (2d), the cells appeared to be healthy based on cell morphology and an intact, dotted endosomal distribution (white arrows). After 3 d (3d), the acceptor cells that contained oAβ1–42–TMR (yellow arrows) displayed a loss of neuronal morphology and increase in the proportion of abnormal endosomal structures that worsened over time. This trend was also observed in donor cells (black arrows). Acceptor cells without incorporated oAβ1–42–TMR appeared to be healthy throughout the incubation period (white arrows; n = 2). Scale bars, 10 μm.
Possible mechanism of transmission
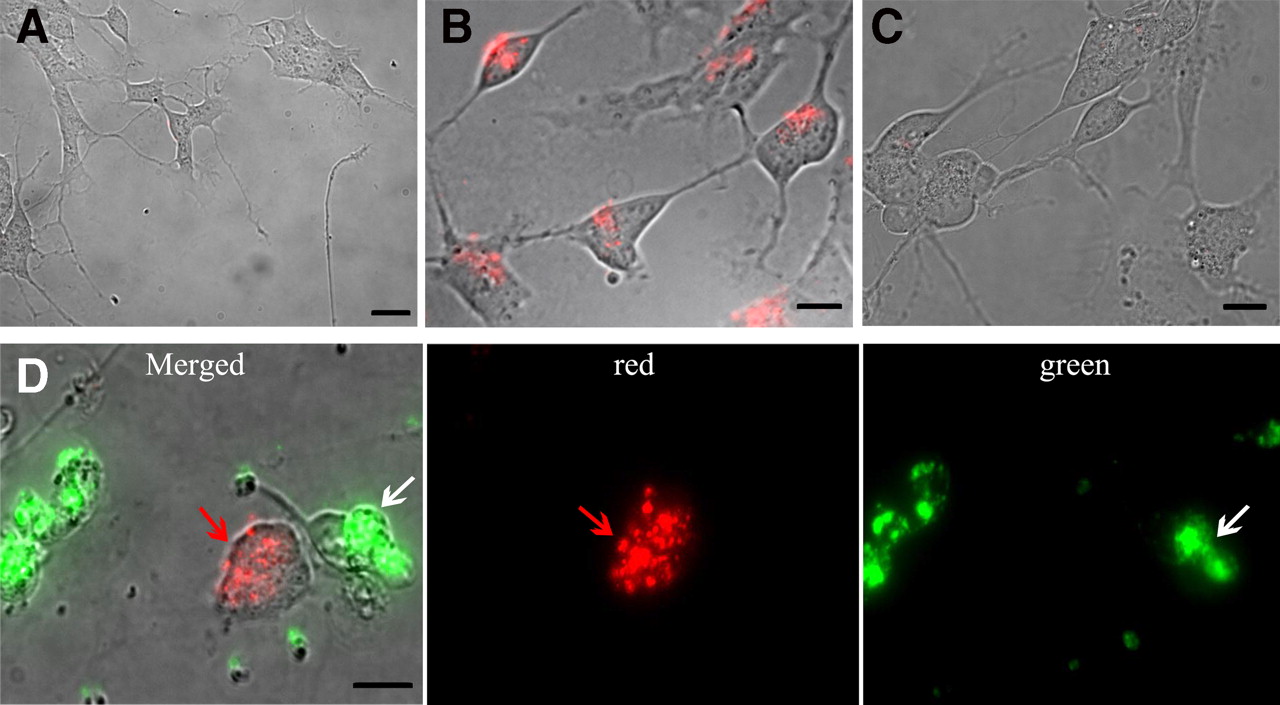
Recent evidence has suggested that oAβ impairs synaptic plasticity and causes cognitive failure in AD before plaque deposition and neuronal cell death (Walsh and Selkoe, 2007; Rönicke et al., 2011). oAβ-mediated spine loss, disruption of synaptic plasticity, and subsequent abnormalities in spine morphology could be mediated by action potentials, nAChRs, NMDA receptors, or AMPA receptors (Wei et al., 2010). Furthermore, it has been proposed that Aβ binds to the PrPc receptor, although this notion has been debated (Kessels et al., 2010). RA-differentiated SH-SY5Y cells have been reported to express NMDA receptors, AMPA receptors, and nAChRs (Xie et al., 2010), although it is not known whether they function as they do in synapses of neurons. However, oAβ transmission was not affected by either previously established concentrations of NMDA receptor, AMPA receptor, nAChR, and PrPc receptor inhibitors or by a full blockade of electrical activity using TTX (Fig. 6A) in our highly differentiated SH-SY5Y cells. Thus, the mechanism for Aβ transfer between neurons remains undiscovered, although potential receptor modulation effects might differ in primary neurons. However, some of our observations suggest the possible involvement of the endosomal–lysosomal pathway. Most of the exogenous oligomers colocalized with lysosome or late endosome markers (Fig. 6B,C) in both donor (as demonstrated by immunocytochemistry) and acceptor (shown using Lysotracker) cells. The colocalization of oAβ1–42–TMR and early endosomes was occasionally observed in the neurites that connected donor and acceptor cells (Fig. 6D); together, these observations suggest that the lysosomal–endosomal system may be involved in the transfer of oAβ1–42–TMR. The effects of common endosomal inhibitors, even with low concentrations of phenyl arsine oxide (1 μm) and sucrose solution (50 μm), had adverse and toxic effects on the differentiated acceptor cells after 24 h, which is consistent with reports of the toxic effects that endosomal inhibitors produce in primary neurons (Macia et al., 2006). Thus, we cannot yet confirm the role of endocytosis in oAβ1–42–TMR transmission.

The transmission mechanism appears to involve exocytosis coupled with endocytosis. A, Application of inhibitors of voltage-gated sodium channels, PrPc receptors, nAChRs, NMDA receptors, or AMPA receptors has no significant effect on oAβ1–42–TMR transmission versus the application of a control substance (p > 0.05), when studied using differentiated human SH-SY5Y coculture. B, In partially differentiated SH-SY5Y donor cells, most of the oAβ1–42–TMR (red) colocalized with lysosomes and late endosomes labeled with Lysotracker (green; n = 4). C, Immunocytochemical staining of differentiated SH-SY5Y cells fed with FITC-Aβ (green) with an antibody against LAMP2 also showed that exogenous Aβ colocalized with lysosomes and late endosomes (n = 2). D, Superimposed red and green confocal channels show that oAβ1–42–TMR (red) from donor cells (red arrow) colocalizes with Rab5a (green; yellow arrow), an EGFP-labeled early endosomal marker, at connected neurites in acceptor cells (white arrowheads). Scale bars, 10 μm.
Characterization of the used Aβ oligomers
oAβ1–42 and oAβ1–42–TMR were prepared by incubation (24 h at 4°C) of 100 μm Aβ1–42–TMR or oAβ1–42 peptides dissolved in Me2SO and HEPES buffer, pH 7.4, as described previously. This preparation method is well known to result in oligomers, not fibrils (Stine et al., 2003; Catalano et al., 2006; Rönicke et al., 2011). Transmission electron microscopy (TEM) images of the oAβ preparations directly after incubation (0 h), that is the time point the oAβ1–42 were otherwise added to the cells, identified them (90–95%) as being primarily heterogeneous assemblies of oligomers, with diameters ranging from 1 to 10 nm; 5–10% of the oligomers had diameters > 10 nm (Fig. 7A). The TEM images revealed the formation of similar oligomeric structures for both oAβ1–42–TMR (Fig. 7Aa) and oAβ1–42 (Fig. 7Ab); neither protofibrils nor fibrillar structures were detected. The oligomer solutions did not show any insoluble precipitation after centrifugation at 15,000 rpm for 15 min. Furthermore, no insoluble aggregates were detected by changes in the absorbance of 340 nm light before and after the centrifugation (15,000 rpm for 15 min) (Martins et al., 2006). The stability of the oligomer preparations were investigated over time; after dilution (500 nm) in MEM media, the solution was kept at 37°C. After 24 h, ∼15–25% small protofibrils were formed (Fig. 7Ba), and gradually (48 and 72 h) the oligomers aggregated to these. The formation of some fibrils also appeared (Fig. 7Bb). It should be noted that the donor cells were incubated with 500 nm oligomers at 37°C only for 3 h, thus before apparent formation of larger protofibrils or fibrils. Using Congo red staining (Westermark et al., 1999), no intracellular fibrillar structures were detected in either the donor cells fed with oAβ1–42–TMR or the acceptor cells after 24 or 48 h of coculture with donor cells. After 72 h of incubation, few cells (< 1%) showed some nonspecific fluorescence that could not conclusively be interpreted as fibril formation. Positive immunostaining with an anti-oligomer-specific antibody (A11) indicated that the oligomer structures was retained in both the oAβ1–42–TMR-fed donor cells and cocultured acceptor cells even after 24 h (Fig. 7C). Cells that did not contain oAβ1–42–TMR had no detectable endogenous oligomers (Fig. 7C, white arrows). The SDS-PAGE showed that oAβ1–42–TMR forms SDS-stable dimers and trimers (Fig. 7D, lane 3), whereas unlabeled oAβ1–42 forms SDS-stable trimers and tetramers (Fig. 7D, lane 2) at 0 h. No larger SDS-stable oligomers were observed at this time point. In native-PAGE, the oligomer preparations formed a trail of Aβ (Fig. 7E), which suggests the formation of oligomeric species assemblies at 0 h. Despite differences in their associated SDS-stable species, the XTT assays of both unlabeled oAβ1–42 and labeled oAβ1–42–TMR revealed slow but significant cytotoxic effects with increasing time. The percentages of cell death among donor cells affected by unlabeled oAβ1–42 is 19.3 ± 6.5% and labeled oAβ1–42–TMR is 11 ± 1.5% after 72 h. Thus, it can be concluded that the Aβ1–42 preparations are in an oligomeric state during the incubation of the donor cells (0–3 h). It is plausible that some protofibrills and eventually larger fibrils might form intracellularly over time. Whether these larger structures could also be transferred remains to be investigated.

Characterization of oAβ preparations. A, TEM images show that both Aβ1–42–TMR (a) and Aβ1–42 (b) only form oligomers during oligomer preparation; no protofibrils or fibrils were formed at 0 h. A layer of oligomers was produced on a copper grid using 10 μl of a 10 μm solution. B, The stability of the oligomer preparations were investigated over time, after dilution (500 nm) in MEM at 37°C. After 24 h, some small protofibrils were formed (white arrow in a), and gradually to 72 h, oligomers aggregated with formation of some fibril-like structure (b). C, Immunostaining with an anti-oligomer-specific antibody (A11) (stained with an Alexa Fluor 594-tagged secondary antibody that is shown in blue; a) suggests that oligomer structures were retained in the donor cells (black arrows) fed with oAβ1–42–TMR (red; b) and cocultured acceptor cells (yellow arrows), even after 24 h. Cells that did not contain oAβ1–42–TMR (red) had no detectable A11-specific (blue) endogenous oligomers (white arrows). Nonreducing SDS-PAGE (D) and native-PAGE (E) Western blot of prepared Aβ1–42 and Aβ1–42–TMR samples at 0 h using anti-Aβ (clone WO2; Millipore). D, SDS-PAGE shows a clear 4 kDa band for a monomeric preparation of Aβ1–42 (lane 1), SDS-stable trimers and tetramers of oAβ1–42 (lane 2), and SDS-stable dimers and trimers of oAβ1–42–TMR (lane 3). E, The trailing of oAβ1–42 and oAβ1–42–TMR in native-PAGE suggests the formation of oligomeric assemblies. Scale bars: A, B, 200 nm; C, 10 μm.
Discussion
The data presented here show that oAβ can be transmitted neuron to neuron and that this transfer depends on direct neuritic connections. In the AD brain, neurofibrillary tangles progress in a fixed pattern in which pathology likely begins in the transentorhinal cortex and spreads to the hippocampus and cortical areas (Braak and Braak, 1991). In contrast, amyloid plaques are first found in the neocortex and do not follow the same strict spatiotemporal pattern. There is also a poor correlation between the amyloid plaque load and severity of disease symptoms (Braak and Braak, 1991). Intracellular soluble Aβ oligomers have received increasing attention and may be an important factor in the AD pathogenesis (LaFerla et al., 2007; Gouras et al., 2010). The mechanisms behind the specific spatiotemporal pattern of neuropathology in AD are not well understood. It has been suggested that the neuropathology begins in small neuronal populations and then progressively spreads to connected brain regions by propagation between neighboring cells and via spread along axonal pathways (Hardy and Selkoe, 2002). However, the mechanism for this transmission is not understood. Experiments in different animal models have demonstrated the spread of pathological changes but have not provided evidence for any specific pathological carrier (Kane et al., 2000; Meyer-Luehmann et al., 2006; Harris et al., 2010; Langer et al., 2011). Our findings point to a possible mechanism responsible for this spread of AD pathology between connected brain areas.
Studies of Aβ are complicated by the inherent properties of the protein. Characterization with electron microscopy and Western blot analysis shows that our prepared oAβ consists of oligomers at the time of application. Immunocytochemistry indicates that the tracked, fluorescent oAβ1–42–TMR retains its oligomeric form after transmission to the acceptor cell. Because some protofibrils and eventually fibrils formed over time when the oligomer preparation was incubated in vitro, it cannot be excluded that this could also happen in the cells. However, there was no evidence of this being a substantial event in the current experiments. Although the use of fluorescent tagging was indispensable in the present study, it also raises some concern as to the in vivo relevance of the finding. However, because both labeled and unlabeled oAβ had similar cytotoxic effects on the cells, it is highly likely that neither the transfer nor cytotoxic effect of oAβ is dependent on the fluorescent tag. In this aspect, it should also be pointed out that the continuous transmission of oAβ shown here is not explained by endogenous Aβ because only the internalized oAβ1–42–TMR, which was originally exogenous, is visualized.
To study the transfer of oAβ shown in rat hippocampal neurons in greater detail, a 3D cell culture model was used. We have established previously this model as an in vitro model for studying human neurons (Agholme et al., 2010). When cultured according to this method, SH-SH5Y neuroblastoma cells differentiate into neuronal phenotypes according to both morphology and neuronal markers. Still, the possibility of some remaining tumor cell-dependent phenotype cannot be excluded. However, because transfer was also established in primary rat neurons, it is unlikely that the current finding depends on a tumor cell phenotype. As shown in the donor–acceptor coculture, the transfer appears to depend on specific, neuritic-like cellular connections. Under differentiated conditions, transmission was mainly observed between cells that had identified connections, and no transfer was observed if undifferentiated cells without neurites were used. Thus, transmission most likely requires these neuritic connections. This is consistent with previous findings showing that uptake of extracellular Aβ is most efficient in the axon (Saavedra et al., 2007). This phenomenon is thus different from the unspecific uptake used to load the donor cells. The possibility that similar unspecific transfer occurred between the differentiated cells was carefully assessed; no remaining oAβ1–42–TMR was detected in the medium of the cultured donor cells that had been washed, trypsinized, and replated, and no uptake was observed among the acceptor cells cultivated with pullout medium from the donor cells. Thus, the possibility that the observed transfer of oligomers was attributable to nonsynaptic extracellular re-secretion of these oligomers is unlikely. In apparent contrast to the specific transfer described above, oAβ uptake was also observed in the supporting glial cells after microinjection of oAβ1–42–TMR into a defined rat hippocampal neuron. However, it has been well established that glia absorb various materials and clear them from the synaptic cleft and that various forms of Aβ are taken up and cleared by glia (Lee and Landreth, 2010). Thus, the finding of oAβ in the glia cells was expected.
Having established the neuron-to-neuron transfer of oAβ, we sought to determine its cellular mechanism. Exosome trafficking is one potential mechanism by which disease-related proteins, such as prions and α-synuclein, could be transmitted between cells (Fevrier et al., 2004; Alvarez-Erviti et al., 2011). It has also been reported that prion transmission occurs by means of tunneling nanotubes (Gousset et al., 2009); this mode of transfer has also been suggested for Aβ monomer and protofibrils (Wang et al., 2011). However, it has been shown that Aβ is secreted by the exosomes of neuronal cells in vitro (Rajendran et al., 2006). In the donor–acceptor coculture, the internalized oligomers collect in lysosomal vesicles, and, in the neurites of the connected acceptor cells, it colocalized with early endosomes. Directly after the injection, oAβ1–42–TMR is evenly distributed in the cytoplasm of primary hippocampal neurons but subsequently accumulates into grain-like structures that likely correspond to vesicles. A possible mechanism for this is macroautophagy, in which cytosolic Aβ can gradually accumulate into lysosomes (Zheng et al., 2011). In the current study, we found that the velocities of the oligomers in the neurites were comparable with the speed of lysosomes (Fuerst et al., 2011). This finding supports macroautophagy as a potential mechanism of disease spread. Prion-containing endolysosomal compartments have been shown to fuse with multivesicular bodies, which results in the extracellular release of exosomes containing prions (Fevrier et al., 2004), and lysosomal exocytosis functions to promote cellular clearance (Medina et al., 2011). Based on both the accumulating evidence and our endosomal–lysosomal data, we propose that the transfer of oAβ might use exocytosis or a similar mechanism. Similarly, the fast, unitary transfer of oAβ1–42–TMR between connected neurites suggests that this transfer may be mediated by a mechanism in which exocytosis and endocytosis are rapid and coupled, such as the ones suggested previously for tau and α-synuclein (Desplats et al., 2009; Frost et al., 2009; Hansen et al., 2011). It could not be determined whether oAβ1–42–TMR transfer depended on a specific receptor because the application of previously established concentrations of known inhibitors of voltage-gated sodium channels, PrPc receptors, nAChRs, NMDA receptors, or AMPA receptors, did not have significant effects on oAβ1–42–TMR transmission, at least not in these highly differentiated, neuronal-like, neuroblastoma cocultures. Additional studies are required to establish the exact mechanisms of transfer.
There is increasing evidence of correlations between intraneuronal oAβ1–42 and synaptic dysfunction, subcellular pathology, and cell death before the development of extracellular plaque pathology (LaFerla et al., 2007; Gouras et al., 2010). We now show that the oAβ1–42 transfer results in a subsequent, slowly developing neurodegeneration that is not observed in cells without oAβ1–42. The progressive tubulin beading after oAβ transfer shown here could be a sign of axonal disruption (Shah et al., 2009), indicating that oAβ can cause axonal damage. Other researchers have proposed that axonal damage is part of the process that leads to neurodegeneration in AD (Pigino et al., 2009), and in concordance with this, a gradually increasing disruption of endosomes, a sign of cytotoxicity (Song et al., 2011), was also observed. This observation not only reinforces the role of intracellular Aβ in AD pathogenesis but also establishes the disease relevance associated with the cell-to-cell transfer of intracellular oAβ1–42. Prion-like intercellular spread and amplification of protein misfolding can also occur for proteins associated with other neurodegenerative diseases, such as tau (Clavaguera et al., 2009; Frost et al., 2009), α-synuclein (Desplats et al., 2009; Hansen et al., 2011), huntingtin (Ren et al., 2009), and superoxide dismutase-1 (Ilieva et al., 2009). Internalized Aβ oligomers can increase the accumulation of newly synthesized Aβ (Bahr et al., 1998; Glabe, 2001) and act as seeds for aggregation of endogenous proteins (Hu et al., 2009; Gaspar et al., 2010). Thus, it is possible that the currently described Aβ transfer could not only cause additional seeding of Aβ oligomerization but might also result in subsequent tau pathology, thereby spreading the neurodegenerative pathology to anatomically connected brain areas. This might be one possible mechanism for the clinical progression of AD.
The present demonstration that accumulated intracellular Aβ oligomers can be transmitted via direct neuritic connections might explain the manner in which AD spreads progressively through anatomical connections. This would also explain how Aβ could be responsible for the disease progression despite the low correlation between the extracellular plaque load and cognitive decline, thereby shedding important light on a possible pathophysiological mechanism of AD progression. Additional elucidation of the detailed mechanisms behind this transmission will facilitate the development of new strategies to stop or inhibit the progression of this dreadful disease.
Footnotes
This research was made possible by funding from the Östergötland County Council, the Linköping University Neurobiology Centre, the Swedish Alzheimer's Foundation, Swedish Research Council Grants 3050 and 2862 (B.G.), Gustav V and Queen Victoria's Foundation, and the Hans-Gabriel and Alice Trolle-Wachtmeisters Foundation for Medical Research. We thank Prof. Katarina Kågedal for valuable discussions, Dr. Johan Brask, and Sofie Sundberg for making hippocampal cultures and patch-clamp injections, Dr. Karin Björnström-Karlsson for supplying rat cortical neurons, and Bengt-Arne Fredriksson for assistance with electron microscopy.
The authors declare no competing financial interests.
- Correspondence should be addressed to either of the following Sangeeta Nath, Division of Geriatrics, Department of Clinical and Experimental Medicine, Faculty of Health Sciences, Linköping University, SE-581 85 Linköping, Sweden, Sangeeta.Nath{at}liu.se; or Martin Hallbeck, Experimental Pathology, Department of Clinical and Experimental Medicine, Faculty of Health Sciences, Linköping University, SE-581 85 Linköping, Sweden. Martin.Hallbeck{at}liu.se
This article is freely available online through the J Neurosci Open Choice option.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}