

Abstract
Hydrocephalus formation is a frequent complication of neuropathological insults associated with neuroinflammation. However, the mechanistic role of neuroinflammation in hydrocephalus development is unclear. We have investigated the function of the proinflammatory acting inhibitor of κB kinase (IKK)/nuclear factor κB (NF-κB) signaling system in neuroinflammatory processes and generated a novel mouse model that allows conditional activation of the IKK/NF-κB system in astrocytes. Remarkably, NF-κB activation in astrocytes during early postnatal life results in hydrocephalus formation and additional defects in brain development. NF-κB activation causes global neuroinflammation characterized by a strong, astrocyte-specific expression of proinflammatory NF-κB target genes as well as a massive infiltration and activation of macrophages. In this animal model, hydrocephalus formation is specifically induced during a critical time period of early postnatal development, in which IKK/NF-κB-induced neuroinflammation interferes with ependymal ciliogenesis. Our findings demonstrate for the first time that IKK/NF-κB activation is sufficient to induce hydrocephalus formation and provides a potential mechanistic explanation for the frequent association of neuroinflammation and hydrocephalus formation during brain development, namely impairment of ependymal cilia formation. Therefore, our study might open up new perspectives for the treatment of certain types of neonatal and childhood hydrocephalus associated with hemorrhages and infections.
Introduction
Hydrocephalus is a frequent, heterogeneous neuropathological disorder characterized by increased pressure of the CSF, resulting in a dilation of the cerebral ventricles. The neuropathological consequences of this increased intracranial pressure are lethal in 20–50% of patients if left untreated (Poca and Sahuquillo, 2005). Main causes of hydrocephalus are infections and injury of the CNS (e.g., a large proportion of patients with subarachnoid hemorrhages and tuberculous meningitis develop hydrocephalus) (Garg, 2010; Germanwala et al., 2010). During brain development, susceptibility to hydrocephalus formation seems especially high (e.g., in congenital toxoplasmosis) (Mack et al., 1999). Observations from patients and mouse models depict some general mechanisms leading to an increase of CSF pressure, mainly an impaired CSF transport due to obstructions of the ventricular system or ependymal cell malfunction, or altered production or resorption of the CSF (Zhang et al., 2006). However, the molecular mechanisms and the specific contribution of these pathological processes to human hydrocephalus remain unclear.
Many neurological diseases, including hydrocephalus, are associated with neuroinflammation, yet its role therein is incompletely understood. In the CNS, microglia and astrocytes are key regulators of inflammation (Glass et al., 2010). Astrocytes regulate the blood–brain barrier, have neurotrophic functions, and act directly on synaptic signaling (Sofroniew and Vinters, 2010), and astrocyte-related radial glia serve as neural stem and scaffolding cells (Kriegstein and Alvarez-Buylla, 2009). Under pathological conditions, astrocytes are activated, produce proinflammatory mediators involved in the recruitment and activation of immune cells, change their neurotrophic functions, and can form scars (Sofroniew and Vinters, 2010).
Major regulators of inflammation are the nuclear factor κB (NF-κB) transcription factors RelA (p65), RelB, c-Rel, p50/p105, and p52/p100. In the resting state, NF-κB dimers are bound to inhibitor of κB (IκB) proteins preventing their nuclear translocation. A great variety of proinflammatory stimuli activate the IκB kinase complex (IKK) that phosphorylates IκB proteins, thereby targeting them for proteasomal degradation. This allows the release and translocation of NF-κB dimers to the nucleus, where they regulate the transcription of target genes involved in inflammation, proliferation, cell survival, and diverse CNS functions (Mattson and Meffert, 2006). IKK2 is the pivotal kinase in canonical NF-κB signaling that is essential for the regulation of inflammation (Hayden and Ghosh, 2008).
Due to the fact that astrocytes are important mediators of CNS inflammation, the functional analysis of NF-κB signaling in astrocytes is of great interest for the understanding and treatment strategies of diseases associated with neuroinflammation. Indeed, transgenic mice with suppressed NF-κB signaling in astrocytes (GFAP.IκBα-dn) have limited inflammatory responses in the context of spinal cord injury and experimental autoimmune encephalitis (Brambilla et al., 2005, 2009).
To understand the consequences of NF-κB activation in astrocytes, we generated a mouse model that allows the tetracycline-dependent expression of a constitutively active IKK2. Interestingly, these animals develop global neuroinflammation and an early postnatal hydrocephalus caused by impaired ependymal cilia formation. Thus, our findings provide the first direct link of ciliogenesis and IKK2/NF-κB signaling, giving a potential mechanistic explanation for the association of neuroinflammation with development of childhood hydrocephalus.
Materials and Methods
Transgenic mice.
GFAP.tTA/(tetO)7.IKK2-CA (short GFAP/IKK2-CA) double-transgenic mice were generated by direct crossing of GFAP.tTA mice from a C57BL/6 background (Pascual et al., 2005) with (tetO) 7.IKK2-CA mice (with a luciferase reporter gene regulated by the same bidirectional promoter) from a NMRI background (Herrmann et al., 2005). For transgene inactivation during complete development, doxycycline (0.5 g/L; MP Biomedicals) in 1% sucrose was given in the drinking water to the mothers during pregnancy till weaning (age, 4 weeks). For transgene inactivation from postnatal day 0 (P0), a single dose of doxycycline was injected in the mother immediately after birth (100 μg/g body weight, i.p.), and then doxycycline was administered with the drinking water as described above.
For a rapid control of transgene expression, luciferase activity in tissue protein extracts from the medulla oblongata and spinal chord was measured, as described previously (Baumann et al., 2007). Animals with very low or absent luciferase activity (<1000 RLU/μg) were excluded. As control animals, single transgenic (tetO)7.IKK2-CA littermates were used, if not indicated otherwise. For additional control experiments, a second founder line with the (tetO)7.IKK2-CA transgene was used (line B), which was backcrossed for 9–10 generations to a C57BL/6 background, and a mouse line with a similar transgene construct with a dominant-negative IKK2 allele, named (tetO)7.IKK2-DN (Herrmann et al., 2005). All animal experiments were approved by the Regierungspräsidium Tübingen (Tübingen, Germany).
Primary astrocyte culture.
Primary astrocytes were obtained from newborn GFAP/IKK2-CA and control mice (P2), which received doxycycline to inactivate the transgene (see above). Cortex tissue was digested with trypsin (0.05%)/EDTA for 20 min, and then trypsin was inactivated with serum containing culture medium (DMEM, 20% FBS, 1% penicillin/streptomycin). The digested tissue was mechanically dissociated, and residual tissue was removed with a 100 μm cell strainer. Cells were seeded on laminin-coated culture dishes and were grown in culture medium in the presence of doxycycline for 1 week. To reactivate transgene expression, doxycycline was removed and carefully washed out. Then the cells were once passaged and after 3 days transgene expression was confirmed by luciferase measurement and staining for human IKK2. Microglia content was <5% as controlled by CD11b staining. The cells were either directly used for experiments or cryopreserved in liquid nitrogen in medium containing 10% DMSO, 50% FBS, and 40% DMEM. After thawing, the cells were passaged once before using them for experiments. The IKK2 inhibitor N1-(1,8-dimethylimidazo[1,2-a]quinoxalin-4-yl)ethane-1,2-diamine hydrochloride (BMS-345541) used in primary astrocyte cultures was purchased from Axon Medchem.
Scanning electron microscopy.
Brains were fixed by immersion for at least 24 h with 2.5% glutaraldehyde and cut to 0.5 mm sagittal sections with a vibratome to expose parts of the lateral walls of the lateral ventricles. The sections were then postfixed with OsO4, critical point dried, and vaporized with Au/Pd. Images were acquired with the Zeiss DSM 962 microscope.
Immunoblotting.
Native tissue protein extracts were prepared as described previously (Baumann et al., 2007). Twenty to 50 μg of protein were separated by SDS-PAGE, transferred to PVDF membranes, and blocked with 5% dry milk in TBS buffer. Transgene expression was detected with antibodies against IKK1/2 or human IKK2, which detects only the transgene. Erk2 was detected as loading control.
All antibodies used for immunoblotting were obtained from Santa Cruz Biotechnology. Luminescence signals were detected with the “Intelligent Dark Box” (Fuji).
RNA extraction, microarray, and qPCR.
RNA was extracted with the Ambion mirVana kit or the Peqlab Trifast kit, and cDNA was synthesized with the Roche Transcriptor High-Fidelity cDNA synthesis kit according to manufacturer's instructions. Microarray analysis was performed with the Mouse Gene 1.0 ST Array (Affymetrix) and evaluated with the “Genesifter” software (Geospiza). Quantitative real-time PCR assays were done with the Roche LightCycler 480 with primers and hydrolysis probes designed by the Roche Universal Probe Library system.
Histology and immunostaining.
Brains were fixed by immersion with 4% PFA (3–4 h on ice), dehydrated, and embedded in paraffin, and cut to 7 μm coronal sections. After rehydration, heat-mediated antigen retrieval was performed with sodium citrate (10 mm, pH 6, 0.05% Tween 20) or Tris-EDTA (10 mm Tris, 1 mm EDTA, pH 9, 0.05% Tween 20), and for full permeabilization sections were incubated with 0.5% Triton X-100 for 30 min. Sections were washed with PBS and blocked with 5% BSA for 1 h. Incubation with the primary antibodies (in 5% BSA) was performed overnight at 4°C; secondary antibodies were applied for 1 h at room temperature with DAPI for nuclear counterstaining. For CD45/CD11b and F4/80/CD11b staining, 10 μm cryosections from natively frozen brains were fixed with cold methanol (−20°C). Blocking and staining were performed as described above; PE-labeled CD11b antibody was applied for 2 h after careful washing after the secondary antibody.
Fluorescence pictures were acquired with the Zeiss Axiovert 200M microscope with filters for DAPI, FITC/Alexa Fluor 488, DsRed/PE, and Texas Red/Alexa Fluor 568/594 and the Zeiss AxioVision software. For each channel, exposure times were separately adjusted and kept for the complete session. Adjustment of contrast and brightness was performed for each channel separately, but in all compared pictures equally.
Antibodies for immunofluorescence.
Rabbit anti-IKK1/2, goat anti-human IKK2, rabbit anti-RelA, mouse anti-glial fibrillary acidic protein (GFAP), rabbit anti-Erk2, and HRP-conjugated goat anti-rabbit were obtained from Santa Cruz Biotechnology. Mouse anti-Aldh1l1, rabbit anti-fibronectin, and rabbit anti-laminin were obtained from Abcam. Rat anti-CD45 and mouse anti-β-catenin were obtained from BD Biosciences. The following further primary antibodies were used in immunofluorescence: rabbit anti-cleaved caspase 3 (Cell Signaling Technology), PE-conjugated rat anti-CD11b, and rat anti-F4/80 (eBioscience).
Alexa Fluor-labeled secondary antibodies and Oregon Green 488-labeled wheat germ agglutinin (WGA) were obtained from Invitrogen.
Quantification of nuclear RelA staining.
All samples for quantification of DAPI/hIKK2/RelA staining were stained simultaneously, and pictures were taken in one session with the same settings with a 20× objective. The raw images were analyzed with the ImageJ software. For each animal, three fields of the midbrain region with ∼400–600 nuclei (or ∼40–80 nuclei of transgenic cells) were measured. Nuclei were detected semiautomatically with the “particle analysis” tool in the DAPI layer, and the fluorescence intensity of the corresponding areas of the RelA layer was measured for each nucleus. To measure only nuclei of transgene-positive cells, these cells were selected in the hIKK2 layer before definition of the nuclei as above. With the Prism software (GraphPad), RelA intensity histograms were generated to set a threshold above which cells were regarded as nuclear RelA positive.
Statistical analysis.
Statistical analysis was performed with the Prism software (GraphPad). The applied tests are indicated in the specific figure.
Results
Expression of constitutively active IKK2 in astrocytes induces NF-κB activation and results in early postnatal hydrocephalus formation
To investigate the role of astrocytic NF-κB signaling in neuroinflammation, we generated a gain-of-function mouse model based on the astrocyte-specific expression of a constitutively active allele of human IKK2 (IKK2-CA). For this purpose, we established the GFAP/IKK2-CA mouse model by combining the responder mouse line (tetO)7.IKK2-CA (Herrmann et al., 2005) with the GFAP.tTA driver mouse line (Pascual et al., 2005). In this system, the expression of the tetracycline-dependent transactivator (tTA) is controlled by the promoter of the GFAP, which is used in various mouse models to achieve astrocyte-specific expression.
The astrocyte-specific expression of the IKK2-CA transgene can be reversibly switched off by application of doxycycline (Fig. 1A), thus allowing the conditional activation of the canonical NF-κB signaling pathway, the master regulator of the inflammatory response. In addition, transgene expression can be monitored by analysis of the coexpressed luciferase reporter gene (Fig. 1A). Unexpectedly, if the transgene is expressed during development, the animals suffer from postnatal growth retardation and develop a hydrocephalus (Fig. 1B). Histological sections of the brains at P16 revealed a massive enlargement of the lateral ventricles in double-transgenic GFAP.tTA × (tetO)7.IKK2-CA animals (Fig. 1C). To exclude that hydrocephalus formation is not an IKK2-CA transgene integration artifact, we confirmed this finding with a second independent GFAP.tTA × (tetO)7.IKK2-CA founder line combination [GFAP/IKK2-CA-(B)]. With this line, GFAP/IKK2-CA double-transgenic animals also displayed dilated ventricles, whereas both single-transgenic GFAP.tTA and (tetO)7.IKK2-CA animals have ventricle sizes in a normal range (Fig. 1C). Ventricular enlargement is most prominent in the caudal part of the lateral ventricles and is more pronounced in founder line A correlating with more severe defects of adjacent brain regions, most prominently the hippocampus. We detected a stronger transgene expression in this founder line compared with line B, suggesting a gene dose effect in phenotype development (Fig. 1D). As both founder lines show hydrocephalus formation, but transgene expression and the overall phenotype was more pronounced in line A, all further experiments were performed with this founder line.

Expression of constitutively active IKK2 in astrocytes results in hydrocephalus formation. A, Conditional expression of the IKK2-CA-transgene and a luciferase reporter gene controlled by a bidirectional tTA-dependent promoter [(tetO)7] is induced in GFAP-expressing cells by the GFAP.tTA transgene and can be repressed by doxycycline (Dox). B, A case of severe hydrocephalus (arrow: enlarged skull) and reduced growth at P16 and a control littermate [only (tetO)7.IKK2-CA transgenic]. C, Nissl staining shows massive enlargement of the lateral ventricles (LV) at P16 in IKK2-CA-expressing mice from two different (tetO)7.IKK2-CA founder lines (IKK2-CA A/B), but not in either GFAP.tTA or (tetO)7.IKK2-CA single-transgenic control animals. Scale bar, 1 mm. D, Ventricular enlargement is most pronounced in the caudal part of the lateral ventricles and is associated with a hippocampal malformation; line A shows a more severe hydrocephalus, which correlates with a stronger transgene expression; Nissl staining (scale bar, 1 mm) and immunoblot for human IKK2 (transgene) in medulla oblongata; Erk2 is detected as loading control. E, Expression of IKK2-CA is required for hydrocephalus formation: doxycycline blocks IKK2-CA expression and hydrocephalus formation; an inactive/dominant-negative allele (DN) of IKK2 cannot induce hydrocephalus despite higher expression levels; Nissl staining (scale bar, 1 mm) and immunoblot for human IKK2 (transgene) in medulla oblongata; Erk2 is detected as loading control.
To further confirm that active expression of the IKK2-CA transgene is required for the observed hydrocephalus formation, we applied doxycycline in the drinking water during pregnancy and weaning time. In this setup, the phenotype was completely rescued at P7 when compared with untreated IKK2-CA animals, and transgene expression was virtually absent in doxycycline-treated animals (Fig. 1E). Finally, we showed that this phenotype is not an artifact of protein overexpression, as the analog expression of a dominant-negative allele of IKK2 in another mouse model (GFAP/IKK2-DN) could not induce hydrocephalus formation, although a higher transgene expression level was achieved (Fig. 1E). Together, these findings prove that increased levels of active IKK2 are responsible for hydrocephalus formation.
We next tested whether transgene is indeed specifically expressed in astrocytes and does function as NF-κB inducer in these cells. Analysis of transgene expression by immunoblotting assays specific for human IKK2 confirmed (Fig. 2A) strong IKK2-CA expression in all brain regions (i.e., the forebrain, the midbrain/thalamus/hypothalamus region, the cerebellum, and the medulla oblongata), but IKK2-CA protein was not detected in kidney and spleen where the GFAP promoter is not active (Brenner et al., 1994). In immunofluorescence stainings, we found transgene expression at P2 mainly in the hippocampus and the subventricular zone (SVZ), which spreads over the whole brain at P7 (data not shown). At the cellular level, transgene staining colocalizes with a subpopulation of Aldh1l1-expressing cells (Fig. 2B). Aldh1l1 was found to be a novel marker for a wide range of astrocyte populations in a systematic screening approach (Cahoy et al., 2008). Interestingly, the transgene is also expressed in a subpopulation of cells of the postnatal ependymal layer and subventricular zone (Fig. 2C). To evaluate the functional consequences of IKK2-CA expression in transgenic astrocytes, we measured nuclear localization of RelA that is indicative for NF-κB activation by immunostaining. In GFAP/IKK2-CA mice, the total numbers of cells with nuclear staining of RelA are increased in brain sections. In particular, ∼80% of transgene-positive cells show nuclear localization of RelA (Fig. 2D). Finally, we investigated whether IKK2-CA can induce expression of known NF-κB target genes and whether this is mediated by IKK2 kinase activity as expected. For this purpose, we prepared primary astroglial cultures from transgenic and control pups born in the presence of doxycycline and reactivated the transgene ex vivo. Reactivation of the transgene expression system in response to doxycycline withdrawal was assessed by measurement of the coexpressed reporter gene luciferase (Fig. 2E). In three independent preparations derived from GFAP/IKK2-CA pups, we found a prominent upregulation of the NF-κB target genes CCL5 and Lcn2, which could be significantly reduced by treatment with the IKK2-specific kinase inhibitor BMS-345541 (Fig. 2E). These results indicate that the GFAP/IKK2-CA transgenic system induces NF-κB signaling in astrocytes via canonical IKK2-dependent NF-κB activation and pharmacological interference with IKK2 activity is able to revert the effects of IKK2-CA overexpression.

The GFAP/IKK2-CA transgene is specifically expressed in astrocytes and a subset of ependymal cells and induces NF-κB activation in astrocytes. A, IKK2-CA is expressed in all brain parts of GFAP/IKK2-CA animals (Tg+) at P7, but not in kidney and spleen, shown by immunoblot for human IKK2 (transgene); no promoter leakiness is observed in (tetO)7.IKK2-CA single-transgenic mice (Tg−); Erk2 is detected as loading control. Fo, Forebrain; Mi, midbrain/thalamus/hypothalamus; Ce, cerebellum; Me, medulla oblongata; Ki, kidney; Sp, spleen. B, Variable transgene expression in astrocytes (Aldh1l1 positive); hIKK2/Aldh1l1 double staining at P7; filled arrows show IKK2-CA-expressing astrocytes; open arrows shows IKK2-CA-negative astrocytes. C, The transgene is also expressed in a subpopulation of cells in the ependymal layer (arrows) and subventricular zone (P2); hIKK2 staining. D, IKK2-CA induces nuclear localization of RelA in most transgene-expressing cells (DAPI/hIKK2/RelA costaining, arrows); quantification shows the fraction of nuclei with a RelA staining intensity above a threshold value for all nuclei (DAPI positive) in control and transgenic animals and nuclei of transgene-positive cells; values show mean ± SD; statistical analysis was performed by t test (two-tailed, with Welch correction; n = 5–6 animals; **p < 0.01; ***p < 0.001). E, IKK2-CA induces expression of NF-κB the target genes CCL5 and Lcn2 in primary astrocytes from GFAP/IKK2-CA mice (measured by qPCR), which can be largely reduced by the pharmacological IKK2 inhibitor BMS-345541 (5 μm; 24 h); each n = 3 independent preparations of GFAP/IKK2-CA and control animals were left untreated and treated with BMS-345541; transgene reexpression in primary astrocyte cultures was monitored by luciferase activity before and after doxycycline withdrawal; statistical analysis: Co versus IKK2-CA unpaired two-tailed t test (#p < 0.05), in the absence or presence of BMS treatment paired one-tailed t test (*p < 0.05; **p < 0.01). Scale bars: B, D, 10 μm; C, 20 μm. All merged images shown with DAPI costaining.
Hydrocephalus formation depends on postnatal IKK2-CA expression
After validation of the transgenic system, we characterized the chronological order of events involved in hydrocephalus formation in more detail. Hydrocephalus formation and growth retardation both evolve early postnatally. While the ventricles look almost normal at P2 (Fig. 3A), at P7 the ventricles of GFAP/IKK2-CA animals are already prominently dilated compared with control animals (Fig. 1E). This corresponds to the onset of growth reduction, which also starts between P2 and P7. While the animals appear normal until P2, many show a variably reduced weight from P7 onward, with nearly no weight gain in severe cases (Fig. 3B). Finally, most animals die during the first postnatal weeks, with the exception of some low IKK2-CA-expressing animals exhibiting no or only a mild phenotype (data not shown).

Hydrocephalus depends on IKK2 activity and develops early postnatally. A, No prominent dilatation of the lateral ventricles in GFAP/IKK2-CA mice at P2; Nissl staining. B, Postnatal growth is impaired in GFAP/IKK2-CA animals, shown by body weight gain between P2 and P12; the diagram displays single values for each animal, showing the variability of growth impairment, and mean ± SEM; statistical analysis: Mann–Whitney test (**p < 0.01; ***p < 0.001). C, Expression of the IKK2-CA transgene becomes detectable by Western blot around birth in GFAP/IKK2-CA transgenic animals (Tg+); the transgene is detected with an IKK1/2 antibody, which also shows a weak band of endogenous IKK; Erk2 is used as loading control. D, No expression of the transgene is detectable by immunofluorescence at embryonic stage E18.5, shown for hippocampus and lateral ventricle/subventricular zone; hIKK2 staining; an image of a hippocampus/dentate gyrus section at P2 was taken with same exposition time as a positive control; DAPI costaining was performed for the visualization of nuclei. E, Repression of IKK2-CA expression by doxycycline application from P0 largely prevents hydrocephalus formation and hippocampal defects, shown by Nissl staining at P16; doxycycline was administered by a single injection to the mother and then continuously in the drinking water. Scale bars: A, 1 mm; D, 50 μm; E, 1 mm.
To investigate whether hydrocephalus formation is an immediate consequence of transgene expression or whether there are preceding early changes induced by IKK2-CA, we characterized the prenatal and early postnatal transgene expression. As the GFAP promoter is described to be active to some extent in other cell types than astrocytes, including neural stem cells (Sofroniew and Vinters, 2010), we also wanted to address the possibility that IKK2-CA expression in embryonic neural stem cells initiates the development of this severe phenotype, since stem cell defects often are associated with hydrocephalus (He et al., 2009; Talos et al., 2010). In immunoblot analysis, we did not detect transgene expression in the embryonic brain at E13.5 and E17.5, whereas from P2 onward a strong transgene expression was found (Fig. 3C). We confirmed the absence of prenatal transgene expression at the cellular level by immunostaining for human IKK2 at E18.5 (Fig. 3D).
Finally, we could demonstrate that repression of the transgene by administration of doxycycline from birth onward could largely prevent hydrocephalus formation, although there are still some moderate abnormalities of the hippocampus and a slight dilatation of the ventricles, indicating that there might be some prenatal transgene activity that we could not detect (Fig. 3E). Nevertheless, this analysis revealed that postnatal transgene expression is required for hydrocephalus formation, whereas a potential prenatal IKK2-CA-dependent NF-κB activation has only a minor contribution to the development of hydrocephalus. Since there is ongoing neurogenesis and major gliogenesis in the hippocampus and SVZ at P2, we cannot completely rule out the involvement of neural stem or progenitor cells in the development of the phenotype.
Astrocytic NF-κB activation causes a hippocampal malformation but does not affect the aqueduct of Sylvius and the subcommissural organ
To identify the mechanism of hydrocephalus formation in the GFAP/IKK2-CA model, we investigated possible anatomical alterations in the brains of these mice by Nissl staining at P7 (i.e., after onset of hydrocephalus). Hydrocephalus formation can originate from defects of the aqueduct of Sylvius, which is a narrow part of the ventricular system that is often obstructed or fused in hydrocephalus, thereby preventing the flow of the CSF (Rekate, 2008). We did not find any obstruction or obvious malformation of the aqueduct (Fig. 4A, top panels), thus classifying this phenotype as a communicating hydrocephalus. The subcommissural organ, whose malformation or loss is thought to be responsible for hydrocephalus formation in some mouse models (Huh et al., 2009), also did not show obvious defects (Fig. 4A, bottom panels). However, as mentioned before, we found a severely disturbed organization of the hippocampus, in particular the dentate gyrus (Fig. 4B). This defect, like hydrocephalus formation, is strongly suppressed by postnatal transgene repression (Fig. 3E). In the cerebellum, a mild delay in the regression of the cerebellar external granule layer was observed (Fig. 4C). This indicates that activation of NF-κB in GFAP-expressing cells impairs maturation of these late developing brain regions.

Hydrocephalus is not caused by obstruction of the aqueduct or agenesis of the subcommissural organ, but is associated with abnormal development of the hippocampus and the cerebellum (Nissl staining). A, No obvious anatomical explanation for hydrocephalus formation: the aqueduct of Sylvius is not obstructed or malformed at P7 (top panels), the subcommissural organ is present and appears morphologically normal at P7 (bottom panels, arrow). B, Malformation of the hippocampus with severe distortion of dentate gyrus granule layer (arrow). C, Impaired loss of the cerebellar external granule layer (arrow). Scale bars: A, C, 200 μm; B, 500 μm.
IKK activation induces a proinflammatory gene expression profile in astrocytes
After having excluded these well documented anatomical explanations for hydrocephalus formation, we generated a global gene expression profile of P7 forebrains by microarray analysis to address potential mechanisms involved in hydrocephalus formation in the GFAP/IKK2-CA mouse model. In this analysis, we found 399 transcripts that were differentially regulated (threshold, 1.5-fold; p < 0.05 in t test with Benjamini–Hochberg correction), of which 388 were upregulated (selected genes; see Table 1), and 11 downregulated (no more than twofold). The expression of several upregulated genes was validated by qPCR (data not shown).
Regulation of selected genes by GFAP/IKK2-CA in forebrain tissue at P7 and in primary astrocytes
To distinguish genes expressed in an astrocyte-specific manner from genes indirectly upregulated by pathological consequences of NF-κB activation in astrocytes, we also analyzed gene expression in primary astrocyte cultures by qPCR. To prevent secondary effects in the primary astrocyte culture due to a possibly varying ratio of contaminating cells, we prepared astrocytes from newborn animals that had received doxycycline during pregnancy and preparation. Transgene expression was activated by doxycycline withdrawal ex vivo 3 d before analysis. Among the most prominently upregulated genes in the microarray of P7 forebrain, we found several chemokines, most prominently Rantes/CCL5, IP-10/CXCL10, and MCP-1/CCL2 (Table 1). These genes are well known NF-κB target genes regulating immune cell infiltration. All three chemokines were also highly upregulated in primary astrocytes (Table 1), indicating a direct link to NF-κB activation in astrocytes. Other upregulated factors important for immune cell recruitment are cell adhesion molecules like ICAM-1 and Madcam-1. Of those, Madcam-1 is most highly upregulated in vivo (Table 1). In primary astrocytes, a direct overexpression of Madcam-1 and Icam-1 was confirmed (Table 1). Together, these findings indicate that the enhanced expression of most chemokines and cell adhesion molecules depends on astrocyte-specific NF-κB activation.
Two other inflammation/immune response-related systems are also highly upregulated, and at least partially directly expressed by astrocytes. Several factors of the complement system are elevated in the brains of GFAP/IKK2-CA animals, most prominently C3, but also C4b as well as components of the C1 complex (Table 1). Whereas C3 is highly upregulated in primary astrocytes, C4b and C1s mRNAs do not show any significant increase (Table 1). The second immune-related system is the antigen presentation machinery, of which various components of the MHC class II complex are upregulated. In this context, the invariant chain CD74 was directly induced in primary astrocytes (Table 1). In contrast to the previously mentioned factors, classical proinflammatory cytokines like TNFα, IL-1β, and IFNγ were not found upregulated in the microarray analysis. Although TNFα and IL-1β were found slightly elevated in the qPCR analysis of forebrain samples at P7 (6.0- and 2.4-fold), they are not induced in primary astrocytes (Table 1).
Apart from these classical inflammatory and immune response-related genes, another less well characterized gene, lipocalin 2, shows an outstanding expression. It is indeed the most highly upregulated gene in the microarray, and it is also massively induced in primary astrocytes on mRNA level (Table 1). Lcn2 was shown to be regulated by NF-κB (Li et al., 2009) and to mediate activation and apoptosis of both astrocytes and microglia in vitro (Lee et al., 2007, 2009).
IKK2-CA induces neuroinflammation
Because the gene expression analysis reveals a strong induction of proinflammatory factors involved in the recruitment of immune cells, we asked whether IKK2-CA expression induces infiltration of immune cells and whether these inflammatory alterations can explain hydrocephalus formation. Indeed, in mouse models as well as in human cases of hydrocephalus, the activation and/or infiltration of macrophages in the ventricles is observed (Wagner et al., 2003; Domínguez-Pinos et al., 2005).
Of note, we found a prominent accumulation of immune cells in the lateral ventricles of GFAP/IKK2-CA mice (Fig. 5A), consisting completely of CD45/CD11b double-positive (i.e., myeloid) cells that attach to the ventricle walls (Fig. 5B). Most of these CD11b-positive myeloid cells are also positive for F4/80 (Fig. 5C), confirming that these cells are macrophages. Remarkably, this activation/infiltration of macrophages precedes hydrocephalus formation, as it is already visible at P2. It is part of a global neuroinflammatory response characterized by prominent myeloid cell accumulation also in the meninges, the dentate gyrus and the cerebellum at P7 (Fig. 5D). These findings suggest that the activation/infiltration of macrophages in the ventricles is not a consequence of hydrocephalus formation, but rather has a causative role in this model. As inflammatory processes can initiate fibrosis, which can lead to hydrocephalus formation by impairing resorption of CSF in the subarachnoid space (Wyss-Coray et al., 1995), we searched for alterations in the deposition of extracellular matrix. In immunostainings, we did not observe any increased levels of fibronectin or laminin in the cortex and meninges (Fig. 5E), arguing against a fibrosis-related CSF resorption defect.

IKK2-CA induces global neuroinflammation with prominent accumulation of intraventricular macrophages. A, Massive accumulation of myeloid cells in the posterior part of the lateral ventricles at P2 (CD45/CD11b costaining). B, A magnification of the marked area in A shows complete colocalization of the general hematopoietic cell marker CD45 and the myeloid cell marker CD11b (i.e., only myeloid cells are found in the ventricles of the GFAP/IKK2-CA animals). C, The intraventricularly accumulating myeloid cells (CD11b+) are mainly macrophages, as shown by costaining with the macrophage marker F4/80 at P2. D, Immune cell infiltrates at P7 in dentate gyrus, cortex/meninges, and cerebellum consist of myeloid cells, shown by CD45/CD11b costaining. E, Deposition of fibronectin and laminin in the subarachnoid space/meninges is not increased. Scale bars: A, D, 100 μm; B, C, 20 μm; E, 50 μm. All merged fluorescent images except E show also DAPI costaining.
Enhanced IKK2 signaling impairs ependymal ciliogenesis
An important biological structure for CSF transport is the ependyma, a layer of epithelial-like cells that cover the ventricles. Ependymal cells possess bundles of motile cilia, which promote the flow of the CSF through the ventricular system. Defects of these cilia or a loss of the entire ciliated ependymal cells can cause hydrocephalus in mouse models and is also observed in human hydrocephalus (Del Bigio, 2010). To analyze the ultrastructure of the ependyma, in particular the ependymal cilia, we analyzed the walls of the lateral ventricles by scanning electron microscopy and immunofluorescence stainings. Strikingly, at P12, scanning electron microscopy (Fig. 6A, bottom panels) shows an almost complete absence of cilia at the lateral walls and the roof of the lateral ventricles, whereas the ventricle walls of control animals are completely covered with cilia bundles. In the transgenic animals, only in small areas single cilia bundles remain at the lateral walls; instead the ependyma is covered with large numbers of irregularly shaped cells, most likely the macrophages that were already observed by CD45/CD11b staining (Fig. 5). Interestingly, the lack of cilia is not found in the whole ventricular system, as in the choroid plexus and the medial wall of the lateral ventricles cilia are not affected (data not shown). This emphasizes findings, that the ependyma is not a homogenous cell layer, but shows regional cellular diversity (Jiménez et al., 2001; Wagner et al., 2003).

IKK2-CA impairs ependymal cilia formation before onset of ventricular dilatation but does not a cause loss of ependymal cells. A, Scanning electron microscopy of the surface of the lateral walls of the lateral ventricles reveals lack of ependymal cilia in GFAP/IKK2-CA animals. The ependyma of control animals is covered with immature cilia bundles at P2, which are mature at P12 (left panels), while the ependyma of transgenic animals is largely devoid of cilia and covered with irregularly shaped cells that most likely resemble macrophages (middle and right panels); only small areas show a few cilia bundles (right panels). B, Lack of cilia is not due to loss of ependyma integrity as shown by β-catenin staining. C, Sialic acid-rich glycocalix of the ependyma appears normal before and after onset of hydrocephalus (P2, P7), shown by WGA staining. D, No increase of apoptosis is found in the ependyma/subventricular zone before and after onset of hydrocephalus (P2, P7), shown by staining for cleaved caspase 3 (P7 images not shown); quantification does not show a significant difference in the number of cleaved caspase 3-positive cells per area; statistical analysis: unpaired t test (n = 3–6); values are shown as mean ± SEM. Scale bars: A, 10 μm; B, C, 20 μm; D, 200 μm. All merged fluorescent images show also DAPI costaining.
The lack of ependymal cilia can be seen already at P2 (i.e., before onset of hydrocephalus formation) when in control animals the ependyma is covered with immature cilia (Fig. 6A, top panels). This strongly suggests that the lack of ependymal cilia at the lateral wall of the lateral ventricles is responsible for hydrocephalus formation.
To investigate whether only the cilia are missing, or whether the whole ependymal cell layer detaches or does not properly differentiate, we performed a staining for β-catenin (Fig. 6B) previously used to depict loss or impaired polarity of ependymal cells (Chae et al., 2004; Ma et al., 2007). We did not find gross alterations in the β-catenin staining pattern, indicating a proper organization of the ependymal cell layer. In contrast to the hyh model (Wagner et al., 2003), we did not find a disruption of the sialic acid rich glycocalix of the ependyma, assessed by WGA staining (Fig. 6C). Moreover, we did not detect any elevated apoptosis in the subventricular/ependymal zone analyzed by cleaved caspase 3 stainings (Fig. 6D).
NF-κB activation in astrocytes causes hydrocephalus only in the developing brain
The lack of ependymal cilia without other observed alterations of the ependyma raises the question, whether this is a specific defect of ependymal cilia development or whether the transgene causes a loss of previously developed cilia. The latter issue would suggest that, in an adult animal, NF-κB activation in GFAP-expressing cells could also induce loss of ependymal cilia and hydrocephalus formation. In our microarray screen, genes that are known to cause hydrocephalus by impairing ependymal ciliogenesis, like Foxj1, Celsr2, and Myo9a, were not deregulated (Table 1) (see also Discussion).
Thus, to confirm that hydrocephalus formation in the GFAP/IKK2-CA model is indeed a developmental defect, we took advantage of our conditional model system and repressed the IKK2-CA transgene by application of doxycycline and thereby shut off constitutive NF-κB activation during brain development. Doxycycline was applied to the drinking water of pregnant mice and to their offspring up to the age of 4 weeks. Thereafter, doxycycline was withdrawn to activate transgene expression. Animals treated in this schedule survived and showed no obvious abnormalities. Mice analyzed at the age of 8 weeks (i.e., 4 weeks after transgene activation) show no prominent dilation of the lateral ventricles (Fig. 7A). We also found no evidence for hydrocephalus formation in older animals (data not shown). To confirm transgene expression in this reactivation approach, we measured luciferase activity to detect reporter gene expression (data not shown) and performed an immunoblot analysis for IKK1/2. Transgene expression was comparable with the expression in young mice that never received doxycycline (Fig. 7B) and more importantly also caused activation and infiltration of immune cells in the brain (Fig. 7C). However, in contrast to the young animals with permanently active transgene, there is no intraventricular accumulation of ventricular macrophages (Fig. 7C) and the ependymal cilia of such animals showed no alterations after 6 weeks of transgene activation (Fig. 7D), further supporting the hypothesis that the lack of cilia is caused by impaired ciliogenesis and is responsible for the hydrocephalus formation in young animals. However, there is still the possibility that ependymal cilia are formed but are destroyed by infiltrating immune cells in an especially vulnerable period of early postnatal development. For this reason, we performed another approach in which IKK2-CA expression was activated already at P7 (i.e., early after completion of the period of ciliogenesis) and studied the consequences at P28. Also in these conditions, we did not find evidence for a dilatation of the ventricles, even in severely diseased animals with high transgene expression and massive neuroinflammation (Fig. 7E). These data further strengthen the hypothesis that indeed only the developmental period of ependymal ciliogenesis is the critical time frame for hydrocephalus formation.

Astrocyte-specific IKK2-CA expression is sufficient to induce neuroinflammation without hydrocephalus formation after completion of ependymal ciliogenesis. Transgene expression was suppressed by application of doxycycline in the drinking water during pregnancy and until the age of 4 weeks (A–D) or 1 week (E, F). A, Animals do not show prominent dilatation of the lateral ventricles at the age of 8 weeks (Nissl staining). Scale bar, 1 mm. B, Transgene expression in forebrain tissue at 8 weeks of age is similar to expression in young animals (P12) without doxycycline treatment; Western blot for IKK1/2 and Erk2 (loading control). C, Transgene expression induces neuroinflammation (CD45/CD11b costaining at the age of 8 weeks), most prominently indicated by infiltration of immune cells (CD45+) in the thalamus (th); increased CD11b staining in CD45-low cells shows activation of microglia; no accumulation of macrophages (CD11b+) is seen in the lateral ventricle (lv); merged images include DAPI costaining. Scale bar, 100 μm. D, Ependymal cilia are normally developed at the age of 10 weeks, shown by scanning electron microscopy. Scale bar, 10 μm. E, Early transgene reexpression from P7 induces neuroinflammation, but no ventricle dilatation; CD45/CD11b/DAPI costaining shows strong myeloid cell activation in thalamus and hippocampus at 4 weeks of age, but not in the lateral ventricles (lv), as seen in animals with constitutive transgene expression at P2. Scale bar, 200 μm. F, Transgene expression at 4 weeks of age (P28) after doxycycline withdrawal at P7 is comparable with transgene expression in young animals without doxycycline treatment; immunoblot for human IKK2 for transgene expression and Erk2 as loading control.
Discussion
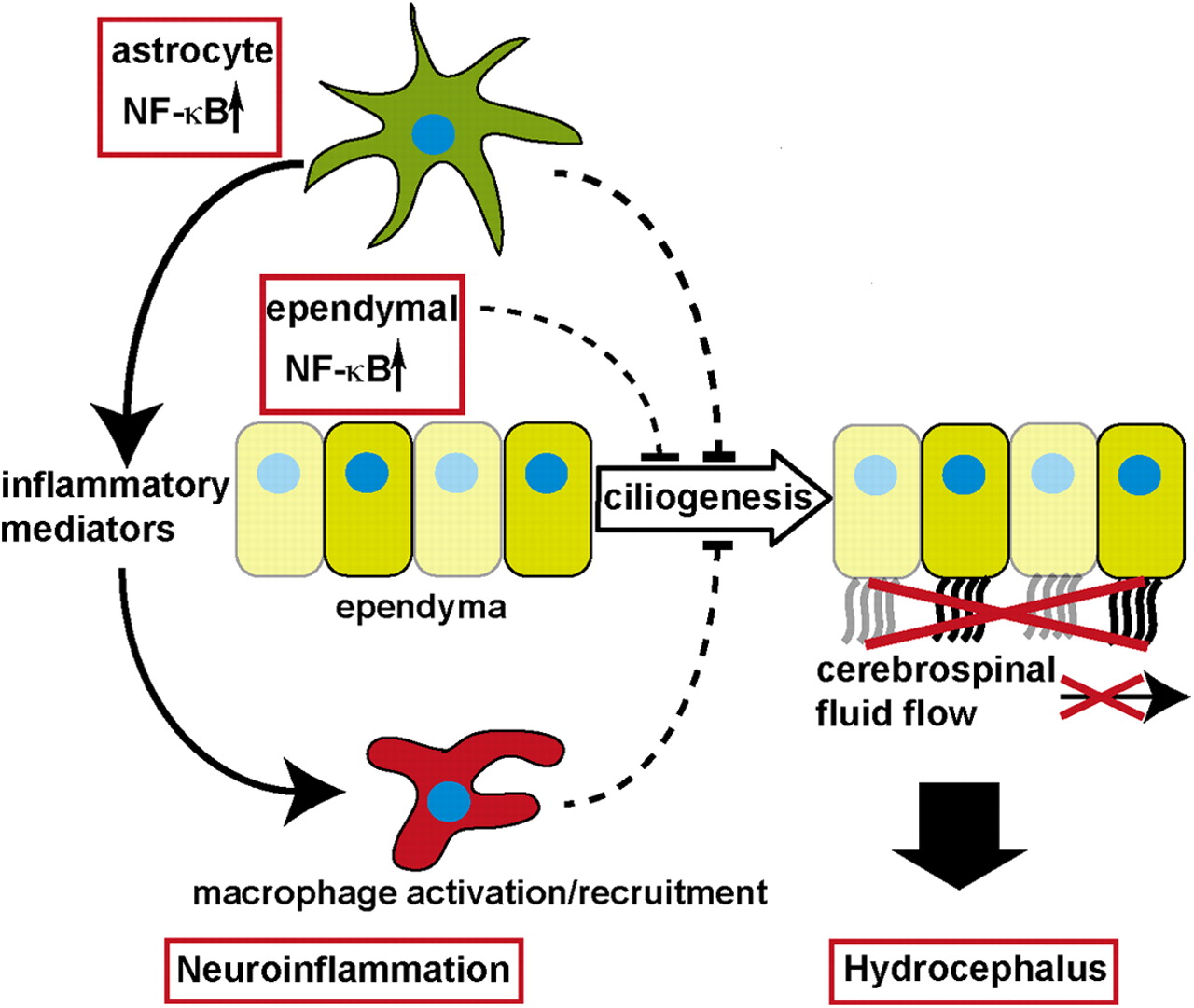
We show for the first time a direct link between NF-κB signaling and hydrocephalus development and reveal a novel role of NF-κB signaling in blocking ependymal cilia formation (Fig. 8). Our results give interesting insights into neuroinflammatory pathologies and their consequences for brain development, in particular hydrocephalus formation. Furthermore, we could show that NF-κB activation in astrocytes is sufficient to induce neuroinflammation, demonstrating that astrocytes are not only passive modulators, but rather active players in the innate immune response of the brain and are able to set the brain into an inflammatory alarm state.

Model of GFAP/IKK2-CA-induced hydrocephalus formation: NF-κB activation in astrocytes induces expression of proinflammatory mediators leading to macrophage recruitment; these neuroinflammatory conditions inhibit ependymal ciliogenesis, which impairs CSF transport through the ventricular system, resulting finally in hydrocephalus formation.
Hydrocephalus, whose etiology is only partially understood, is a frequent complication of brain injuries and CNS infections. Remarkably, both pathological insults share a strong neuroinflammatory component. In such inflammatory conditions, the IKK/NF-κB system is activated (e.g., via TNFα, IL-1, and TLR signaling) (Hayden and Ghosh, 2008). Interestingly, a recent study shows nuclear translocation of RelA in ependymal cells in a mouse model of intraventricular hemorrhages under conditions that induce hydrocephalus (Simard et al., 2011).
In our model, we show that NF-κB activation in astrocytes and subsets of ependymal cells results in a lack of ependymal cilia and hydrocephalus formation in a distinct developmental window, in which ependymal ciliogenesis takes place. The time of onset of hydrocephalus is in line with observations in mice deficient in Celsr2 or Celsr2/3, which also have defective ciliogenesis resulting in early postnatal hydrocephalus (Tissir et al., 2010).
Whether this mechanism plays a role in human hydrocephalus remains to be investigated. While obstructions of narrow parts of the ventricular system are well known causes of noncommunicating hydrocephalus (Rekate, 2008), the molecular mechanisms underlying communicating hydrocephalus like in our GFAP/IKK2-CA model are less well understood. In posthemorrhagic hydrocephalus, fibrosis of the subarachnoid space and inflammation seem to play a critical role (Poca and Sahuquillo, 2005).
A loss of ependymal integrity is observed frequently in human developmental hydrocephalus (Domínguez-Pinos et al., 2005; Sival et al., 2011). This process is usually accompanied by activation of ventricular macrophages (Ulfig et al., 2004; Domínguez-Pinos et al., 2005), an issue that is phenocopied in our model system. Several mouse models provide evidence that ependymal defects, in particular impaired cilia function, are involved in hydrocephalus formation (Huh et al., 2009; Del Bigio, 2010). However, the relevance of these models for human hydrocephalus is still under debate, as there are cases known showing on the one hand a partial loss of ependymal integrity and ciliopathies but on the other hand are rarely associated with hydrocephalus (Huh et al., 2009; Del Bigio, 2010). So far, it is unclear whether defects in ependymal ciliogenesis are involved in the development of human hydrocephalus that is associated with elevated IKK/NF-κB signaling in course of infections or intracerebral hemorrhages. Interestingly, intracerebral mumps virus infection causes prominent hydrocephalus in newborn Syrian hamsters with ultrastructural alterations similar to the GFAP/IKK2-CA model, whereas at later developmental stages hydrocephalus formation in this model is much less pronounced (Takano et al., 1993; Uno et al., 1997). This supports the idea that an increased susceptibility to inflammation or infection-induced hydrocephalus during development might be due to impaired ciliogenesis as consequence of elevated IKK/NF-κB signaling in ependymal cells. So far, the interpretation of our data do not allow a definite discrimination between an ependymal cell-intrinsic ciliogenesis defect or a scenario in which activated ventricular macrophages disrupt nascent cilia formation between P2 and P7 or the combination of both mechanisms as the cause of hydrocephalus formation. However, our findings clearly define an early postnatal time frame (P2–P7) of high susceptibility to hydrocephalus formation.
Our microarray analysis did not show deregulated genes with known functions in ciliogenesis and hydrocephalus formation, but there is indirect evidence that supports the idea of pathological NF-κB signaling in hydrocephalus formation. As mentioned above, intraventricular hemorrhages cause activation of NF-κB in the ependyma. Inactivation of MyD88, an upstream regulator of the IKK-complex in IL-1 and TLR signaling (Hayden and Ghosh, 2008), reduces intracranial pressure despite an increased bacterial load in a mouse model of pneumococcal meningitis (Koedel et al., 2004). Thus, repressed NF-κB activation might counteract hydrocephalus formation, a frequent complication in this disease (Jit, 2010).
Interestingly, inactivation of Foxj1 prevents ependymal ciliogenesis and causes hydrocephalus (Jacquet et al., 2009). At least in lymphoid cells, Foxj1 inhibits NF-κB activation by induction of IκB proteins (Lin et al., 2004, 2005); thus, its inactivation might activate NF-κB like the GFAP/IKK2-CA model. Ciliogenesis via Foxj1 needs tight control of RhoA activation (Pan et al., 2007). Abouhamed et al. (2009) revealed that loss of the myosin family protein Myo9a impairs ependymal differentiation and induces hydrocephalus, which is mediated by the Rho effector kinase ROCK. In an epithelial cell line, the authors demonstrate that repression of Myo9a induces NF-κB activity and changes the epithelial phenotype, which both is reverted by ROCK inhibition (Abouhamed et al., 2009). Likewise, the Rho/ROCK pathway might also be affected in our GFAP/IKK2-CA model. The most prominently upregulated gene in our microarray analysis, lipocalin 2, can activate the Rho/ROCK pathway (Lee et al., 2009). Furthermore, chemokines, like the highly expressed CCL2, CCL5, and CXCL10, can act on the cytoskeleton via Rho family proteins (Thelen and Stein, 2008). Thus, these factors might disturb the Rho-mediated cytoskeletal rearrangements required for cilia development. Independent of that, Lcn2 was recently shown to be critically involved in the regulation of neural cell migration and morphology (Lee et al., 2011, 2012). Whether increased Lcn2 levels also affect the morphology of ependymal cells and thereby impair ciliogenesis remains open.
While these pathways might explain a potential ependymal cell-intrinsic ciliogenesis defect, the upregulation of complement components, most prominently C3, might implicate macrophage-mediated mechanisms in the disruption of cilia formation. Indeed, C3 is also indirectly linked to hydrocephalus formation as its ablation results in a similar outcome in pneumococcal meningitis like the inactivation of MyD88 mentioned above (i.e., decreased intracranial pressure despite an increased bacterial load) (Rupprecht et al., 2007).
Beyond the new link of NF-κB signaling and hydrocephalus formation, our findings strengthen the emerging view that, beside microglia, astrocytes are active key players in neuroinflammation. It is well established that astrocytes can amplify neuroinflammation in pathological situations by the permeabilization of the blood–brain barrier and the production of chemokines and cytokines (Glass et al., 2010; Wilson et al., 2010). Inactivation of NF-κB signaling reduces this proinflammatory function of astrocytes (Brambilla et al., 2005, 2009; Dvoriantchikova et al., 2009).
Here, we demonstrate that NF-κB activation in astrocytes is itself sufficient to induce an inflammatory response of the brain. With the recruitment of macrophages by the induction of chemokines and cell adhesion molecules, the production of complement factors and the upregulation of MHC proteins, astrocytes provide defense tools for a rapid reaction to pathological insults. The final activation of these defense systems seems to require additional signals, as the major proinflammatory cytokines TNFα and IL-1β were not upregulated in astrocytes and only moderately increased in brain tissue.
Our findings also emphasize the interference of inflammation with CNS development, which is well documented for prenatal and postnatal neuroinflammatory insults in humans, like intrauterine cytomegalovirus infections or perinatal stroke (Tsutsui et al., 2005; Stolp and Dziegielewska, 2009; Vexler and Yenari, 2009). The neuroinflammatory state induced by GFAP/IKK2-CA causes additional distortions of CNS development beyond impaired ciliogenesis. These affect late developing brain regions that depend on postnatal neurogenesis and progenitor migration, in particular the dentate gyrus and, to a lesser extent, the cerebellum. This is most likely caused by deregulation of chemokines, which are important guidance cues for neural progenitors. In this context, the CXCL12/CXCR4 axis was intensively studied. Deficiency in either ligand or receptor causes a severe distortion of the dentate gyrus and alterations in the cerebellar maturation similar to our model (Li and Ransohoff, 2008). This system seems unaffected in the GFAP/IKK2-CA model, but likely neural progenitor cells can also respond to other chemokines, as they express several chemokine receptors. These include CCR2, CCR5, and CXCR3, the receptors for CCL2, CCL5, CXCL10 (Tran et al., 2007), which are prominently upregulated in our model. As chemokine-dependent migration is controlled by chemokine gradients, a homogenous high chemokine level could interfere with migration in the same manner as its absence. For CCL2, there is evidence that it can regulate migration and differentiation of neural progenitor cells in the SVZ (Liu et al., 2007), which might affect the immature ependyma and might thus be involved in hydrocephalus formation.
In summary, the present study gives new insights in the mechanisms by which neuroinflammation can impair brain development. This is still a major problem in pediatrics, when dealing with brain infections or injury in young children, which often lead to lasting disabilities or death. By showing the critical interference of astrocytic NF-κB activation with brain development, our study opens up new perspectives for potential therapeutic strategies (e.g., the use of specific chemokine inhibitors) to avoid developmental complications in the treatment brain infections and injury, especially with regard to hydrocephalus formation.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grant DFG KFO167-P5 (B.B.). We thank T. Böckers and S. Liebau for helpful discussions, and U. Leschik, B. Ries, and Melanie Gerstlauer for excellent technical assistance.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Bernd Baumann, Institute of Physiological Chemistry, Ulm University, Albert Einstein Allee 11, 89081 Ulm, Germany. bernd.baumann{at}uni-ulm.de





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}