

Abstract
The formation of neuronal circuits is a key process of development, laying foundations for behavior. The cellular mechanisms regulating circuit development are not fully understood. Here, we reveal Psidin as an intracellular regulator of Drosophila olfactory system formation. We show that Psidin is required in several classes of olfactory receptor neurons (ORNs) for survival and subsequently for axon guidance. During axon guidance, Psidin functions as an actin regulator and antagonist of Tropomyosin. Accordingly, Psidin-deficient primary neurons in culture display growth cones with significantly smaller lamellipodia. This lamellipodial phenotype, as well as the mistargeting defects in vivo, is suppressed by parallel removal of Tropomyosin. In contrast, Psidin functions as the noncatalytic subunit of the N-acetyltransferase complex B (NatB) to maintain the number of ORNs. Psidin physically binds the catalytic NatB subunit CG14222 (dNAA20) and functionally interacts with it in vivo. We define the dNAA20 interaction domain within Psidin and identify a conserved serine as a candidate for phosphorylation-mediated regulation of NatB complex formation. A phosphomimetic mutation of this serine showed severely reduced binding to dNAA20 in vitro. In vivo, it fully rescued the targeting defect but not the reduction in neuron numbers. In addition, we show that a different amino acid point mutation shows exactly the opposite effect by rescuing only the cell number but not the axon targeting defect. Together, our data suggest that Psidin plays two independent developmental roles via the acquisition of separate signaling pathways, both of which contribute to the formation of olfactory circuits.
Introduction
The development of functional nervous systems relies on the formation of appropriate synaptic connections. The specification of the right number and type of different neurons and the guidance of their axons to their target cells are essential in this process. During targeting, growth cones at axon tips provide a dynamic platform for integration of attractive and repulsive guidance cues (Lowery and Van Vactor, 2009). Through binding to membrane receptors displayed on growth cones, guidance cues orchestrate dynamic changes of the cytoskeleton, in particular of the actin and microtubule networks (Driessens et al., 2001; Gordon-Weeks, 2004; Kalil and Dent, 2005; Evans et al., 2007; Lowery and Van Vactor, 2009). The growth cone consists of actin-rich structures, finger-like filopodia, and veil-like lamellipodia. Dynamic changes of the actin networks are mediated by actin-binding proteins, such as disassembly factors (e.g., cofilin) and stabilizers (e.g., Tropomyosin, Fascin) (Pak et al., 2008). Their activity is regulated through common effectors of signaling events involved in pathfinding (Huber et al., 2003; Ng and Luo, 2004; Hall and Lalli, 2010). However, how these molecular pathways operate and are integrated at the growth cone level to implement predictable morphogenetic changes is not fully understood.
To gain this understanding, the olfactory system of Drosophila provides an efficient experimental platform to study different aspects of neuronal specification, axon guidance, and synapse formation (Rodrigues and Hummel, 2008; Brochtrup and Hummel, 2011). Two peripheral olfactory organs, the third segment of the antenna and the maxillary palp (MP), contain olfactory receptor neurons (ORNs) located mostly in groups of two to four neurons inside chemosensory sensilla. ORNs develop from the eye-antennal disc in a series of asymmetric divisions and regulated cell death to reach their final sensilla type-dependent composition of different ORN classes plus several kinds of supporting cells (Rodrigues and Hummel, 2008). All ORNs project through the antennal or labial nerves into the antennal lobe (AL) in the brain, where they form stereotypic synaptic contacts. Each ORN expresses the general olfactory receptor named ORCO (Or83b) together with a specific odorant-binding receptor (OR) out of a set of ∼60 genes (Rodrigues and Hummel, 2008; Vosshall and Hansson, 2011). ORNs expressing the same OR innervate the same glomerulus, a structural AL sub-compartment, where they form synaptic contacts with local interneurons and projection neurons (PN) at stereotypic positions. This innervation pattern relies on a complex hierarchical guidance process using ORN axon–axon interactions and axon–target interactions (ORN-PN) (Brochtrup and Hummel, 2011).
We identified the actin-binding protein Psidin (Brennan et al., 2007; Kim et al., 2011) as a novel regulator of olfactory system development. We provide in vitro and in vivo evidence that Psidin is required during two independent steps of circuit development: as a component of the N-acetyltransferase complex B (NatB) complex to maintain neuron number and as an actin-binding protein during axonal targeting. During targeting, we suggest that Psidin influences growth cone responsiveness to guidance cues by regulating actin dynamics. In the olfactory system, this translates into a class-specific Psidin requirement correlating with the specific routes and distances the ORN growth cone travels.
Materials and Methods
Fly genetics
Fly stocks were raised at 25°C on standard cornmeal medium. The ethyl methanesulfonate (EMS) screen was performed as described previously (Cayirlioglu et al., 2008). ORN analysis was performed using the MARCM (mosaic analysis with a repressible cell marker) technique with flies of the following genotype: Or-GAL4 UAS-syt-GFP (or UAS-mCD8-GFP)/+; FRT82 mutation/FRT82 gal80 (E2F). ORNs were labeled using GAL4 or direct fusion of the respective OR promoter elements. All analyses were done in eyFlp mosaic animals. The following alleles of Psidin were used: psidinIG978 (this study), psidin1 (Brennan et al., 2007), and psidin55D4 (Kim et al., 2011). All psidin mutations were lethal and noncomplementing. Act-GAL4 was used to drive the expression of various UAS elements in eyFlp clones. UAS-Psidin-HA was generated and generously provided by D. Montell (Johns Hopkins School of Medicine, Baltimore, MD). Transgenic flies carrying RNAi against dNAA20 were ordered from the Kyoto and Vienna Drosophila RNAi Center (VDRC) stock centers. Flies carrying overexpression constructs of LimK and Cofilin were obtained from the Bloomington Stock Center. Three different RNAi lines [VDRC (lines 1 and 2), NIG-Kyoto (line 2)] were used to knock down dNAA20. We saw similar results for all three lines.
Detailed genotypes.
For all genotypes, X stands for FRT82B, FRT82B psidinIG978, FRT82B psidin55D4, or FRT82B psidin1.
MARCM genotypes.
eyflp; Or-GAL4 UAS-syt-GFP (or UAS-mCD8-GFP)/+; FRT82,CL,gal80/X.
Reverse MARCM genotypes.
eyflp; Or-GAL4 UAS-syt-GFP (or UAS-mCD8-GFP)/+; FRT82/X gal80.
Rescue experiment.
eyflp; Or59c-mCD8-GFP,act-gal4/+; FRT82,CL, gal80/X, eyflp; Or59c-mCD8-GFP,act-gal4/UAS-Psidin-HA; FRT82,CL,gal80/X, eyflp; Or59c-mCD8-GFP,act-gal4/UAS-p35; FRT82,CL,gal80/X, eyflp; Or59c-mCD8-GFP,act-gal4/UAS-PsidinIG978; FRT82,CL,gal80/X, eyflp; Or59c-mCD8-GFP,act-gal4/UAS-PsidinS678A; FRT82,CL,gal80/X, eyflp; Or59c-mCD8-GFP,act-gal4/UAS-PsidinS678D; FRT82,CL,gal80/X.
Tropomyosin experiment.
eyflp; Or59c-mCD8-GFP/+; FRT82,CL,gal80/FRT82 psidin1, eyflp; Or59c-mCD8-GFP/+; FRT82,CL,gal80/FRT82 psidin55D4, eyflp; Or59c-mCD8-GFP/+; FRT82,CL,gal80/FRT82 tm1ZCL0722, eyflp; Or59c-mCD8-GFP/+; FRT82,CL,gal80/FRT82 tm1ZCL0722 psidin55D4.
RNAi experiments.
eyflp; Or59c-mCD8-GFP,act-gal4/UAS-RNAi; FRT82,CL,gal80/X, eyflp; Or42a-mCD8-GFP,act-gal4/UAS-RNAi; FRT82,CL, gal80/X, eyflp; Or59c-mCD8-GFP,act-gal4/+; FRT82,CL,gal80/X, eyflp; Or42a-mCD8-GFP,act-gal4/+; FRT82,CL,gal80/X.
Overexpression experiments.
eyflp; Or59c-mCD8-GFP,act-gal4/UAS-Limk(wt); FRT82,CL,gal80/X, eyflp; Or59c-mCD8-GFP,act-gal4/UAS-Limk(KI); FRT82,CL,gal80/X, eyflp; Or59c-mCD8-GFP,act-gal4/UAS-Limk(RNAi); FRT82,CL,gal80/X, eyflp/UAS-TsrS3A; Or59a-mCD8-GFP,act-gal4/+; FRT82,CL,gal80/X, eyflp/UAS-TsrS3E; Or59a-mCD8-GFP,act-gal4/+; FRT82,CL,gal80/X, eyflp; Or59c-mCD8-GFP,act-gal4/+; FRT82,CL,gal80/X.
Histology
Adult fly brains were dissected to analyze the ORN targeting pattern as described previously (Hartl et al., 2011). For each experiment, a minimum of 20 brains from male and female flies were analyzed per condition (i.e., n = 20–36). Pupal stages were determined by collecting white pupae and incubating them at 25°C until they reached the correct age. The cell numbers of ORNs were determined in adult antennae (ANTs) and MPs. Confocal pictures were taken using the Olympus FV1000 and Leica SP2. Images were processed in ImageJ and Adobe Photoshop.
In situ hybridization
A 671 bp fragment (antisense probe) and a 682 bp fragment (sense probe) of psidin cDNA (GenBank accession number LD30731) was cloned into the pCRIITOPO vector. Hybridization was performed at 55°C using standard methods (Green and Sambrook, 2012). Probes were detected using anti-DIG antibody coupled to alkaline phosphatase (Roche). Staining was developed with NBT/BCIP substrate. Pictures were taken using the Leica MZ500.
Quantification of targeting phenotype
We used three different categories to describe the phenotype of Or59c neurons. Normal wild-type targeting displays a strongly innervated Or59c glomerulus and no additional innervation on the dorsal area of the AL. Mild mistargeting shows a weaker innervation of the glomerulus and additional mistargeting and axon dispersal across the AL. Strong mistargeting was scored when there was no concentrated innervation in the Or59c target glomerulus, but rather random innervations across the entire AL. Statistical analysis was done using one-way ANOVA and Bonferroni's posttest (*p < 0.05; **p < 0.01; ***p < 0.001). Error bars depict the SEM.
Coimmunoprecipitation experiments
S2 cells were transfected using Effectene (QIAGEN). Ubiquitin-GAL4 (ub-GAL4) was used to drive the expression of UAS-Psidin-HA, UAS-PsidinIG978-HA, UAS-PsidinS678X-HA, UAS-PsidinΔNatBX-HA, and UAS-dNAA20-myc. All constructs were N-terminally tagged with HA or myc. Cells were harvested after 3 d and lysed in lysis buffer [50 mm Tris, pH 7.4, 150 mm NaCl, 2 mm EDTA, 1% Triton X-100, and protease inhibitor (Sigma-Aldrich)]. Lysate was cleared, and supernatant was used for coimmunoprecipitation. Proteins were incubated with rat anti-HA (1:1000; Sigma-Aldrich) and Sepharose beads [50% (v/v)] for 2 h at 4°C. Afterward, samples were boiled in SDS loading buffer and analyzed using Western blot. dNAA20 cDNA from clone LD30731 was subcloned into a pUAST vector and fused to a myc tag. Five repetitions were performed for each condition.
Western blot quantification
Western blots were quantified using ImageJ. The ratio of dNAA20 to PsidinX was quantified measuring the intensity of the respective bands. We compared the amount of dNAA20 pulled down with wild-type Psidin versus the amount of dNAA20 pulled down with a Psidin variant. We measured the respective bands of HA-captured Psidin and pulled-down dNAA20. Only the HA-bound fraction of Psidin can effectively pull down dNAA20. The same was done for wild-type Psidin and dNAA20, which was used as a reference on each blot. We used the wild-type control to normalize the pulldown of dNAA20 with a Psidin variant on each individual blot.
RNAi knock down in S2 cell culture
S2 cells were transfected using Effectene (QIAGEN). Ub-GAL4 was used to drive the expression of the RNAi against dNAA20 and UAS-dNAA20-myc. Cells were harvested after 3 d and lysed in lysis buffer [50 mm Tris, pH 7.4, 150 mm NaCl, 2 mm EDTA, 1% Triton X-100, and protease inhibitor (Sigma-Aldrich)]. Protein samples were loaded on SDS gels and detected with anti-tubulin and anti-myc antibodies. Western blots were quantified by measuring the intensity of the tubulin loading control and the amount of Psidin in each lane using ImageJ software. Experimental bands (anti-myc band) of one lane were normalized to their respective tubulin control of the same lane. A single transfected lane (UAS-dNAA20-myc only) was used as the control for RNAi efficiency. Each condition was repeated four times.
Generation of Psidin isoforms
PsidinIG978, PsidinS678A, and PsidinS678D were generated using the QuikChange Lightning Site-Directed Mutagenesis kit (Agilent Technologies). Wild-type UAS-Psidin-HA (provided by D. Montell) was used as a template. PsidinΔNatBX constructs were generated using PCR-mediated generation of deletions.
Drosophila primary neuron cultures
Drosophila primary neuron cultures were generated as described previously (Sánchez-Soriano et al., 2010). In brief, cells were removed with micromanipulator-attached capillaries from stage 11 embryos (6–7 h after egg laying at 25°C) (Campos-Ortega and Hartenstein, 1997), treated for 5 min at 37°C with dispersion medium, washed, and dissolved in 30–40 ml of Schneider medium (Schneider, 1964). Then, the aliquots were transferred to coverslips and kept as hanging drop cultures in air-tight, special culture chambers (Dübendorfer and Eichenberger-Glinz, 1980) usually for 6 h at 26°C. Cultured Drosophila neurons were analyzed 6 h after plating. They were fixed (30 min in 4% paraformaldehyde/0.05 m phosphate buffer, pH 7.2) and washed in PBS and 0.1% Triton X-100 (PBT). Incubation with antibodies was performed in PBT. Microtubules were stained with anti-tubulin (1:1000; Sigma-Aldrich) and FITC- or Cy3-conjugated secondary antibodies (1:200; Jackson ImmunoResearch). Filamentous actin was detected with TRITC-conjugated phalloidin (Sigma-Aldrich). Stained Drosophila neurons were mounted in Vectashield (Vector Laboratories). Primary neuron culture images were taken using an AxioCam camera mounted on an BX50WI microscope (Olympus). Lamellipodia area was quantified using ImageJ. Statistical analyses were performed with SigmaStat software using a t test or Mann–Whitney rank sum test.
Antibodies
The following primary antibodies were used: rabbit anti-GFP (1:1000; Clontech), mouse anti-NC82 (1:20; DSHB), rat anti-HA (1:1000; Roche), and rabbit anti-myc (1:1000; Abcam). Secondary antibodies were used as follows: anti-mouse Cy5 (1:200; Dianova), anti-rabbit 488 (1:200; Dianova), anti-rat HRP (1:1000; Jackson ImmunoResearch), anti-rabbit HRP (1:1000; Jackson ImmunoResearch), anti-Psidin (1:50; Kim et al., 2011), anti-Elav (1:50; Hybridoma Bank), rat anti-tubulin (1:1000; Abcam).
Yeast experiments
Saccharomyces cerevisiae strain YZ1143 (mdm20 deletion was kindly provided by Dr. F. S. Sherman, University of Rochester Medical Center, Rochester, NY) carrying a complementing plasmid, p[CEN URA3 MDM20–3HA], was used for the analysis. Integrative vector pRS405 (LEU2) carrying either a wild-type MDM20-myc or a mutant allele MDM20K304E-myc was digested by XcmI to facilitate integration into the yeast genome. Selected transformants were grown on 5-Fluoroorotic acid plates lacking leucine to remove the complementing plasmid. The presence of integrated allele was confirmed by PCR and subsequent sequencing. Temperature sensitivity was determined at 30°C and 37°C. For actin staining, exponentially growing cells were fixed with formaldehyde for 10 min, washed three times in PBS, and resuspended in Alexa-fluor-phalloidin (1:1000). After a 30 min incubation at room temperature, the cells were washed with PBS, mounted on slides in 70% glycerol/PBS/0.05% paraphenylenediamine, and immediately visualized.
Results
A mutation in the psidin gene affects olfactory neuron circuit formation
Using mosaic histological screening of ∼6000 EMS mutants, we addressed the molecular mechanisms required for ORN axons to navigate to their correct targets within the AL. We used the eyFlp mosaic system, which induces mutant clones in the antenna and MP in addition to in the eye, but not in the central brain (Newsome et al., 2000). These clones usually resulted in ∼50% of all ORNs being homozygous for the respective mutant allele. Using specific reporter lines for distinct ORN classes (see Materials and Methods), we analyzed the effects that the mutations had on the synaptic targeting patterns of different ORN subclasses.
We identified a mutant IG978 displaying ORNs that failed to innervate their correct target glomeruli (Fig. 1A). Using deletion and SNP mapping, we mapped the mutant to the psidin locus (phagocyte signaling impaired; Fig. 1C). Psidin is the homolog of the yeast protein MDM20. MDM20 is the noncatalytic subunit of the N-acetyltransferase complex NatB known in yeast to acetylate ∼60% or more of all cytosolic proteins on specific N-terminal amino acid residues (Polevoda and Sherman, 2003a; Singer and Shaw, 2003). Psidin was identified previously in Drosophila in two independent screens: as a lysosomal protein in blood cells (Brennan et al., 2007; Kim et al., 2011) and as a novel actin-binding protein essential for oocyte migration (Kim et al., 2011). To validate and understand the role of Psidin during olfactory circuit formation, we analyzed three different alleles in parallel: our newly established psidinIG978 allele (harboring the missense mutation E320K) and two expected null alleles, psidin1 (Brennan et al., 2007) and psidin55D4 (Kim et al., 2011) (harboring a STOP codon at K441 and K471, respectively; Fig. 1C). These analyses revealed two phenotypes. First, psidin null alleles revealed an apparent reduction in axon numbers in several, but not all, ORN classes (Fig. 1A,E; Table 1). This phenotype was caused by a reduction in neuron numbers of these ORN classes, which was already visible at around 8 h after puparium formation (APF) in developing pupae (Fig. 1E and data not shown), and therefore seemed to relate to a defect in early neurogenesis or cell survival (see below). PsidinIG978 mutant clones showed a trend similar to psidin1 mutants but no significant change in neuron numbers compared with controls (Fig. 1E).

Psidin is required for pathfinding of a subset of olfactory receptor neurons. Adult brains were stained with anti-GFP (green) and NC82 (magenta) to visualize synaptic innervation of ORNs in the AL. In forward MARCM, only mutant cells are visible selectively labeled in green in a wild-type background. Conversely, wild-type cells are labeled in a mutant background in reverse MARCM. MARCM analysis was done using eyFlp or hsFlp to generate clones. A, Or59c and Or42a mutant axons show a strong defasciculation phenotype in the psidin1 and psidin55D4 background (dashed lines). Or47a psidin mutant axons show ectopic synapse formation in ventral parts of the AL (arrows). Centrally projecting Or22a mutant neurons display no mistargeting phenotype but a weaker innervation (arrows). All ORN classes show a strong cell-loss phenotype in the psidin1 and psidin55D4 background. B, Reverse MARCM analysis of eyFlp clones. Or59c, Or42a, and Or47a wild-type neurons project normally in the psidin1 or psidinIG978 mutant background. Forward MARCM analysis using hsFlp showed that small psidinIG978 clones are sufficient to cause mistargeting in Or47a axons. C, Protein structure of Psidin and the used psidin alleles. Psidin contains a Tpr-domain and coiled coils at the C terminus. A “NatB interaction domain” is predicted in the center of Psidin. D, ORN classes are affected differently in targeting according to their projection route in psidin1 mutants. E, Mutant cells in the adult ANT and MP were counted using Or-gal4 and UAS-mCD8-GFP. The number of neurons is significantly reduced in the psidin1 loss-of-function mutant background in all classes. Although the neuron number is not significantly reduced in psidinIG978 mutants, there is a trend toward a reduction. Counting of Or83b-positive cells (Orco) in the MP revealed a 35% reduction of neurons in the psidin1 background. *p < 0.05; **p < 0.01; ***p < 0.001, one-way ANOVA and Bonferroni's posttest. Error bars ± SEM. Scale bar, 20 μm.
Quantification of the targeting and neuron-loss phenotypes
Second, all three psidin mutant alleles revealed defects in glomerulus targeting, consistently affecting the same subclasses of ORNs (Fig. 1A,D; Table 1). The nature of these phenotypes was comparable regardless of whether ORN numbers were reduced (psidin1, psidin55D4) or unaffected (psidinIG978). Or47a-expressing ORNs target the dorsomedial glomerulus DM3 in control animals (Fig. 1A). In psidin mutants, they frequently innervated a ventromedial glomerulus (Fig. 1A,D). The majority of the ventromedial targeting ORNs (e.g., Or59c, Or42a, and Or92a) displayed strong mistargeting and ectopic synapse formation (Fig. 1A,D; Table 1). In contrast, centrally projecting ORNs like Or22a, Or47b, and Or71a axons, which travel a short distance at a straight angle from the AL entry point, were hardly affected (Fig. 1A,D; Table 1).
Since the targeting tissue in the AL was not genetically manipulated by eyFlp, potential phenotypes were expected to affect the ORNs but not their target cells in the brain. We sought additional evidence that Psidin was required within the mistargeting ORN by using single cell and reverse MARCM analysis to visualize selectively wild-type axons in a mutant background. In these experiments, wild-type Or47a, 42a, and 59c ORNs within nonlabeled mutant ORNs projected normally, confirming that Psidin is required in a cell-autonomous manner in ORNs (Fig. 1B). In addition, hs-Flp-mediated recombination to induce very small mutant ORN clones showed that single mutant axons mistargeted in psidin clones (Fig. 1B). To confirm the requirement of Psidin, we used the GAL4/UAS system to reexpress Psidin in ORNs homozygous for psidin (eyFLP clones; Fig. 2A). In these experiments, targeted expression of Psidin achieved successful rescue of ORN cell numbers and pathfinding phenotypes (Fig. 2A).

Cell-loss phenotype can be rescued independent from the targeting phenotype. The entire ORN population was visualized using a direct-fusion Or-mCD8-GFP. Act-GAL4 was used to drive the expression of wild-type Psidin or p35 in eyFlp clones. A, A mistargeting phenotype is visible in both the psidinIG978 and psidin1 mutant backgrounds, displaying mildly and strongly dispersed (dashed circles) glomerular innervation, respectively. Additionally, in the psidin1 background, axons mistarget to dorsal parts of the AL (arrowheads). The normal targeting pattern is restored after reexpression of wild-type Psidin in the respective mutant background. In contrast, expression of p35 does not rescue the mistargeting phenotype. B, We observed a strong loss of neurons in the psidin1 background. In the psidin1 background, the ORN number was reduced by 36% compared with wild type. Reexpression of wild-type Psidin rescues the ORN cell number in the psidin1 background. Similarly, expression of p35 restores the neuron number in the psidin loss-of-function background. The neuron number was not significantly changed in the psidinIG978 background. C, Quantification of the mistargeting phenotype. In the psidin1 mutant background, 41 and 32% of the neurons display a strong or mild mistargeting phenotype, respectively. Mistargeting phenotype is completely rescued after expression of wild-type Psidin. Expression of p35 reduces the targeting phenotype only to levels comparable to psidinIG978 mutants. Similarly, in the psidinIG978 background, reexpression of wild-type Psidin rescues the phenotype completely. *p < 0.05; **p < 0.01; ***p < 0.001, one-way ANOVA and Bonferroni's posttest. Error bars ± SEM.
To understand the reason for the reduced number of ORNs, we expressed the antiapoptotic and caspase-inhibiting protein p35 (Davidson and Steller, 1998) in psidin1 and psidinIG978 mutant and control clones. In these experiments, the cell number defect of psidin1 mutant ORNs was fully rescued, but axonal targeting defects persisted (Fig. 2). These findings suggest that Psidin prevents apoptosis of ORNs during development. It also strongly supported the conclusion from our anatomical studies that the role of Psidin in axon navigation represents an independent function. Therefore, we conclude that Psidin-deficient defects in olfactory circuit formation are caused by two distinct cellular mechanisms.
Psidin is expressed at the right stages in the developing olfactory organs
We next analyzed the expression of psidin during different developmental stages using in situ hybridization and antibody staining (Fig. 3). Both methods suggested that Psidin is expressed ubiquitously in all cells, including the developing olfactory organs. Using in situ hybridization, psidin expression was detected at 5–10 h APF in the eye-antennal disc, the primordia of MP and antenna (Fig. 3A,B). This expression is consistent with a cell-autonomous role of Psidin in ORNs during neuronal differentiation, since ORNs arise at 6–20 h APF (Rodrigues and Hummel, 2008). At around 24 h APF, expression reached its maximum in both the MP and the antenna (Fig. 3C,D). At 30 and 45 h, expression started to decline in the olfactory organs (Fig. 3E–H). This expression is coherent with a role in the axonal pathfinding of ORNs, known to start around 20 h APF and continue over a period of ∼30 h (Rodrigues and Hummel, 2008). We next asked whether Psidin protein was expressed in ORNs during development. ORs start to be expressed only after development of the olfactory system. Therefore, we used the pan-neuronal marker Elav to label all neurons. Analysis of protein expression using an anti-Psidin antibody showed that Psidin protein was expressed in neuronal marker Elav-positive cells likely to be developing ORNs, judging by location in the antennal disc (Fig. 3I). Thus, we conclude that Psidin is expressed during the relevant developmental times in ORNs both during neuron differentiation and during axonal targeting.

Psidin is expressed in the developing olfactory organs. A–H, In situ hybridization (ISH) for psidin at different developmental stages. A, At 6 h APF, the psidin ISH signal can be detected in the developing eye-antennal discs (arrow). C, E, The expression stays high in the developing antenna and MPs both at 24 and 30 h APF (arrows). G, Psidin expression starts to decline at 45 h APF. B, D, F, H, Control (sense) probes for psidin do not show any signals. I, Staining of a developing antennal disc in 6 h APF pupae. Psidin is widely expressed in the developing antenna at this stage and partially correlates with elav-positive cells. Scale bars, 100 μm.
Psidin promotes lamellipodia of neuronal growth cones
Recent data showed that Psidin is a novel actin-binding protein required for the migration of Drosophila oocytes (Kim et al., 2011). To unravel how Psidin might contribute to the process of axonal navigation and whether similar mechanisms are required as for oocyte migration, we turned to cultures of embryo-derived primary Drosophila neurons that are ideally suited for the genetic dissection of cytoskeletal mechanisms underpinning axonal growth (Matusek et al., 2008; Sánchez-Soriano et al., 2010; Gonçalves-Pimentel et al., 2011). We found that psidin1, psidin55D4, or psidinIG978 mutant neurons displayed significantly smaller lamellipodia compared with wild-type controls (Fig. 4A,B). These findings were consistent with potential roles of Psidin in actin regulation. Furthermore, neurites were significantly longer (Fig. 4B), similar to observations for other actin-regulating proteins with known pathfinding defects in vivo (Sánchez-Soriano et al., 2010). HA-tagged Psidin partially colocalized with actin filaments in neurites and growth cones (Fig. 4C). These data were in agreement with the above-mentioned report that Psidin binds actin and regulates lamellipodia during border cell migration in Drosophila oocytes (Kim et al., 2011). In that study, Psidin was suggested to antagonize the function of the actin-stabilizing protein Tropomyosin 1 (Tm1), thus maintaining actin networks in a dynamic state. To test whether a similar mechanism might apply to Drosophila neurons, we analyzed tm1ZCL0722 mutant neurons and found no detectable phenotypes (Fig. 4A,B). However, when the tm1ZCL0722 mutation was combined with the psidin55D4 null mutant, the Psidin-deficient axon length and lamellipodial phenotypes were reverted to wild-type levels (Fig. 4A,B). These data were consistent with observations in non-neuronal cells and suggested that the psidin mutant phenotype is caused by unrestrained Tm1 activity.

Psidin regulates lamellipodia size in the growth cones. A, B, Growth cones (asterisks, A) are significantly smaller in psidin1, psidin55D4, and psidinIG978 neurons compared with wild type. Specifically, neurons lacking Psidin have significantly smaller lamellipodia compared with wild type. Additionally, psidin mutant axons are longer than wild-type axons. Tm1ZCL0722 neurons displayed no lamellipodia phenotype. Lamellipodia size is restored in the psidin55D4 plus tm1ZCL0722 double mutants. C, Primary culture of 6-h-old neurons from wild-type embryos overexpressing HA-tagged Psidin. It localizes to the growth cone together with actin and is visible in the lamellipodia. Neurons were stained with anti-tubulin, phalloidin, and anti-HA (Psidin). D, F-actin pelleting assay. Arrows indicate F-actin, and arrowheads represent the respective proteins: α-actinin, BSA, Psidin, and PsidinIG978. The indicated proteins were incubated in the presence (+) and absence (−) of F-actin. Reaction mixtures were incubated and centrifuged at 150,000 × g. Supernatant (S) and pellet (P) were then separately loaded onto an SDS gel. The positive control, α-actinin, can be found in the pellet together with F-actin. In contrast, the negative control, BSA, can only be found in the supernatant. Both, wild-type Psidin and PsidinIG978 are only found in the pellet in the presence of F-actin. Both proteins seem to pellet with F-actin at comparable levels. *p < 0.05; **p < 0.01; ***p < 0.001, one-way ANOVA and Bonferroni's posttest. Error bars ± SEM.
We asked whether the point mutation in PsidinIG978 that causes defects in lamellipodia size and axon targeting, but not in cell survival, would interfere with actin binding and thereby explain the observed phenotype. Using an in vitro actin-binding assay, we found that PsidinIG978 bound actin at similar levels as wild-type Psidin (Fig. 4D). Thus, unidentified mechanisms in addition to actin binding must be involved in Psidin's regulation of lamellipodia size and actin dynamics.
Roles of Psidin in maintaining dynamic actin networks can explain its in vivo roles
We next tested whether the role of Psidin in restraining Tm1 activity in growth cones is of relevance for the axon-targeting phenotype of ORNs in vivo (Fig. 5). For this, we analyzed the two independent loss-of-function mutant alleles, tm1ZCL0722 (Morin et al., 2001; Kim et al., 2011) and tm1su(flw)4 (Vereshchagina et al., 2004), using eyFlp analysis. Consistent with our findings in growth cones, both alleles displayed no defects, neither in axonal projection patterns nor in the number of Or59c-expressing ORNs (Fig. 5A–C). Also consistent with our findings in primary neuron cultures, ORN homozygous mutants for both psidin55D4 and tm1ZCL0722 showed a marked reduction in the percentage of brains that display Or59c axonal targeting defects compared with psidin55D4 mutants alone (Fig. 5A,B; 70% vs 20%). However, the psidin55D4, tm1ZCL0722 double-mutant condition did not rescue the reduction in the number of Or59c-expressing ORNs typical of the psidin55D4 or psidin1 single-mutant backgrounds (Fig. 5C). These data suggest that Psidin functions as a regulator of actin dynamics in conjunction with Tm1 during axon targeting (see also Kim et al., 2011). In contrast, it implicates a Tm1-independent mechanism of Psidin during neuronal survival.

Psidin and Tropomyosin have opposing roles during axon targeting. A, Or59c neurons in a psidin1 and a psidin55D4 mutant background showed a complete loss of innervation at their innate glomerulus (dashed circles). The tropomyosin mutant tm1ZCL0722 did not show any targeting defect (dashed circles). The targeting pattern of psidin single mutants was restored in psidin55D4 plus tm1ZCL0722 double mutants (dashed circles). B, Single mutants of tm1ZCL0722 show no targeting defect, whereas psidin1 (72%) and psidin55D4 (71%) single mutants showed a pronounced overall targeting defect. The targeting defect of psidin55D4 mutants was rescued in the double-mutant psidin55D4 plus tm1ZCL0722 (25%). C, The loss of Or59c neurons in the psidin55D4 background (n = 25) was not rescued in the double mutant (n = 20). The tm1ZCL0722 background did not show any significant reduction of Or59c neurons. D, E, Overexpression of constitutively active (const. act.) Cofilin partially rescues the psidin1-targeting phenotype (72% vs 58%). Expression of inactive (inact.) Cofilin aggravates the phenotype (83%). Expression of both cofilin variants leads to a mild phenotype in the wild-type background. F, G, Overexpression of LimK in a wild-type background leads to a strong targeting defect. Axons innervate along the outer rim of the AL (red arrowheads). Knockdown and overexpression of a kinase inactive form leads to a mild phenotype. The overall targeting phenotype in psidin1 (72%) can be further aggravated by the overexpression of LimK (88%). Knockdown or expression of DN LimK (72% vs 31% or 38%) rescues the targeting phenotype, especially the strong targeting phenotype. *p < 0.05; **p < 0.01; ***p < 0.001, one-way ANOVA and Bonferroni's posttest. Error bars ± SEM.
Tm1 acts primarily as a stabilizer of F-actin (Bugyi and Carlier, 2010). Another factor reported to compete with Tm1 is ADF/cofilin (Kuhn and Bamburg, 2008). Therefore, we tested whether Drosophila cofilin (called Twinstar/Tsr) has an impact on Psidin function. For this, we overexpressed a constitutive active (S3A) and a constitutive inactive version (S3E) of Tsr in wild-type and psidin1 mutant clones. Overexpression of either of the two Tsr isoforms in wild-type clones resulted in mild mistargeting of Or59c ORN axons in ∼20% of the brains (TsrS3A, 20%; TsrS3E, 23%; Fig. 5D,E). However, constitutive active TsrS3A overexpressed in psidin1 mutant Or59c ORNs reduced their mistargeting phenotype by 14% compared with psidin1 alone, consistent with a compensatory role of cofilin through competing with Tm1 for actin. Vice versa, inactive TsrS3E aggravated the psidin1 phenotype (8% increase; Fig. 5D,E), likewise consistent with the above idea.
Cofilin function in F-actin disassembly is negatively regulated by phosphorylation through LimK, which was previously implicated in ORN targeting (Ang et al., 2006). We found that expression of wild-type LimK in wild-type ORNs caused 88% of Or59c ORNs to mistarget (Fig. 5F,G). Expression of LimK RNAi or of a kinase-inactive LimK affected the targeting of only 10% of ORNs (Fig. 5F,G). Expression of wild-type LimK in psidin1 mutant ORNs resulted in a comparable degree of targeting defects as the same experiment caused in wild-type neurons (87%). This value was slightly higher than that of psidin1 mutant ORNs without targeted LimK expression (Fig. 5F,G). Consistent with the idea that Psidin promotes actin dynamics and instability, overexpression of LimK RNAi or kinase-inactive LimK (i.e., those conditions that promote cofilin activity) significantly rescued the psidin1 mutant axonal targeting phenotype (Fig. 5F,G). The results confirm that Psidin's function as an actin regulatory protein, originally discovered in non-neuronal cells, can be applied in the context of neuronal guidance. Furthermore, we conclude that Psidin's role in actin dynamics is required for growth cone targeting but is not involved during ORN survival.
Neuron number but not axon targeting depend on Psidin as a subunit of the N-acetyl transferase B complex
We next addressed potential molecular mechanisms of Psidin function during the regulation of neuron number. Psidin is the homolog of yeast MDM20, the noncatalytic subunit of the evolutionarily well conserved NatB complex required for N-terminal protein acetylation (Polevoda and Sherman, 2003a). In budding yeast, natB subunit mutants show defects including mitochondrial inheritance, budding, and cell division (Polevoda and Sherman, 2003b; Singer and Shaw, 2003). A role in cell growth was also found using human cell culture (Starheim et al., 2008). To test whether Psidin's role in ORN survival reflects a function as a constituent of the NatB complex, we investigated the Drosophila gene CG14222 (referred to as dNAA20 in the following) encoding the predicted catalytic subunit of NatB. Because of the lack of dNAA20 mutants or suitable deletions, we used eyFlp in combination with a strong driver (act-GAL4) and expressed three independent RNAi constructs selectively in ORNs (Fig. 6A). All constructs showed comparable phenotypes (Fig. 6A). The RNAi used for the experiments achieves strong knockdown of dNAA20 levels (Fig. 6B), but its expression in ORNs of wild-type flies was insufficient to cause neuron loss or axon-targeting phenotypes in Or59c or Or42a neurons (Fig. 6C,E,F,H). Expression of dNAA20 RNAi in clones mutant for the hypomorphic allele psidinIG978, however, resulted in a significant decrease in ORN numbers, reminiscent of phenotypes observed in psidin1 mutant clones (Fig. 6D,G). This suggests that psidinIG978 provides a sensitized background for knockdown of dNAA20, and it is consistent with the trend in neuron number reduction in hypomorphic mutant clones (see Fig. 1). Or59c and Or42a neuron numbers in psidin1 null mutants were not further decreased by dNAA20 RNAi expression, suggesting that Psidin's role in neuron survival is NatB dependent (Fig. 6D,G). In contrast to the reduction in cell number, Or59c and Or42a ORNs did not show any enhancement in mistargeting or ectopic synapse formation when dNAA20 RNAi was expressed in psidinIG978 or psidin1 mutants (Fig. 6E,H). These results indicate that dNAA20 is dispensable for ORN targeting but is required for the survival of the correct number of ORNs in a Psidin-dependent manner.

Psidin interacts with dNAA20 in vivo A, The effect of dNAA20 on ORN targeting was assessed using RNAi. Act-GAL4 was used to drive the expression of RNAi in eyFlp clones. Three different RNAi lines were used to knock down dNAA20 in Or42a neurons. All three lines had the same effect. B, Quantification of dNAA20 knockdown. S2 cells were transfected with dNAA20-myc and RNAi against dNAA20 (line#2). After coexpression of the RNAi against dNAA20, protein levels were reduced by ∼70–80% compared with expression of dNAA20-myc alone. C, Knockdown using RNAi (line#2) of dNAA20 in the psidinIG978 mutant background showed a loss of glomerular innervation of Or42a neurons similar to the psidin1 mutant background. D, Quantification of the total Or42a cell population: knockdown leads to reduction of Or42a neurons comparable to levels of psidin1 mutants. No further reduction was observed in the psidin1 mutant after dNAA20 knockdown. E, Quantification of the Or42a mistargeting phenotype expression of RNAi had no effect on the targeting phenotype. F, The effect of dNAA20 on Or59c targeting. The targeted glomerulus of Or59c neurons appears not to be innervated in psidin1 mutant flies (dashed circles). Axons display an overall random innervation pattern across the AL (arrowhead). The targeting phenotype does not change after expression of RNAi. Comparably, the expression of RNAi has no effect on psidinIG978 mutant neurons. G, Quantification of the total Or59c cell population: knockdown using RNAi (line#2) of dNAA20 in the psidinIG978 mutant background leads to a significant reduction of Or59c neurons, to levels comparable to psidin1 mutants. No further reduction of the neuron number was observed after knockdown in psidin1 mutants. The knockdown of dNAA20 had no effect on wild-type cells. H, Quantification of the targeting phenotype of Or59c axons expression of RNAi in psidin1 and psidinIG978 had no effect on targeting. *p < 0.05; **p < 0.01; ***p < 0.001, one-way ANOVA and Bonferroni's posttest. Error bars ± SEM.
The central domain of Psidin binds the catalytic NatB subunit dNAA20
We next tested whether dNAA20 and Psidin physically interact. To this aim, we used coimmunoprecipitation assays of HA-tagged Psidin with myc-tagged dNAA20, both coexpressed in S2 cells (Fig. 7A). Pulldown of wild-type Psidin with anti-HA antibody resulted in significant amounts of coimmunoprecipitated dNAA20 (Fig. 7A). These data show that Psidin and dNAA20 form a complex, as has similarly been reported for their yeast and human homologs (Singer and Shaw, 2003; Starheim et al., 2008). Comparable coimmunoprecipitation was achieved with PsidinIG978, suggesting that this mutation does not interfere with NatB complex formation (Fig. 7A). The same physical interaction was observed in the reverse experiment using anti-myc-mediated pulldown (data not shown).

Psidin interacts with dNAA20 in vitro A, S2 cells were cotransfected with Psidin-HA/PsidinIG978-HA and dNAA20-myc. dNAA20 was coimmunoprecipitated with wild-type Psidin (arrowhead) and PsidinIG978 (arrow) isoforms in S2 cells at comparable levels. The protein PsidinIG978 carries the point mutation found in psidinIG978 mutants. B, The position E320 is highly conserved in higher organisms having a glutamate at this position. Interestingly, the yeast wild type resembles exactly the mutation found in the psidinIG978 allele. C, Representative examples of coimmunoprecipitated dNAA20-myc with various Psidin deletions. The top blot shows the different Psidin deletions (anti-HA blot). The bottom blot displays the amount of dNAA20 that was pulled down together with the Psidin variants (anti-myc blot). As a reference, wild-type Psidin pulldown of dNAA20 was used (arrowhead, left lane) to compare the pulldown of dNAA20 with Psidin deletions (arrow, right lane). Deletion of the entire NatB interaction domain in PsidinΔNatBfull leads to loss of dNAA20 (arrow) pulldown. Smaller deletions of this domain (PsidinΔNatB23, PsidinΔNatB2) showed a reduced pulldown of dNAA20 (arrow). Only PsidinΔNatB1 was able to pull down dNAA20 (arrow) at levels close to wild-type Psidin (arrowhead). D, Quantification of the Western blots showing the normalized pulldown of dNAA20 with different Psidin isoforms. PsidinIG978, PsidinS678A, and PsidinΔNatB1 pulled down dNAA20 at wild-type levels. In contrast, PsidinS678D, PsidinΔNatBfull, PsidinΔNatB23, and PsidinΔNatB2 show a reduced or no pulldown of dNAA20, respectively. E, Representative examples of coimmunoprecipitated dNAA20-myc with Psidin phosphomutants. The top blot shows the Psidin phosphomutant (anti-HA blot). The bottom blot displays the amount of dNAA20 that was pulled down together with the phosphomutant (anti-myc blot). As a reference, wild-type Psidin pulldown of dNAA20 was used (arrowhead, left lane) to compare the pulldown of dNAA20 with Psidin deletions (arrow, right lane). PsidinS678A was able to pull down dNAA20 (arrow) at levels similar to wild-type Psidin (arrowhead). In contrast, PsidinS678D showed a reduced pulldown of dNAA20 (30% of wild-type Psidin). F, The serine residue at the position 678 in Drosophila is highly conserved among other organisms. Again, in yeast this residue seems not to be conserved.
Next we sought to map the interaction domain of Psidin with dNAA20 within the Psidin protein. In addition to the Tpr- and coiled-coil domains, a so-called putative NatB interaction domain is predicted by some software programs (Fig. 1B and SMART Heidelberg). We deleted this domain entirely (ΔNatB-full) or in parts (ΔNatB 1–3) from Psidin and used it to test coimmunoprecipitation of dNAA20 from S2 cell lysates (Fig. 7C). The deletion of the entire domain resulted in complete loss of coimmunoprecipitated dNAA20 (Fig. 7C). Subdeletions of the N-terminal, middle, or C-terminal parts of the domain resulted in partial loss of interaction (Fig. 7C,D). The strongest interaction was maintained with a deletion of the region where PsidinIG978 carries the point mutation E320K (ΔNatB 264–378; Fig. 7C). We quantified the level of bound dNAA20 to the Psidin deletions relative to the binding of dNAA20 to the wild-type Psidin protein (Fig. 7D). We conclude that the putative NatB interaction domain is indeed required for the interaction of dNAA20 and Psidin.
Phosphorylation of a conserved serine in Psidin prevents NatB complex formation and interferes with Psidin's function during ORN survival
Our next experiments were designed to more precisely distinguish the dNatB-dependent and -independent roles of Psidin in vivo. Deleting the entire or parts of the NatB interaction domain appeared very crude and not well suited for in vivo experiments. Therefore, we addressed potential mechanisms for regulated interaction between dNAA20 and Psidin. Previously, a conserved serine (S678 in fly; Fig. 7F) has been shown to be phosphorylated in human MDM20 using mass spec analysis (Trost et al., 2009). As this serine is located just downstream to the NatB interaction domain, we generated mutants that would either mimic phosphorylation (S678D) or make phosphorylation impossible at this location (S678A). The nonphosphorylatable Psidin mutant S687A coimmunoprecipitated dNAA20 at similar levels to PsidinIG978 and wild-type Psidin (Fig. 7D,E). In contrast, the phosphomimetic mutant S678D pulled down significantly lower amounts of dNAA20 compared with wild-type Psidin and PsidinIG978, but similar levels to the Psidin deletion mutants (Fig. 7D,E, PsidinS678D vs PsidinΔNatB2). Hence, this amino acid mutation provides an excellent opportunity to ask whether binding to dNAA20 is required for both of the in vivo functions we observed for Psidin. We generated transgenic flies and reexpressed three different point mutations within the Psidin protein in psidin mutant clones (Fig. 8A–C). First, we expressed PsidinIG978 in psidin1 and wild-type clones. UAS-psidinIG978 fully rescued the cell-loss phenotype in psidin1 clones (Fig. 8B). In contrast, the axon-targeting defect remained and was rescued only to the level of psidinIG978 mutant clones (Fig. 8C). This result confirms that the single-point mutation in PsidinIG978 is sufficient to interfere with axon targeting without affecting ORN numbers. Next, we expressed psidinS678A and psidinS678D in mutant and control clones. While expression of PsidinS678A rescued both the cell-loss and axon-targeting phenotypes, expression of PsidinS678D selectively rescued the axon-targeting phenotype but showed no ability to prevent cell loss (Fig. 8A–C). Given that PsidinS678D binds dNAA20 significantly less, these data provide additional strong evidence for a dNAA20-dependent role of Psidin during neuron survival.
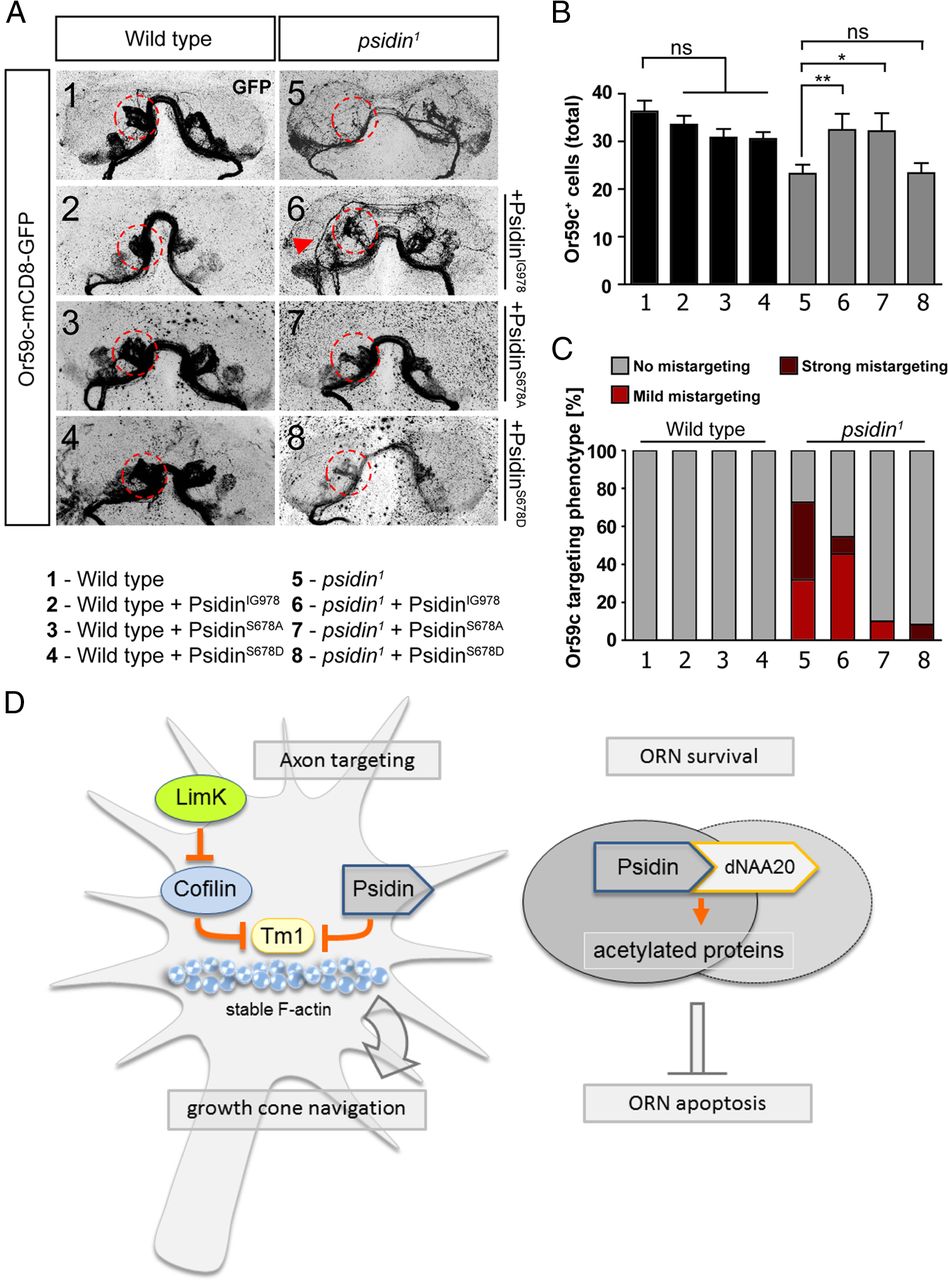
Conserved phosphorylated serine 678 prevents binding of dNAA20 to Psidin. A, Psidin isoforms were expressed in eyFlp clones driven by act-GAL4. Expression of PsidinIG978 (carrying the E320K point mutation), rescued the targeting phenotype of psidin1 mutant neurons, mimicking the psidinIG978-targeting phenotype. Expression of both serine mutant isoforms PsidinS678A and PsidinS678D rescued the targeting phenotype to wild-type levels. B, Quantification of the Or59c cell population: expression of PsidinIG978 and PsidinS678A rescues the cell loss in the psidin1 background. Expression of the phosphomimetic PsidinS678D fails to rescue the cell loss. C, Quantification of the targeting phenotype. Expression of PsidinIG978 in the psidin1 mutant background rescues targeting to psidinIG978 levels. Expression of both serine mutants phosphomutants rescue the targeting to wild-type levels. D, Model of Psidin's two functions during axon targeting and ORN survival. During axon guidance of olfactory neurons, Psidin function as an actin-binding protein, preventing the formation of long and stable filaments in growth cones. It acts as an antagonist to the actin filament stabilizer Tropomyosin. LimK, possibly via inactivation of cofilin, allows actin filament stabilization, and inactivation of LimK rescues Psidin's requirement in targeting. For ORN survival, Psidin is required as a noncatalytic subunit of the NatB complex, in which it interacts with the catalytic subunit dNAA20 to regulate protein acetylation (e.g., during cell cycle). *p < 0.05; **p < 0.01; ***p < 0.001, one-way ANOVA and Bonferroni's post-test. Error bars indicate ± SEM.
Discussion
The formation of a neuronal network depends on the proper development and targeting of neurons within the network. Here, we show that Psidin, in a similar manner to its recently described role in oocyte migration, acts as a constituent part of the actin machinery in the context of neuronal circuit formation in vivo. Furthermore, it functionally links to known actin regulators, in particular LimK and nonmuscle Tropomyosin. In addition, we define a novel role of Psidin in cell survival of neurons during development. Our data strongly suggest that Psidin uses two modes of activity. First, it prevents apoptosis of developing olfactory neurons as a noncatalytic subunit of the NatB complex. Second, it regulates axon targeting via its interaction with cytoskeleton regulators. Regarding the first role as part of the NatB complex, our results prove an evolutionarily conserved role of Psidin that has been predicted so far merely on the basis of in silico comparisons (see below) (Brennan et al., 2007).
For its second role, our findings essentially support the idea that cell migration and axonal growth share important properties (Lowery and Van Vactor, 2009). Axon guidance is coordinated through signaling processes downstream of extracellular biochemical cues and mechanical properties of the growth cone environment. The key effectors of these signaling processes are the actin- and microtubule-associating proteins, which determine the cytoskeletal dynamics underpinning any morphogenetic changes that growth cones undergo (Dent and Gertler, 2003; Lowery and Van Vactor, 2009). We show that loss of Psidin results in smaller growth cones with significantly reduced lamellipodia compared with wild-type controls. The aberrant growth cone morphology is completely rescued by the additional removal of Tm1, suggesting that Psidin promotes actin dynamics of growth cones by restraining Tm1 activity. Similar genetic relationships between Psidin, cofilin, and Tm1, as we found in neurons, were recently demonstrated in the context of oocyte migration (Kim et al., 2011).
We present a point mutation in Psidin (E320K) that selectively affects lamellipodia morphology, and hence axon targeting, but has no significant impact on neuronal cell numbers. Kim et al. (2011) demonstrated previously that Psidin physically binds actin in vitro. We reproduced this result and hypothesized that the E320K point mutation in PsidinIG978 might interfere with Psidin's actin-binding ability, thus explaining the loss of function of this allele during axonal growth. However, PsidinIG978 maintained its actin affinity, suggesting that actin interaction might be required for Psidin function in growth cones but that the key mechanism underlying this role is still to be uncovered. Differential screens for proteins interacting with Psidin in the region around position E320 might clarify this point. For example, Psidin might bind Tm1 directly, and this is not unlikely when considering links between the NatB complex and Tropomyosin in yeast (Singer and Shaw, 2003).
Not all ORN axon types require Psidin equally. While neurons that target dorsal and ventral glomeruli are strongly affected, axons that innervate central glomeruli of the AL show no or only mild mistargeting phenotypes. At first sight, this contradicts the highly penetrant phenotypes in embryonic primary neurons. Given that these primary neurons represent a broad range of different neuron types, we suggest that Psidin is part of the housekeeping machinery of growth cones. We can think of two explanations. First, as suggested by the genetic interactions of Psidin not only with Tm1 but also with cofilin, different signaling pathways operating in distinct ORNs are likely to orchestrate their actin networks through different molecular routes, part of which appear redundant. This would mean that the degree of functional Psidin contributions is neuron and/or context specific. Second, even if Psidin was promoting flexible F-actin structures equally in all ORNs, the requirement for this lamellipodial property could be context dependent, consistent with the idea that intact F-actin networks in vivo are not required for axon extension per se, but for axon navigation (Sánchez-Soriano et al., 2010). For example, neurons using the dorsolateral and ventromedial targeting routes or traveling further distances are likely to be more dependent on dynamic actin networks and therefore more affected by the loss of Psidin than axons that do not turn or meet and adhere to their targets early on (centrally projecting ORNs). Judging from the distribution of the affected glomeruli and the fact that embryonic neurons in culture were equally affected by Psidin deficiency, we propose that the latter explanation is the more likely to be the correct one. Essentially, depending on the neuron's task in targeting to the correct glomerulus, the lack of Psidin and its effect on actin dynamics can be compensated for or turn into a visible phenotype. These data might provide additional evidence for the pivotal role of actin and cytoskeleton dynamics as an intrinsic property of a neuron. They also suggest that neurons with more challenging targeting routes require more complex guidance cues and downstream integrating signaling mechanisms at the level of the cytoskeleton.
Our data indicate that Psidin uses an evolutionarily conserved pathway via its function as a NatB complex subunit and interactor of dNAA20 to ensure ORN survival, likely by inhibiting their apoptosis (Polevoda and Sherman, 2003b; Starheim et al., 2008). However, the interaction domain within MDM20 or Psidin had only been predicted so far. We provide the first experimental evidence that the predicted domain within Psidin is indeed required for the interaction of Psidin and dNAA20. In addition, we identify a conserved serine just downstream of the interaction domain as a putative tool to regulate NatB complex formation in a phosphorylation-dependent manner. Given that this amino acid was found to be phosphorylated in human MDM20, it seems likely that this mechanism applies to humans as well. Our findings suggest that phosphorylation at this amino acid potently interferes with binding between Psidin and dNAA20. Furthermore, the failure of the phosphomimetic mutant to rescue the psidin cell-loss phenotype while rescuing the targeting defect strongly suggests that Psidin is required in two modes: nonphosphorylated as part of the NatB complex to regulate survival of neurons and phosphorylation independent as a regulator of actin dynamics in processes like cell migration and axon targeting. Interestingly, this serine is not conserved in yeast MDM20, indicating that this regulation was not present yet in yeast.
In contrast to psidinS678D, the psidinIG978 mutation selectively impacts on axon targeting but not on neuron survival. Nevertheless, in addition to the targeting phenotype, the psidinIG978 allele provides a sensitized background for knockdown of dNAA20. Although this functional interaction demonstrates a Psidin-dependent requirement of dNAA20, it also suggests that the PsidinIG978 mutation affects a less critical and likely dNAA20-related function of Psidin during neuronal survival. Yet, neither protein stability or expression levels nor protein localization appear affected in psidinIG978 mutants (data no shown). The fact that yeast wild-type MDM20 resembles the PsidinIG978 mutant at this position further indicates that a glutamate or aspartate at this site is not essential for NatB function. A single amino acid mutation that we introduced into the yeast MDM20 to render it into a higher eukaryote-like version (K304E) did not interfere with MDM20's function during cell division and budding (Fig. 9A,B), providing evidence that this amino acid was able to evolve (from positively to negatively charged) without significantly compromising NatB acetyltransferase function.

“Fly-like” yeast MDM20K304E rescues wild-type MDM20 function during cell division. A, Mimicking a fly-like MDM20 by replacing lysine with glutamate did not change the functionality of the yeast MDM20 protein compared with wild type. Expression of MDM20K304E in an mdm20Δ strain rescues the actin cable formation during cell division. B, Expression of MDM20K304E can fully rescue the growth defect of mdm20Δ strain. C, The percentage of polarized yeast cells is not affected in MDM20K304E-expressing cells under different growth conditions.
Finally, whereas we provide mechanistic and in vivo evidence for a role of the NatB complex in neuronal survival, we have not been able to identify a target protein that would be sufficient to explain the N-acetylation-dependent prevention of cell death. Two main reasons have made this endeavor impossible so far: (1) 60% or more of all proteins undergo cotranslational acetylation, and (2) this modification can only be detected by mass spectrometry making cell-specific analysis of developing fly tissues currently unfeasible. And although it would be highly desirable to identify such a key target protein, it is possible that this single protein does not exist but rather that the sheer number of non-N-acetylated proteins does not allow for the cell to function and survive. Future studies and improved methodology will hopefully elucidate this point.
We conclude that Psidin provides an interesting regulator of neuronal development with key functions during neuronal survival and axon targeting. Thus, characterization of the in vivo role and the regulation of Psidin and its homologs could support research aimed at understanding regeneration and degeneration of the nervous system.
Footnotes
This work was supported by an Emmy-Noether Grant from the German Research Foundation, by a career development award from the Human Frontiers Science Organization, and by the Max-Planck Society (I.C.G.K.). The work of N.S.-S. and A.P. was funded by the BBSRC (BB/I002448/1) and funds by the Wellcome Trust supporting the Manchester Fly Facility (087742/Z/08/Z). We are very grateful to D. Montell for sharing data and reagents before publication. We thank V. Schilling and C. Knappmeyer for great technical help. The psidinIG978 allele was found by I.C.G.K. during her postdoctoral stay in the laboratory of S. L. Zipursky. We thank S. L. Zipursky and Pelin C. Volkan for fruitful collaborations and discussions. Budding yeast strain lacking MDM20 was kindly provided by Dr. F. S. Sherman (University of Rochester Medical Center, Rochester, NY).
The authors declare no competing financial interests.
- Correspondence should be addressed to Ilona C. Grunwald Kadow, Max Planck Institute of Neurobiology, Sensory Neurogenetics Research Group, Am Klopferspitz 18, 82152 Martinsried, Germany. ikadow{at}neuro.mpg.de





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}