

Abstract
Cholinergic overactivity in diseases of neuromuscular transmission elicits a retrograde signal resembling homeostatic synaptic plasticity that downregulates transmitter release. Understanding this compensatory pathway could provide insights into novel therapeutic avenues and molecular mechanisms underlying learning and memory. Here we identify nitric oxide as a possible source of this signal in pathological human and mouse muscle samples and link this signaling pathway to changes in synaptic function in the neuromuscular junction. We further show that neuronal nitric oxide synthase is regulated by cholinergic excess through activation of skeletal muscle calpain and its effect on Cdk5 and CaMKII, leading to direct modulation of presynaptic function. Finally, we show that this signaling pathway acts through specific miRNA control of presynaptic vesicle protein expression. The control of presynaptic miRNA levels by postsynaptic activity represents a novel mechanism for the modulation of synaptic activity in normal or pathological conditions.
Introduction
Different forms of synaptic plasticity have been suggested to underlie learning and memory as well as synaptic alterations in neurological diseases such as epilepsy, Alzheimer's disease, and dystonia (Ben-Ari, 2008; Palop and Mucke, 2010; Quartarone and Pisani, 2011). However, the direct evidence of synaptic plasticity in human neurological disease remains largely unknown. Characterization of the primary synaptic defects in several genetic neuromuscular diseases revealed functional and structural abnormalities in both presynaptic and postsynaptic sites of the neuromuscular junction (NMJ). In congenital endplate acetylcholinesterase deficiency (CEAD) due to loss-of-function mutations in the collagen-like tail subunit of endplate AChE (ColQ) that anchors AChE to the postsynaptic membrane, the abnormalities at the NMJ include small nerve terminals, reduced acetylcholine release, and postsynaptic degeneration (Engel et al., 1977; Ohno et al., 1998). The postsynaptic degenerative changes were attributed to excitotoxic damage from Ca2+ overload of the postsynaptic region resulting from excessive ACh activation of the muscle nicotinic ACh receptor (AChR). The presynaptic changes in CEAD, however, were mysterious and explained as a possible “compensatory response” to the effect of excessive cholinergic activation (Engel et al., 2010). Similar changes in the nerve terminus have been seen in pharmacological models of cholinesterase inhibition (Roberts and Thesleff, 1969; Tiedt et al., 1978), and in genetic AChE knock-out (KO) models (Adler et al., 2011).
Slow-channel congenital myasthenic syndrome (SCS) due to mutations in the muscle AChR shows similar postsynaptic defects and presynaptic abnormalities to CEAD, attributed to prolonged openings of the AChR channel and Ca2+ overload at the subsynaptic region (Engel et al., 1982; Engel et al., 1996; Gomez et al., 1996). We have generated a series of SCS transgenic mouse lines (mSCS) that develop similar disease process with respect to impairment of both postsynaptic and presynaptic components (Groshong et al., 2007; Zhu et al., 2011). The presynaptic changes in these postsynaptic disorders strongly suggest that a compensatory response reduces synaptic strength via a retrograde signal resembling homeostatic synaptic plasticity. Our previous studies demonstrated that overexpression of human calpastatin (CPST), the natural inhibitor of calpain, significantly improved strength and neuromuscular transmission in mSCS (Groshong et al., 2007) and strongly suggested that postsynaptic calpain activity negatively regulates a retrograde signal that influences presynaptic nerve function.
In this study, we show that presynaptic alterations in quantal release at the normal and pathological NMJ are dependent on a calpain-activated neuronal nitric oxide synthase (nNOS) pathway. nNOS activity is abnormally increased at the NMJ in SCS mice and in muscle biopsies from patients with SCS or CEAD. Reducing nNOS activity via inhibition of calpain or Cdk5 or via chemical inhibitor improves presynaptic strength in wild-type (WT) and mSCS muscles, demonstrating that calpain acts via nNOS activity to modulate quantal release. Furthermore, presynaptic alterations in quantal release are correlated with miR-124- and miR-142-regulated Rab3a expression in motor nerve. Our findings reveal a novel pathway for retrograde control of presynaptic function in the NMJ that may underlie homeostatic synaptic plasticity and provide the basis for impaired synaptic function and potential therapeutic strategy for neuromuscular disorders.
Materials and Methods
Materials.
Chemical reagents were from Sigma. Cell culture materials were from Invitrogen. Fluorescence-tagged secondary antibody was from Life Technologies. Secondary antibodies with horseradish peroxidase were from GE Healthcare Bioscience. Primary antibodies to the following targets were used: p35/25 (1:1000; Cell Signaling Technology), CaMKII (1:1000; Abcam), p-CaMKII (Thr286; 1:500; Cell Signaling Technology), Cdk5 (1:500; SCBT), CPST (1:500; SCBT), nNOS (1:1000; Abcam), phospho-nNOS ser847 (1:500; Abcam), phospho-nNOS ser1412 (1:500; Abcam), cGMP (1:100; Abcam), Ras-related protein Rab-3a (Rab3a, 1:500; SCBT), Synaptic Vesicle Glycoprotein 2A (SV2a, 1:500; SCBT), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 1:5000; Ambion).
Plasmids and construction.
Full-length human CPST (kind gift from Dr. Masatoshi Maki, Nagoya University, Nagoya, Japan) was inserted into pcDNA3 plasmid. CaMKII-GFP plasmid was a kind gift from Dr. Tobias Meyer, Stanford University, Stanford, CA. nNOS plasmid was a kind gift from Dr. E. Clementi, San Raffaele Scientific Institute, Milan, Italy. CDK5DN plasmid (D144N; K33T) was a kind gift from Dr. Nancy Y. Ip, Biotechnology Research Institute and Molecular Neuroscience Center, Hong Kong University of Science and Technology, Hong Kong, China (Fu et al., 2007). Cdk5-GFP and p25-GFP plasmids were from Addgene. Rab3a 3′-UTR (NM_009001) was amplified with 5′-TTATATATACTAGTGCCACTTCCCTTCCCTGCTGCC-3′ (Forward-SpeI) and 5′-ATATAATATAAGCTTCAGGGTGCGGAAATTCATTAAATTCT-3′ (Reverse-HindIII) and cloned in miRNA reporter (Ambion). miRNAs were cloned and expressed by The BLOCK-iT Pol II miRNA and RNAi Expression Vector Kit (Life Technologies).
Cell culture.
Neuro-2a (a mouse neuroblastoma cell line), C2C12 (a mouse myogenic cell line), and HEK293 (a human embryonic kidney cell line) were grown in standard medium according to the manufacturer's medium protocol (American Type Culture Collection). Cells were cultured on six-well plates and coexpressed with different plasmids, miRNA expression vector, and miRNA reporter using Lipofectamine 2000 (Life Technologies) for 72 h and collected for analysis.
Animals and tissue preparation.
Three- to 4-month-old WT FVB mice and mSCS mice that express the εL269F mutation (Gomez et al., 1997) were used in this study. nNOS KO mice were from The Jackson Laboratory. All drug treatments and surgical procedures followed the animal care and use protocols established by the institutional animal care and use committee. Sciatic nerve axotomy (denervation) was described previously (Zhu et al., 2011). All mice used in this study were age-matched males (3 to 4 months of age). Sciatic nerve (SN) used for investigation was dissected from lumbar region to the proximal terminal of the tibialis anterior (TA) muscle. Mouse lumbar region (L1–6) was selected for investigation of Rab3a mRNA and protein expression level in spinal cord (SC). Table 1 shows the details of human SCS and CEAD biopsies used in our investigation. De-identified muscle biopsies were used in accordance with the respective institutional review boards.
Human SCS and CEAD biopsies used in this study
Drug administration.
7-Nitroindazole (7-NI; 50 mg/kg) was administered intraperitoneally daily for 9 d (Ikeda et al., 1998). Control mice were treated with DMSO or saline (0.9% w/v; ThermoFisher Scientific).
RNA extraction, cDNA synthesis, and quantitative real-time PCR.
RT-PCR and quantitative RT-PCR (qRT-PCR) were performed as described previously (Zhu et al., 2011). To measure miRNA expression, cDNA was synthesized and amplified with the NCode miRNA First-Strand cDNA Synthesis and qRT-PCR kit (Life Technologies), in which U6 was selected as internal control and 100 ng of miRNA-based cDNA was used for investigation (Peltier and Latham, 2008). ImageJ version 5.3 was used for semiquantitative study of Rab3a expression in SN. Table 2 shows the sequences used for qRT-PCR.
Sequences used for qRT-PCR
Western blotting.
Immunoblotting was done as described previously (Zhu et al., 2011). Protein expression levels were quantified by scan densitometry using ImageJ.
Tissue staining and imaging.
Tissue fixation, immunohistochemistry, and confocal fluorescence microscopy on serial TA muscle cryosections were as described previously (Zhu et al., 2011). ImageJ was used to determine the fraction of NMJs labeled with specific antibodies.
Calpain activity assay.
The calpain activity detection kit, buffer, and procedures were as described previously (Zhu et al., 2011).
Cdk5 kinase assay.
Cdk5 activity assay was as described previously (Smith et al., 2006). Tissue was homogenized with lysis buffer. The supernatants (500 μg) were then collected and immunoprecipitated with Cdk5 antibody (5 μg). The Cdk5 immunoprecipitates were washed twice with the lysis buffer and twice with a kinase buffer containing the following (in mm): 50 Tris, pH 7.5, 5 MgCl2, 1 EGTA, 1 EDTA, 40 β-glycerophosphate, 20 p-nitrophenylphosphate, 0.1 sodium vanadate, and 1 DTT. Kinase activity was measured in a total volume of 50 μl of kinase assay buffer with 0.1 mm [γ-32P]ATP (PerkinElmer) and 10 μg of histone H1 (Calbiochem) at 30°C for 60 min. The reaction was stopped by adding SDS sample loading buffer (4×). Samples were separated by SDS-PAGE gel (15%), transferred to nitrocellulose membrane, and autoradiographed to detect phosphorylation of histone H1. Cdk5 activity in different tissue samples was normalized via the ratio of phosphorylated histone H1 to internal Cdk5 expression.
nNOS activity detection via assay and enzyme histochemistry.
Cells and muscle tissue were homogenized via lysis buffer supplemented with protease inhibitor and 5 μm NG-iminoethyl-l-ornithine. Protein samples (10 μg) were analyzed with the Ultrasensitive Colorimetric NOS Assay Kit following the manufacturer's protocol (Oxford Biomedical Research). nNOS activity was detected on muscle slides using the NADPH diaphorase (NADPH-d) reaction as described previously (Grozdanovic et al., 1995). Endplates were labeled using acetyl cholinesterase reaction as described previously (Koelle and Friedenwald, 1949).
miRNA report assay.
Rab3a 3′-UTR was cloned in the pMIR-REPORT system (Ambion) and coexpressed with the miRNA vector system (Life Technologies) in HEK 293 cells in which luciferase activity was used to exhibit miRNA effects on a target gene. β-galactosidase activity was used to normalize the miRNA effect. Luciferase and β-galactosidase assay kits (Promega) were used for activity detection.
Intramuscular electroporation-mediated gene delivery and knock-down.
Electrically mediated gene delivery and siRNA knock-down (KD) in mouse TA muscle were performed as described previously (Zhu et al., 2011).
In vitro electrophysiology.
Voltage-clamp analysis of sciatic-tibialis nerve–muscle preparations was performed as described previously (Zhu et al., 2011).
Quantitative studies in tissue sections.
Quantification of endplate labeling was performed as described previously (Zhu et al., 2011).
Bioinformatics.
miRNA sequences and target gene prediction was conducted using miRNA-focused engine servers (www.mirbase.org and www.targetscan.org). Gene-specific RNAi design was done with siRNA Target Finder (Ambion).
Statistical analysis.
Data are expressed as mean ± SD. Statistical analyses were performed by Student's t test, two-way ANOVA, and the nonparametric Mann–Whitney U test.
Results
nNOS is abnormally activated at the NMJ in congenital myasthenic syndromes
Reduced presynaptic function in the “postsynaptic” congenital myasthenic syndromes such as SCS and CEAD suggests the presence of a retrograde signaling pathway (Engel et al., 1977; Engel et al., 1982; Gomez et al., 1997). The nNOS-NO pathway has been linked to retrograde synaptic signaling in a wide range of species and synapses (Aonuma et al., 2000; Etherington and Everett, 2004; Graves et al., 2004). nNOS activity is modulated by direct Ca2+-calmodulin binding, as well as by the relative extent of phosphorylation at serine residues 847 (inhibition; p-S847) and 1412 (activation; p-S1412) (Hayashi et al., 1999; Rameau et al., 2007). To determine whether the nNOS pathway is activated in SCS and CEAD, we first used antisera specific for p-S847 and p-S1412 nNOS epitopes in immunofluorescence colocalization studies with the NMJ using fresh-frozen muscle sections from diagnostic muscle biopsies from patients with SCS (n = 3) or CEAD (n = 4) (Fig. 1A, left). The genotypes of the patients are shown in Table 1. Compared with normal human muscle (n = 3), both SCS and CEAD have significantly higher proportions of NMJs labeled with p-S1412 antibody (44% and 30% vs 10%, respectively, p < 0.001) and significantly lower proportions labeled for p-S847 antibody (9% and 7% vs 37%, respectively, p < 0.001) (Fig. 1A, right). NO is a highly unstable molecular species that cannot be readily measured in tissue sections (Archer, 1993). However, because one key effect of NO is to activate soluble guanylyl cyclase, which generates cGMP, we used antibody specific for cGMP to localize this molecule in serial sections of these muscle biopsies. We found significantly higher numbers of NMJs labeled for cGMP in SCS and CEAD muscle (45% and 30% vs 10%, respectively, p < 0.001; Fig. 1A). cGMP immunolabeling was colocalized with immunolabeling for the nerve terminal antibody, synaptophysin (data not shown), suggesting that increased postsynaptic nNOS activity in SCS increases nerve terminal cGMP.

nNOS is abnormally activated at NMJs in SCS and CEAD. A, Left: Immunolabeling using antibodies to nNOS p-S847, nNOS p-S1412, and cGMP in cryosections from muscle biopsies from patients with SCS (n = 3) and CEAD (n = 4) compared with control muscle (n = 3). nNOS p-S847 immunolabeling (green) was strongly colocalized with NMJs (labeled with TxR-αBT, red) in normal human muscle, but to a lesser degree in SCS and CEAD (arrows). nNOS p-S1412 (green) and cGMP (green) immunolabeling were highly colocalized with NMJs in SCS and CEAD muscles, but to a lesser degree in normal human muscle. cGMP immunolabeling was associated with the presynaptic neuron at the NMJ and colocalized with synaptophysin (not shown). Right: Fraction of nNOS p-S847-labeled endplates in normal human muscle was significantly greater than in SCS and CEAD muscle. The fraction of nNOS p-S1412-labeled endplates in SCS and CEAD was significantly greater than in normal human muscle. The fraction of cGMP-labeled endplates was higher in SCS and CEAD than in normal human muscle. Scale bar, 15 μm. ***p < 0.001, Student's t test. B, Left: Cryosections of transgenic mSCS TA muscle immunolabeled using antibodies to nNOS p-S847, nNOS p-S1412, and cGMP. nNOS p-S847 immunolabeling (green) was strongly colocalized with NMJs labeled with TxR-αBT (red) in normal mouse muscle, but to a lesser degree in mSCS (arrows). nNOS p-S1412 (green) and cGMP (green) immunolabeling were highly colocalized with NMJs in mSCS, but to a lesser degree in WT muscle. Right: The fraction of nNOS p-S847-labeled endplates in WT muscle was significantly greater than in mSCS. The fraction of nNOS p-S1412-labeled and cGMP-labeled endplates was significantly greater in mSCS muscle than in WT muscle (n = 5). Scale bar, 15 μm. ***p < 0.001, Student's t test. C, Immunoblots of nNOS p-S847 and p-S1412 protein in WT and mSCS TA muscle. The expression level of nNOS p-S847 was higher in WT than mSCS TA muscle, whereas the level of nNOS p-S1412 expression was higher in mSCS than in WT (n = 3). *p < 0.05, Mann–Whitney U test. D, Histochemical measurement of nNOS activity using NADPH-d in WT and mSCS cryosections. Serial TA muscle sections were labeled for cholinesterase (dark brown; AChE) to localize endplates and NADPH-d (dark blue) to identify nNOS activity. Top: Numbers on NADPH-d-labeled fibers correspond to the fibers with AChE-labeled fibers in adjacent sections. Bottom left: The fraction of NADPH-d-labeled endplates in mSCS muscle was significantly greater than in WT muscle (n = 5). ***p < 0.001, Student's t test. Bottom right: nNOS activity in TA muscle homogenates was twice that of WT muscle, as measured by colorimetric assay (n = 5). Scale bar, 90 μm. **p < 0.01, Mann–Whitney U test. E, Top: Immunolabeling using antibodies to nNOS p-S847, nNOS p-S1412, and cGMP in cryosections of ColQ KO muscle. nNOS p-S847 immunolabeling (green) was strongly colocalized with NMJs (red) in normal mouse muscle, but to a lesser degree in ColQ KO (arrows). nNOS p-S1412 (green) and cGMP (green) immunolabeling were highly colocalized with NMJs in ColQ KO, but to a lesser degree in WT. Bottom: The fraction of nNOS p-S847-labeled endplates in WT muscle was significantly greater than in ColQ KO. The fraction of nNOS p-S1412-labeled and cGMP-labeled endplates was significantly greater in ColQ KO muscle than in WT muscle (n = 3). Scale bar, 50 μm. ***p < 0.001, Student's t test.
We tested for similar changes using a well characterized transgenic mouse model for SCS expressing the εL269F mutation (Gomez et al., 1997; Groshong et al., 2007; Zhu et al., 2011) Figure 1B shows that in WT TA, muscle only ∼33% of NMJs show p-S1412 immunoreactivity, whereas 86% label for the p-S847 antibody. mSCS NMJs, however, show the reverse pattern, with ∼81% showing p-S1412 immunoreactivity and 46% labeling for the p-S847 antibody (p < 0.001). Similarly, only ∼33% of NMJs label for cGMP, whereas in SCS mice, 56% of NMJs contain detectable cGMP immunoreactivity (p < 0.001), which colocalized with synaptophysin (data not shown). We found similar changes in the relative proportions of the phosphorylated forms of nNOS in immunoblot studies (Fig. 1C). p-S847 was readily detectable in WT muscle homogenates, but only faintly present in mSCS muscle, whereas p-S1412 was absent in WT muscle and present at high levels in SCS muscle. The relative levels of nNOS protein in muscle were not different between the mouse groups. Increased amounts of activated forms of nNOS should correspond to changes in nNOS activity. We used nitro blue tetrazolium to histochemically detect nNOS activity in muscle sections (Grozdanovic et al., 1995) and colocalized to NMJs in serial sections using a cholinesterase histochemical stain (Fig. 1D, top). In WT muscle, only ∼6% of NMJs were labeled for activated nNOS, whereas in mSCS muscle, ∼39% had histochemically detectable nNOS activity (Fig. 1D, bottom left). These measurements were also consistent with colorimetric assay of nNOS activity in muscle homogenates, which showed that nNOS activity in mSCS muscle was approximately twice that of WT muscle (Fig. 1D, bottom right). These findings correlate well with the more sensitive immunolabeling for nNOS phospho-epitopes and show that in mSCS muscle, nNOS activity is increased at the NMJ (Fig. 1B).
Finally, we studied the pattern of nNOS activity in muscle sections from a mouse model of CEAD generated by targeted disruption of the gene encoding ColQ. In homozygous ColQ−/− (ColQ KO) TA muscle sections, only 35% of NMJs labeled for the p-S847 antibody compared with 83% of WT mice, whereas 70% of ColQ KO NMJs labeled with the p-S1412 antibody and 60% of NMJs labeled with the cGMP antibody compared with 33% of WT NMJs (Fig. 1E). These findings demonstrate that the NO-nNOS signaling pathway is abnormally activated at the NMJ, leading to an increase in presynaptic cGMP in clinical and experimental conditions associated with cholinergic overactivity.
nNOS activity negatively regulates neurotransmitter release in normal and mSCS NMJ
To test for a direct correlation between the changes in nNOS activity and quantal release in mSCS and WT muscle, we used voltage-clamp analysis of spontaneous and evoked synaptic activity in the NMJ to study mice deficient in nNOS by siRNA-mediated nNOS gene silencing. Figure 2A shows the KD effect on nNOS expression in TA muscle 9 d after electroporation with 6 nNOS-specific siRNA sequences, as we described previously for silencing type 1 IP3 receptor (Zhu et al., 2011). Segments S5 and S6 led to 90% reduction at nNOS protein levels compared with little or no effect of control (scrambled) RNA (ctRNA) or other nNOS siRNAs. Nine days after electroporating segments S5 and S6, combined siRNA-mediated nNOS silencing in mSCS muscle led to a significant increase in miniature endplate current (MEPC) frequency (1.31 ± 0.15 s−1 vs 0.74 ± 0.10 s−1, respectively, n = 18, p < 0.05) and quantal content (QC) (25.83 ± 3.74 vs 10.28 ± 2.31, respectively, n = 13, p < 0.01) to normal levels compared with the effect of ctRNA. This suggests that the changes seen in nNOS activation markers in mouse and human SCS and CEAD muscle may contribute to the impairment of neuromuscular transmission. We next investigated whether modulation of nNOS activity plays a similar role in normal neuromuscular transmission (Fig. 2B). siRNA silencing of nNOS in WT mouse muscle also led to an increase in MEPC frequency (1.90 ± 0.13 s−1 vs 1.29 ± 0.13 s−1, respectively, n = 18, p < 0.01) and QC (32.04 ± 2.23 vs 23.53 ± 3.35, respectively, n = 9, p < 0.05) compared with ctRNA. Therefore, skeletal muscle nNOS activity negatively regulated presynaptic strength. MEPC amplitude, a measure of postsynaptic strength, was also significantly increased in both mSCS and WT muscle after nNOS silencing by 70% and 55%, respectively, most likely due to an additional effect on AChR expression. This is consistent with the finding that nNOS silencing in muscle also led to an increased expression of AChR subunit mRNAs (data not shown). The resultant amplitudes of EPCs were significantly increased by approximately 2-fold in both mSCS and WT mice after nNOS KD.

nNOS activity negatively regulates neuromuscular transmission in normal and mSCS muscles. A, nNOS silencing in WT TA muscle in vivo. Comparison of nNOS protein levels by immunoblot after electroporation of WT TA muscle with six nNOS-specific siRNA sequences (S1–S6) and scrambled RNA as ctRNA. Nine days after electroporation, nNOS protein levels were significantly decreased by sequences S5 and S6. #Normalized to untreated WT (n = 3). *p < 0.05, Mann–Whitney U test. B, nNOS silencing increases synaptic strength in SCS and WT mouse NMJ. Representative traces of spontaneous synaptic activity measured as MEPCs (voltage clamped at −60 mV) from excised, intact TA muscle-nerve preparations 9 d after electroporation of siRNA. Raw data show spontaneous MEPCs recorded in mSCS muscle treated with ctRNA (upper) or nNOS siRNA sequence S5 and S6. nNOS silencing produced an increase in spontaneous synaptic activity. Graphs: nNOS silencing in TA muscle caused a >40% increase in MEPC frequency (n = 18, *p < 0.05) in mSCS TA and a >30% increase in WT TA (n = 18, **p < 0.01). nNOS silencing in TA caused a 150% increase in QC (n = 13, **p < 0.01) in mSCS TA and a 25% increase in WT TA (n = 9, *p < 0.05). nNOS silencing in TA caused a 40% increase in MEPC amplitude in mSCS TA (1.48 ± 0.17 nA, n = 18, vs 0.87 ± 0.08 nA, respectively, n = 10, *p < 0.05) and a >35% increase in WT TA (2.86 ± 0.3 nA, n = 10, vs 1.84 ± 0.08 nA, respectively, n = 7, **p < 0.01). EPC amplitudes were increased >2-fold in mSCS TA muscle (16.71 ± 10.07 nA, n = 14, vs 7.29 ± 1.13 nA, n = 7, respectively, **p < 0.01) and nearly 2-fold in WT TA (82.08 ± 11.74 nA, n = 9, vs 42.09 ± 5.79 nA, n = 7, respectively, **p < 0.01) by nNOS KD. Traces: Average MEPCs and EPCs recorded from excised muscle. C, Expression of nNOS in TA muscle of nNOS KO mice reduced synaptic strength in NMJ. Voltage-clamp study of excised, intact TA muscle-nerve preparations 5 d after plasmid electroporation. Recordings of MEPCs taken from TA muscle of nNOS KO mice treated with empty vector (top) or nNOS expression plasmid (bottom). Graphs: nNOS expression in TA muscle of nNOS mice reduced MEPC frequency and QC by >2-fold (n = 8, ***p < 0.001) compared with the effect of empty vector (n = 7). nNOS expression in TA muscle of nNOS mice reduced MEPC amplitude >40% (n = 8, ***p < 0.001) and EPC amplitude >70% (n = 7 control; n = 8, nNOS plasmid, ***p < 0.001). Traces: Average MEPCs (spontaneous) and evoked EPCs (evoked release) indicates an increase in MEPC and EPC amplitude after nNOS expression in contralateral TA muscle.
Long-term genetic ablation of nNOS in KO mice may lead to other compensatory changes. Therefore, to test for changes attributable to nNOS alone in mice with targeted disruption of the nNOS gene (nNOS KO), we performed voltage-clamp analysis of TA muscle of nNOS KO mice after electroporating TA muscles with either nNOS expression plasmid or control plasmid 5 d previously (Fig. 2C). TA muscle of nNOS KO mice treated with control plasmid had a normal to increased MEPC frequency and QC compared with WT mice. However, expression of nNOS in the contralateral nNOS KO TA muscle led to a significant reduction in MEPC frequency (0.75 ± 0.08 s−1 vs 1.55 ± 0.16 s−1, respectively, n = 8, p < 0.001) and QC (7.74 ± 1.65, n = 8, vs 18.35 ± 1.68, respectively, n = 7, p < 0.001) compared with control plasmid (Fig. 2C). These results demonstrate that in both normal and diseased muscle, nNOS activity negatively regulates quantal activity of neuromuscular transmission, presumably by production of the messenger, NO. Consistent with the nNOS-silencing results, after electroporation of nNOS plasmid to restore nNOS expression in nNOS KO mice, MEPC amplitude was decreased in TA muscle compared with the side treated with control plasmid (1.09 ± 0.12 nA vs 1.88 ± 0.2 nA, respectively, n = 8, p < 0.01), suggesting a postsynaptic effect of NO as well. In agreement with this finding, AChR subunit mRNA levels were increased in nNOS KO mice relative to WT mice and there was a trend for a decrease after transient expression of nNOS plasmid (data not shown). Not surprisingly, the combined effect of restoring nNOS expression in nNOS KO TA muscle on neuromuscular transmission was a significant decrease in EPC amplitude (7.74 ± 1.65 nA, n = 8, vs 33.66 ± 1.46, respectively, n = 7, p < 0.001) due to the decrease in QC and MEPC amplitude. These findings suggest that inhibition of the nNOS pathway in muscle may be an appropriate therapeutic target to increase synaptic strength in some neuromuscular disorders.
Calpain modulates quantal release through a Cdk5 pathway in normal and mSCS muscles
To explore the mechanism of nNOS activation in SCS and CEAD, we investigated the role of the Ca2+-dependent protease calpain, which is highly activated in skeletal muscle in conditions of cholinergic excess (Groshong et al., 2007; Zhu et al., 2011). Transgenic expression of human CPST in skeletal muscle corrects impaired neuromuscular transmission in mSCS and significantly increases quantal release in WT mice, presumably through a retrograde effect on presynaptic nerve function, in a pattern similar to the effect of nNOS modulation (Groshong et al., 2007). Cdk5 is believed to negatively regulate presynaptic transmitter release in the NMJ (Fu et al., 2005) and calpain is known to modulate Cdk5 activity by cleavage of the Cdk5 coactivator p35 to its constitutively active form, p25 (Lee et al., 2000; Smith et al., 2006). We investigated whether acute inhibition of skeletal muscle calpain by electroporation of CPST expression plasmid into TA muscle affected presynaptic motor nerve function and whether this effect was correlated with changes in Cdk5 activity. As shown previously (Zhu et al., 2011), calpain activity is elevated in mSCS muscle (Fig. 3A, top). Five days after electroporation of CPST, CPST protein levels were significantly greater than in control muscle (Fig. 3A, bottom). As expected, this treatment reduced calpain activity levels in mSCS muscle to the level of WT mice. Using voltage-clamp analysis of excised TA muscle 5 d after electroporation, we observed that transient expression of CPST had a pronounced effect on presynaptic transmitter release in neuromuscular transmission in both mSCS and WT muscle (Fig. 3B). In CPST-treated mSCS TA muscle, MEPC frequency increased >2-fold compared with the control plasmid-treated TA muscle (1.38 ± 0.13 s−1 vs 0.67 ± 0.07 s−1, respectively, n = 12, p < 0.05). QC was also increased by 2-fold (26.48 ± 2.65 vs 13.09 ± 4.29, respectively, n = 12, p < 0.01). This is similar to the findings with transgenic expression of CPST in skeletal muscle (Groshong et al., 2007). MEPC amplitudes, which are controlled predominantly by postsynaptic factors, were slightly decreased, as noted previously, presumably because of persistent activation of other proteases (0.54 ± 0.10 nA vs 0.84 ± 0.18 nA, respectively, n = 17, p < 0.001; Groshong et al., 2007). The net effect of these quantal changes was still a significant increase in EPC amplitudes (15.98 ± 2.76 nA vs 10.33 ± 2.2 nA, respectively, n = 12, p < 0.05; Fig. 3B). Furthermore, as we found with transgenic expression of CPST and inhibition of nNOS, acute inhibition of calpain had an equivalent effect on neuromuscular transmission in normal mice, increasing MEPC frequency (1.64 ± 0.12 s−1 vs 1.18 ± 0.09 s−1, respectively, n = 16, p < 0.05) and QC by >2-fold (44.74 ± 3.66 vs 20.05 ± 3.03 nA, respectively, n = 10, p < 0.01), with no effect on MEPC amplitude. In WT mice with acutely inhibited muscle calpain, EPCs were increased by >2-fold (82.75 ± 9.9 nA vs 30.10 ± 3.11 nA, respectively, n = 13, p < 0.001). This confirms our previous observations with transgenic expression of CPST that the activity of calpain within skeletal muscle negatively regulates synaptic strength through an effect on vesicular transmitter release in both physiological and pathological conditions.

Calpain and Cdk5 activities negatively regulate neuromuscular transmission in normal and mSCS muscles. A, Top: Calpain levels measured in TA muscle homogenates of WT mice, mSCS mice, and mice treated with by electroporation of plasmid expressing CPST, the dominant-negative form of Cdk5 (Cdk5DN), or empty vector (pcDNA3) 5 d previously. Calpain activity in mSCS muscle are 4× those of WT. CPST decreased calpain activity in mSCS muscle, but neither Cdk5DN nor pcDNA3 had any effect. #Normalized to WT calpain activity (n = 5). **p < 0.01, Mann–Whitney U test. Bottom: Immunoblots of CPST and calpain expression in WT and mSCS TA muscle homogenates. CPST was markedly increased in CPST-treated WT and mSCS TA muscle 5 d after electroporation of CPST. Treatment of WT and mSCS TA muscles with CPST and Cdk5DN had no effects on calpain expression (n = 3). *p < 0.05, Mann–Whitney U test. B, Voltage-clamp study of excised, intact TA muscle-nerve preparations 5 d after electroporation of CPST expression vector. Representative traces spontaneous synaptic activity (MEPCs) recorded from TA muscle of mSCS mice treated with empty vector (top) or CPST expression plasmid (bottom). Graphs: CPST expression in TA muscle of mSCS mice increased MEPC frequency (n = 27) and QC (n = 13) by more than 2-fold (***p < 0.001) compared with the effect of empty vector (n = 12). CPST expression in TA muscle of WT mice increased MEPC frequency by 28% (n = 27, *p < 0.05) and QC by more than 2-fold (n = 13, *p < 0.01) compared with the effect of empty vector (n = 16, MEPC frequency; n = 10, QC). CPST expression in TA muscle of mSCS mice reduced MEPC amplitude by 33% (n = 17, ***p < 0.001) compared with control plasmid (n = 23). CPST expression in WT TA muscle had no significant effect on MEPC amplitude. CPST expression in TA muscle of mSCS mice increased EPC amplitude by >2 fold (n = 13, *p < 0.05) compared with control plasmid (n = 12). CPST expression in TA muscle of WT mice increased EPC amplitude by >2-fold (n = 13, ***p < 0.001) compared with control plasmid (n = 10). Traces: Illustration of average MEPCs (spontaneous) and EPCs (evoked release) indicates an increase in MEPC and EPC amplitude after CPST expression in the contralateral side of the TA muscle. C, Immunoblots (top) and quantitation (bottom) of p35 and 25 protein levels in WT and mSCS TA muscle homogenates from untreated mice or mice treated 5 d previously with CPST or control plasmid. The p25 polypeptide is present in mSCS and in mSCS treated in control plasmid, but not seen in WT muscle. The p25 polypeptide is not present in mSCS treated with CPST. #Normalized to WT levels (n = 3). *p < 0.05, Mann–Whitney U test. D, Left: Cryosections of WT or mSCS TA muscle immunolabeled with antibody to Cdk5 (green) show that Cdk5 colocalized with NMJ labeled with TxR-αBT (red) in both mouse types (n = 5). Scale bar, 25 μm. Right: Cdk5 activity in TA muscle homogenates estimated as the phosphorylating activity for the substrate histone H1 was significantly elevated in mSCS and mSCS mice treated with pcDNA3, but significantly reduced in mSCS TA muscle treated with CPST or Cdk5DN plasmids 5 d previously. #Normalized to untreated WT (n = 3). *p < 0.05, Mann–Whitney U test. E, Voltage-clamp study of excised, intact TA muscle-nerve preparations 5 d after electroporation of Cdk5DN expression vector. Recordings of MEPCs were taken from TA muscle voltage-clamped at −60 mV. Raw data show spontaneous MEPCs recorded in TA muscle of mSCS mice treated with empty vector (top) or Cdk5DN expression plasmid (bottom). Graphs: Cdk5DN expression in TA muscle of mSCS mice increased MEPC frequency (n = 8, ***p < 0.001) by nearly 2-fold and QC (n = 10, *p < 0.05) by more than 2-fold (***p < 0.001) compared with effect of empty vector (n = 8). Cdk5DN expression in TA muscle of WT mice increased MEPC frequency by almost 2-fold (n = 10, *p < 0.05) and QC by 36% (n = 8, **p < 0.01) compared with the effect of empty vector (n = 8, MEPC frequency; n = 6, QC). Cdk5DN expression in TA muscle of mSCS mice increased MEPC amplitude by >60% (n = 10, **p < 0.01) compared with control plasmid (n = 8). Cdk5DN expression in WT TA muscle increased MEPC amplitude by 75% (n = 7, **p < 0.01) compared with control plasmid (n = 6). Cdk5 expression in TA muscle of mSCS mice increased EPC amplitude by >2 fold (n = 8, ***p < 0.001) compared with control plasmid (n = 8). Cdk5 expression in TA muscle of WT mice increased EPC amplitude by more than 2-fold (n = 8, **p < 0.01) compared with control plasmid (n = 6). Traces: Illustration of average MEPCs (spontaneous) and EPCs (evoked release) indicates an increase in MEPC and EPC amplitude after CDK5 expression in the contralateral side of TA-muscle.
Because calpain modulates the activity of Cdk5 by cleavage of its coactivator, p35, to the constitutively active form as p25 (Lee et al., 2000), we investigated whether the Cdk5 system is activated in mSCS. Figure 3C shows that both the p35 and p25 forms are present in mSCS muscle, whereas there is no detectable p25 in WT muscle. Electroporation of CPST plasmid into mSCS muscle prevents the formation of p25 (Fig. 3C), which is consistent with inhibition of calpain (Fu et al., 2005). Cdk5 protein highly localized to the NMJ in WT and mSCS muscle (Fig. 3D, left) and its distribution was not affected by CPST expression (data not shown). However, we found that Cdk5 activity, measured using histone 1 as a substrate (Smith et al., 2006), was increased in mSCS muscle and normalized by inhibition of calpain through electroporation of CPST plasmid into muscle (Fig. 3D, top right). Overexpression of CPST in WT muscle reduced basal Cdk5 activity to 67% of the normal level (n = 3, p < 0.05; Fig. 3D, bottom right).
Using voltage-clamp analysis of synaptic events in excised TA muscle, we investigated whether quantal release of ACh at the NMJ in mSCS or WT muscle was affected directly by Cdk5 activity. Figure 3E shows that direct inhibition of Cdk5 in mSCS muscle by electroporation of a dominant-negative form of Cdk5 (Cdk5DN) 5 d previously increased MEPC frequency by nearly 2-fold (0.99 ± 0.04 s−1 vs 0.56 ± 0.07 s−1, respectively, n = 8, p < 0.001) and QC by 2-fold (23.52 ± 3.7 vs 11.46 ± 2.94, respectively, n = 10, p < 0.05), to nearly normal levels, with a resultant increase in EPCs (30.38 ± 3.81 nA vs 11.23 ± 3.5 nA, respectively, n = 10, p < 0.01). Interestingly, Cdk5DN had similar effects on neuromuscular transmission in normal mice, increasing MEPC frequency nearly 2-fold (2.28 ± 0.33 s−1 vs 1.20 ± 0.12 s−1, respectively, n = 10, p < 0.05) and significantly increasing QC (27.02 ± 2.07 vs 19.76 ± 1.51, respectively, n = 8, p < 0.01). This suggests that, like NO and calpain, postsynaptic Cdk5 activity plays a role in modulating synaptic transmission in normal mice. These findings are similar to previous studies in which Cdk5−/− mice showed increased quantal release (Fu et al., 2005). In the present study, however, Cdk5 or calpain were inhibited only in muscle and apparently exerted a retrograde effect on presynaptic function. These studies suggest that muscle calpain activity negatively regulates ACh release through its effect on muscle Cdk5 activity. Interestingly, in both mSCS and WT mice, MEPC amplitudes were also increased by ∼40%, suggesting that Cdk5 activity also influences postsynaptic factors, perhaps through an effect on AChR gene expression. Our findings suggest that calpain modulates quantal release through a Cdk5 pathway in normal and mSCS muscles.
The calpain/Cdk5 pathway modulates nNOS activity through CaMKII in normal and mSCS muscles
To investigate the correlation between increased calpain and Cdk5 activities and NO production in mSCS muscle, we examined the nNOS system in mSCS muscle treated by electroporation of CPST or Cdk5DN plasmids. Figure 4A shows that inhibition of calpain or Cdk5 locally in mSCS TA muscle by expressing CPST or Cdk5DN reverses phospho-nNOS ratios of p-S847 and p-S1412 at the NMJ to the proportions similar to those of WT mice, with greater numbers of NMJs showing labeling for p-S847, the inhibitory nNOS marker, than p-S1412, the activated nNOS marker, in treated mice compared with control plasmid-treated mice (p < 0.001). Similarly, after inhibition of either calpain or Cdk5, the number of NMJs labeled with cGMP antibody was reduced to WT levels. In mSCS muscle homogenates, the levels of p-S847 nNOS were significantly increased, whereas p-S1412 nNOS levels were decreased by inhibition of calpain or Cdk5 (p < 0.05; Fig. 4B). Finally, NMJ-localized nNOS activity (Fig. 4C, top) and the relative levels of nNOS in muscle homogenates (Fig. 4C, bottom) were decreased by local inhibition of either calpain or Cdk5. Electroporation of control plasmid, pcDNA3, had no effect on nNOS activity markers in these experiments. These results suggest that the retrograde effect of NO on motor nerve quantal activity is mediated through a calpain-Cdk5 pathway.
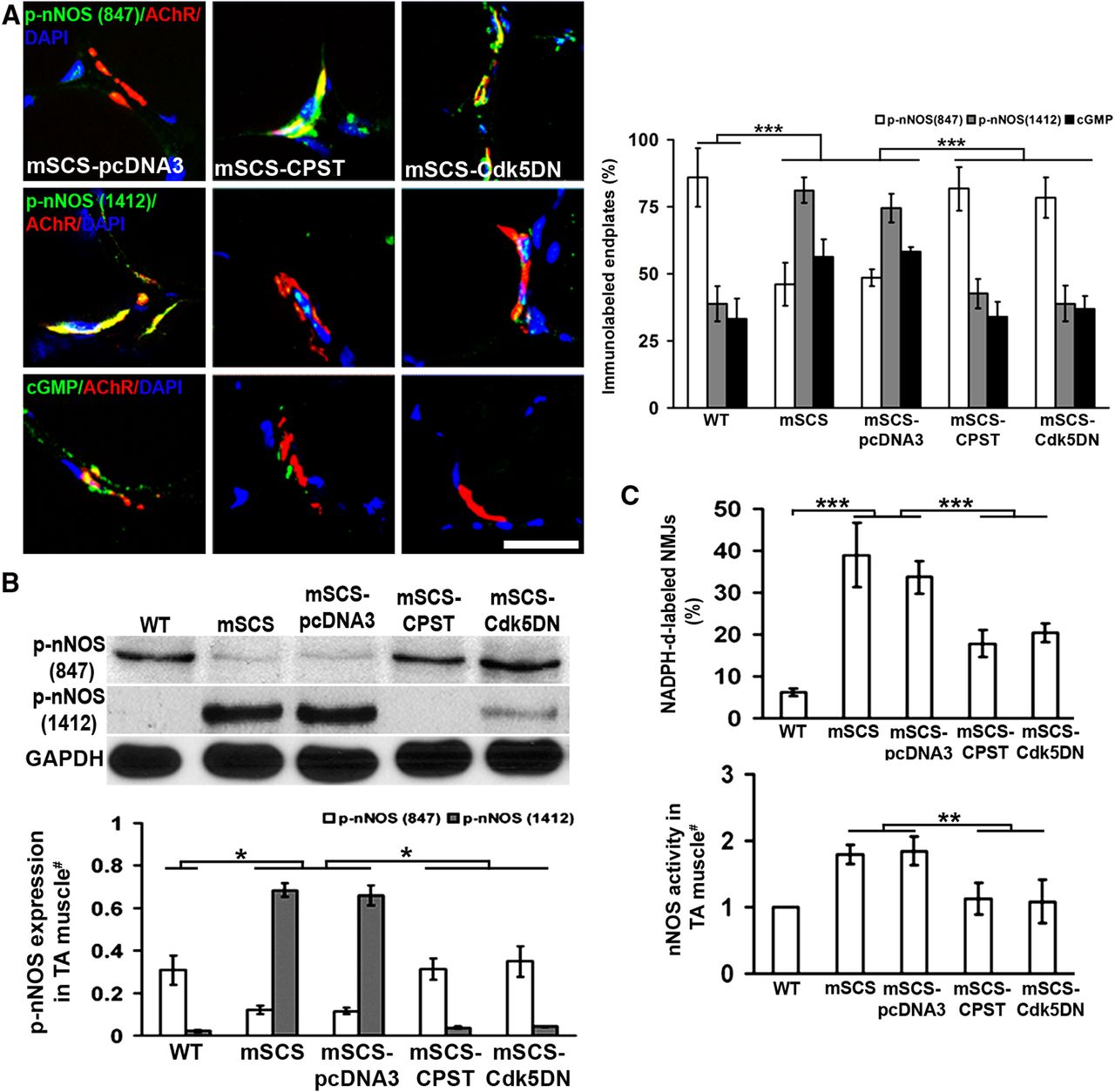
Calpain and Cdk5 increase nNOS activity in mSCS muscle. A, Left: Cryosections of mSCS TA muscle immunolabeled (green) with antibodies to nNOS p-S847, nNOS p-S1412, and cGMP, colocalized with NMJs labeled with TxR-αBT (red) after electroporation with CPST or CDK5DN expression vectors 5 d previously. nNOS p-S847 immunolabeling (green) strongly colocalized with NMJs (red) in mSCS muscle electroporated with CPST or Cdk5DN compared with NMJs in mSCS treated with pcDNA3. nNOS p-S1412 (green) and cGMP (green) immunolabeling was strongly colocalized with NMJs in mSCS plasmid control compared with mSCS treated with CPST or Cdk5DN. Right: Fraction of nNOS p-S847-labeled endplates in mSCS muscle pretreated with CPST or Cdk5DN was significantly greater than in untreated mSCS or control-treated mSCS. The fraction of nNOS p-S1412-labeled and cGMP-labeled endplates was significantly greater in untreated mSCS muscle and control-treated mSCS muscle than in WT and mSCS muscle with CPST or Cdk5DN treatment (n = 5). Scale bar, 15 μm. ***p < 0.001, Student's t test. B, Immunoblots (top) of p-S847 and p-S1412 phospho-epitopes on nNOS in WT, mSCS, and treated mSCS TA muscles. The expression level (bottom) of nNOS p-S847 was significantly higher in mSCS treated with CPST or Cdk5DN than in untreated mSCS TA muscle, whereas the level of nNOS p-S1412 expression was higher in untreated mSCS than in mSCS with treated with CPST or Cdk5DN (n = 3). *p < 0.05, Mann–Whitney U test. C, Top: Histochemical measurement of nNOS activity at NMJs in cryosections using NADPH-d in WT, mSCS, and mSCS treated with CPST or Cdk5DN. NMJs were localized using cholinesterase in adjacent slides, as in Figure 1D. The fraction of NADPH-d-labeled endplates in mSCS muscle was significantly greater than in mSCS muscle treated with CPST or Cdk5DN (n = 5). ***p < 0.001, Student's t test. Bottom: nNOS activity in TA muscle homogenates was significantly higher in untreated mSCS mice and significantly reduced in mSCS muscle electroporated with CPST or Cdk5DN plasmid (n = 5). **p < 0.01, Mann–Whitney U test.
Regulation of nNOS activity can be attributed in part to inhibitory phosphorylation of nNOS at S847 by CaMKII (Hayashi et al., 1999). To test whether increased calpain and Cdk5 activities were associated with a reduction in CaMKII-mediated nNOS phosphorylation in the NMJ, we examined cryosections and homogenates of TA muscle treated as shown in Figure 4 by electroporation with CPST or Cdk5DN vectors or with control plasmid to determine the expression level of the activated form of CaMKII, autophosphorylated at Thr-286 (p-T286 CaMKII), relative to total CaMKII. Immunostaining for CaMKII protein highly localized to WT mouse NMJs, as shown previously (Martinez-Pena y Valenzuela et al., 2010), as well as at mSCS NMJs, and was not altered by expression of CPST or Cdk5DN (Fig. 5A, left). However, using antibody to p-T286 CaMKII, we found that no NMJs from untreated SCS mice had detectable p-T286 CaMKII labeling compared with extensive p-T286 labeling in WT mice. Treatment of mSCS TA muscle with CPST or Cdk5DN increased the NMJ labeling for p-T286 CaMKII compared with the effect of control plasmid (Fig. 5A, right).

Calpain and Cdk5 act through CaMKII to modulate nNOS in neuromuscular disorders. A, Left: Cryosections of mouse TA muscle immunostained (green) for CaMKII protein colocalized with NMJs TxR-αBT (red) in WT, mSCS, and treated mSCS mice. CaMKII colocalized with NMJs in all muscle sections (n = 9). Scale bar, 15 μm. Right: Cryosections of mouse muscle immunostained for CaMKII p-T286 phospho-epitope showing that CaMKII p-T286 colocalized with NMJs in WT, but was markedly reduced in mSCS. Treatment of TA muscle with CPST or Cdk5DN plasmid restored CaMKII p-T286 immunostaining (n = 5). Scale bar, 50 μm. B, Immunoblots of CaMKII and CaMKII p-T286 phospho-epitopes in WT, mSCS, and treated mSCS TA muscles. #Values for CaMKII p-T286 are normalized to the level in WT muscle. The CaMKII p-T286 level is significantly reduced in mSCS muscle and control treated mSCS muscle, but partially restored 5 d after electroporation with CPST or Cdk5DN plasmids (n = 3). *p < 0.05, Mann–Whitney U test. C, Top: Cryosections of human muscle biopsies from normal, SCS, and CEAD patients showing immunostaining (green) for CaMKII (top) and CaMKII p-T286 (bottom). CaMKII colocalizes with NMJs in all sections. Immunostaining for CaMKII p-T286 is present in WT, but markedly reduced in SCS (n = 3) and CEAD (n = 4) muscles. Bottom: The fraction of CaMKII p-T286-labeled endplates in normal human (n = 3) was significantly greater than in human SCS (n = 3) and CEAD (n = 4) diseased muscle. #Normalized to CaMKII-stained endplates. Scale bar, 15 μm. **p < 0.01. D, Left: Immunoblots of CaMKII p-T286 phospho-epitope in HEK293 and C2C12 cell lines transiently coexpressing CaMKII plasmid with p25, Cdk5, and Cdk5DN plasmids. Transfected plasmids are indicated below. Right: Quantitation shows that in HEK293 and C2C12, the CaMKII p-T286 phospho-epitope level was increased in cells cotransfected with p25 and Cdk5DN plasmids, but was significantly less in cells cotransfected with p25 and Cdk5 plasmids. #Normalized to CaMKII plasmid-only transfection (n = 3). *p < 0.05, Mann–Whitney U test.
In TA muscle homogenates, total CaMKII protein levels were no different in WT, mSCS, and treated mSCS muscle (Fig. 5B). However, using the phospho-epitope-specific antibody to p-T286 CaMKII, the amount of p-T286 CaMKII in mSCS muscle was approximately 3-fold less than in WT muscle, as indicated by the plotted ratios of p-T286 CaMKII to total CaMKII (p < 0.05; Fig. 5B). Moreover, expression of CPST or Cdk5DN in mSCS TA muscle by electroporation of plasmid vectors increased p-T286 CaMKII to levels slightly higher than in WT, presumably correcting CaMKII activity in mSCS muscle (Fig. 5B). This change would have the predicted effect of increasing phosphorylation of nNOS at S847 and reducing NOS activity at the NMJ, as shown in Figure 1. These studies suggest that increased activities of calpain and Cdk5 are responsible for the reduced activity of CaMKII in mSCS muscle, the resultant reduced numbers of p-S847 nNOS-labeled versus p-S1412 nNOS-labeled NMJs, and the increased NO production at NMJs in mSCS muscle.
To look for a similar pattern of CaMKII labeling in the SCS and CEAD biopsies shown in Figure 1A, we used specific antibodies to CaMKII and p-T286 CaMKII to determine the fraction of endplates labeled in each disorder. Figure 5C shows that p-T286 CaMKII is localized to human NMJs and more readily detectable in normal human muscle compared with SCS and CEAD biopsies. We found that 48 ± 6% of endplates labeled for p-T286 CaMKII in normal human muscle, nearly 2-fold over that seen in human SCS (27 ± 7%, p < 0.001) and CEAD muscle (24 ± 8, p < 0.001). These findings suggest that a reduced activation of CaMKII in SCS and CEAD muscle might underlie the changes in nNOS activity seen in these diseases.
The inhibitory effect of Cdk5 on CaMKII phosphorylation may be facilitated by the fact that Cdk5 coactivators p35 and p39 interact with CaMKII and that Cdk5 phosphorylates protein phosphatase-1, which is known to dephosphorylate CaMKII (Bradshaw et al., 2003). To test for an effect of Cdk5 activity on CaMKII in a cellular model, we transiently overexpressed CaMKII in HEK293 and C2C12 myoblast cells in the presence of control plasmid or the plasmids expressing Cdk5, Cdk5DN, and p25. Figure 5D shows that expression of p25 alone or Cdk5 with p25 significantly decreased p-T286 CaMKII expression, whereas coexpression of Cdk5DN with p25 increased p-T286 CaMKII expression. These findings indicate that CaMKII autophosphorylation is influenced by the activity of Cdk5, presumably by an effect on an intermediate regulatory enzyme, suggesting an additional potential regulatory pathway for CaMKII in the NMJ. These findings strongly suggest that a calpain/Cdk5/CaMKII pathway modulates quantal release through the nNOS-NO pathway at NMJ.
The calpain/Cdk5/nNOS pathway modulates presynaptic expression of synaptic vesicle proteins in normal and SCS muscles
To look for structural correlates for the calpain-nNOS-dependent regulation of quantal activity, we measured the changes in expression of the presynaptic proteins SV2 and Rab3a by immunolocalization and immunoblotting (Fig. 6A,B). Rab3a is involved in exocytosis by regulating a late step in synaptic vesicle fusion through an effect on membrane flow in the nerve terminal (Matteoli et al., 1991). SV2 is a synaptic vesicle protein that interacts with synaptotagmin (Janz et al., 1999). Both SV2 (Fig. 6A, left) and Rab3a (Fig. 6B, top) showed strong colocalization with αBT labeling at NMJ in WT muscle and severely reduced expression at NMJ in untreated mouse mSCS muscle. However, after electroporation of mSCS muscle with CPST or Cdk5DN plasmids, expression of both SV2 and Rab3a at the NMJ was restored, consistent with the effect of expression of CPST and Cdk5DN on restoration of quantal activity in mSCS muscle. Similarly, in homogenates of TA muscle protein, the levels of both SV2 (Fig. 6A, right) and Rab3a (Fig. 6B, bottom) were reduced in SCS mice compared with WT. Expression of CPST and Cdk5DN increased the SV2 and Rab3a protein levels in mSCS TA muscle compared with control. Treatment of mSCS muscle either by siRNA-mediated nNOS KD in TA muscle or by intraperitoneal injection with 7-NI, a relatively specific nNOS inhibitor, increased Rab3a levels (Fig. 6B). CPST, nNOS KD, and 7-NI also increased Rab3a in WT muscle, although the modest increase after Cdk5DN treatment was not significant. Figure 6C shows that Rab3a level in nNOS KO TA muscle was increased 1.5-fold over WT. Electroporation of nNOS plasmid into nNOS KO TA muscle reduced Rab3a protein to WT levels. Electroporation of nNOS plasmid into WT TA muscle reduced the Rab3a level to 50% of WT. Reduced presynaptic Rab3a protein levels in mSCS mice are attributable to increased cholinergic activity. Also, cholinergic blockade is known to lead to increased presynaptic function. Therefore, we investigated whether blockade of cholinergic activity at the NMJ altered Rab3a expression in muscle. Seven days after intraperitoneal administration of αBT to WT mice, we found that Rab3a expression levels in TA muscles were 2-fold greater than in saline-treated controls (p < 0.05; Fig. 6D). These findings suggest that the calpain-Cdk5DN-nNOS pathway provides retrograde feedback from changes in cholinergic input by influencing structural components of vesicular transmitter release and function of the innervating motor nerve.

The activity-related, calpain-Cdk5-nNOS pathway modulates vesicle protein expression in skeletal muscle. A, Left: Cryosections of WT and mSCS muscle immunostained for SV2a protein (green) and NMJ (TxR-αBT, red) show that SV2a is colocalized with NMJ in WT muscle, but markedly reduced in mSCS muscle. Treatment of TA by electroporation with CPST or Cdk5DN 5 d previously caused a notable increase in SV2a immunolocalization at NMJ. Right: Immunoblots (top) of SV2a protein in homogenates of TA muscle from WT, mSCS, or pretreated mSCS. Quantitation (bottom) shows that SV2a was reduced in mSCS and mSCS treated with control pcDNA3 plasmid relative to WT, but SV2a levels in mSCS treated with CPST or Cdk5DN were significantly increased (n = 3). #Normalized to WT. Scale bar, 15 μm. *p < 0.05. B, Top: Cryosections of WT and mSCS muscle immunostained for Rab3a protein (green) and NMJ (TxR-αBT, red) show that Rab3a is colocalized with NMJ in WT muscle, but markedly reduced in mSCS muscle. Treatment of TA by electroporation with CPST or Cdk5DN for 5 d previously caused a marked increase in Rab3a immunolocalization at NMJ. Bottom: Immunoblots of Rab3a protein in homogenates of TA muscle from WT, mSCS, or pretreated WT or mSCS. Quantitation shows that Rab3a level was reduced in mSCS and mSCS treated with control pcDNA3 plasmid relative to WT, but Rab3a levels in mSCS treated with CPST or Cdk5DN, as well as siRNA for nNOS KD and the nNOS inhibitor 7-NI were significantly increased. #Normalized to WT (n = 3). Scale bar, 15 μm. *p < 0.05. Treatment of WT muscle with CPST and siRNA to nNOS KD also significantly increased Rab3a expression relative to WT. Electroporation of Cdk5DN in WT TA muscle had no effect on Rab3a expression (n = 3). Scale bar, 15 μm. *p < 0.05. C, Immunoblots (top) of Rab3a protein in homogenates of TA muscle from WT and nNOS KO muscle after treatment with control plasmid or with nNOS expression plasmid. Quantitation (bottom) shows that the Rab3a protein level was higher in nNOS KO TA muscle compared with WT. Five days after electroporation of nNOS plasmid into nNOS KO, the Rab3a level was significantly reduced relative to that of nNOS KO. After expression of nNOS in WT muscle, the level of Rab3a was significantly less than in WT levels. #Normalized to WT (n = 3). *p < 0.05. D, Immunoblots (top) of Rab3a protein in homogenates of TA muscle from untreated WT and after systemic treatment saline or αBT. Thirty and 60 μg of sample were loaded into separate gels. Quantification (bottom) shows that saline treatment did not affect Rab3a protein level, but pretreatment with αBT led to a significant increase in Rab3a level (n = 3). #Normalized to WT. **p < 0.05, Mann–Whitney U test.
The Rab3a detected in muscle presumably originates from the intramuscular peripheral nerves. To confirm this, we used qRT-PCR to compare Rab3a mRNA levels in normal TA muscle and denervated TA muscle. The Rab3a mRNA level in 3-day denervated muscle was reduced to 10% of normal muscle, which confirmed its neural origin (Fig. 7A, left). Rab3a mRNA was also more abundant in the SN and muscle of WT mice compared with mSCS muscle, although there were no differences between WT and mSCS in SC. To investigate whether the changes of Rab3a at mRNA level were accompanied by alterations at the protein level, we compared the Rab3a protein expression in TA muscle, SN, and SC in WT and SCS mice. We found that relative expression of Rab3a protein (vs GAPDH) in the TA muscle in both WT and SCS mice was ∼2-fold higher than in SC and 7-fold higher than in SN (Fig. 7A, right). Consistent with the changes in Rab3a mRNA in muscle, the Rab3a protein level in mSCS TA muscle was significantly lower than in WT TA muscle. Interestingly, Rab3a protein levels in SN and SC showed no difference between WT and SCS mice. These results suggest that the differences in Rab3a protein levels between mSCS and WT muscles result from gene products arising from motor nerves, as opposed to either cross-reacting proteins or aberrant muscle transcripts. More importantly, these findings are consistent with the view that reduced Rab3a in SCS mice results from an effect on mRNA stability and protein synthesis limited to the nerve terminus. Finally, we investigated whether the changes in the calpain-nNOS pathway that influence neuromuscular transmission presynaptically in mSCS and WT muscle act through an effect on Rab3a mRNA levels in SN. Figure 7B shows that there are significant differences in the abundance of a 168 bp amplimer corresponding to Rab3a after the pretreatments that modulate the calpain-Cdk5-nNOS pathway (as shown in Fig. 7C), as compared by semi-qRT-PCR and qRT-PCR. In mSCS muscle, CPST, Cdk5DN, and 7-NI significantly increased Rab3a mRNA levels, whereas pcDNA3 and saline injection had no effect. Expression of CPST in WT muscle significantly increased Rab3a mRNA levels. The increase after 7-NI treatment was not statistically significant. These results suggest that calpain, Cdk5, and nNOS activities in skeletal muscle negatively regulate presynaptic quantal activity through an effect on the mRNA levels of at least one presynaptic protein, Rab3a.

The calpain-Cdk5-nNOS pathway influences Rab3a mRNA expression in SN. A, Left: Rab3a mRNA levels in TA muscle (TA), 3-d-denervated TA muscle (Dv), SN, and SC of WT and mSCS mice. #Normalized to untreated WT TA muscle. After 3 d denervation, the Rab3a mRNA level in denervated WT TA muscle (WT-Dv) was significantly reduced. In WT or mSCS mice, the Rab3a mRNA level was higher in SN and SC but lower in TA (n = 5). **p < 0.01. Right: Immunoblots (top) of Rab3a protein from homogenates of TA, SN, and SC. Quantification (bottom) in the WT or mSCS mice showed that the relative Rab3a protein level in SN and SC was significantly lower in SN and SC than in TA, #Normalized to untreated WT TA muscle (n = 3). *p < 0.05, Mann–Whitney U test. B, Top: Rab3a mRNA levels in SN by semi-qRT-PCR. Rab3a PCR product (168 bp) was amplified from SN in WT or mSCS mice with different treatments. Bottom: #Values expressed as a ratio of GAPDH signal normalized to untreated WT SN. CPST increased Rab3a mRNA in both WT and mSCS. Cdk5DN and the nNOS inhibitor 7-NI increased Rab3a in mSCS. 7-NI had no effect on WT. Saline had no effect in mSCS (n = 5). *p < 0.05. C, Measurement of Rab3a mRNA level in SN by qRT-PCR showed similar results as in B. #Normalized to untreated WT SN (n = 5). **p < 0.01, Mann–Whitney U test.
Neural miRNAs control calpain/Cdk5/nNOS-dependent alterations in synaptic vesicle protein levels in normal and mSCS muscles
Changes in Rab3a mRNA levels in peripheral nerve without changes in SC (Fig. 7A) suggest that the SCS mice have altered mRNA stability within peripheral nerve, which may be modulated in WT and SCS mice through the calpain-Cdk5-nNOS pathway. miRNAs, small, noncoding RNA segments (20–24 bp) function by binding with 3′-UTR of target mRNA, reducing mRNA stability, leading to mRNA degradation (Nielsen et al., 2009). miRNA-mediated mRNA and protein changes have been widely reported in injured SN and SC studies (Strickland et al., 2011; Yu et al., 2011) and in presynaptic and postsynaptic regulation of homeostatic synaptic plasticity in the CNS (Cohen et al., 2011). To test for a possible miRNA-mediated mechanism, we selected seven candidate miRNAs using the following rationale: miR-1a, miR-133a, and miR-206 principally regulate muscle differentiation and are expressed in both murine skeletal muscle and neural tissue (Chen et al., 2006; Kim et al., 2006; Bak et al., 2008). miR-124, miR-142, miR-370, and miR-486 are primarily identified in murine neural tissue (Bak et al., 2008) and are predicted to act on Rab3a 3′-UTR based on miRNA-specified target-finding engine (Griffiths-Jones et al., 2006; Kozomara and Griffiths-Jones, 2011).
We used qRT-PCR to investigate whether the levels of any of these miRNAs are affected in WT or SCS mice. Figure 8A shows the relative miRNA expression levels of the seven miRNAs in SN. Of the muscle-specific miRNAs, miR-1, miR-133a, and miR-206 (Fig. 8A, top), only miR-206 was significantly reduced in mSCS SN (to 75% of WT, p < 0.01). However, miR-206 was not increased by CPST, Cdk5DN, 7-NI, or control treatments in either mSCS or WT mice. In contrast, of the four predicted nerve-specific miRNAs, miR-124, miR-142, miR-370, and miR-486 (Fig. 8A, bottom), only miR-124 was significantly increased in mSCS SN compared with WT SN (nearly 2-fold, p < 0.01). Pretreatment of SCS mice by electroporation of CPST or Cdk5DN reduced the miR-124 level to 47% and 51% of WT, respectively, whereas no change was found in control plasmid (mSCS-pcDNA3, 1.6-fold, p < 0.01). Pretreatment of SCS mice with 7-NI reduced the miR-124 level in SN to 46% of WT, whereas no change was identified in saline control treatment (1.7-fold; p < 0.01). Pretreatment of WT mice with CPST reduced miR-124 level to 15% of untreated WT, whereas treatment with 7-NI reduced miR-124 to 17% of WT SN (p < 0.01). Moreover, the miR-124 level in SN of nNOS KO mice was reduced to 53% of WT, whereas electroporation of nNOS plasmid to nNOS KO mice increased the miR-124 level in SN 1.3-fold over WT. Reduced expression of miR-124 in SN was correlated closely with the increase of Rab3a expression and the increased quantal activity in WT and mSCS muscle pretreated with CPST, Cdk5DN, or siRNA for nNOS. Interestingly, although the level of miR-142 expression was no different between WT and mSCS SN, miR-142 showed effects similar to those of miR-124 in SN after these same pretreatments to mSCS and WT TA muscle. Furthermore, the level of miR-142 in SN of nNOS KO was reduced to 54% of WT (p < 0.05), whereas electroporation of nNOS plasmid to nNOS KO mice increased the miR-142 level in SN to 82% of WT (p < 0.05; Fig. 8B). There were no differences in the levels of the other miRNAs in untreated or pretreated WT and SCS mice.

Calpain-Cdk5-nNOS pathway modulates Rab3a expression through miR-124 and 142 in SN. A, Expression levels of seven miRNAs in WT and mSCS SN with different treatments measured by qRT-PCR. Top: miR-1a, miR-133a, and miR-206 expression levels in WT, mSCS, and treated mSCS mice. The levels of miR-1, miR-133a, and miR-206 in SN showed no significant differences between WT and mSCS mice or before and after treatments. Bottom: miR-124, miR-142, miR-370, and miR-486 expression levels in WT, SCS, and treated SCS mice. The miR-124 expression level in SN was significantly higher in mSCS than in WT mice. Treatment of mSCS TA by electroporation of CPST or Cdk5DN expression plasmids or by systemic administration of 7-NI caused a significant reduction of miR-124 levels. Treatment of WT mouse TA with CPST expression plasmid or by systemic administration of 7-NI caused a significant reduction of miR-124 levels. Treatment of mSCS or WT TA with pcDNA3 or with saline injection had no effect on miR-124 expression. Levels of miR-142 expression were no different between WT and mSCS mice. However, inhibition of the calpain-Cdk5-nNOS pathway in mSCS had similar effects on miR-142. Treatment of mSCS mouse TA by electroporation of CPST or Cdk5DN expression plasmids or by systemic administration of 7-NI caused a significant reduction of miR-142 levels. Treatment of WT mouse TA with CPST expression plasmid or by systemic administration of 7-NI caused a significant reduction of miR-142 levels. Treatment of mSCS or WT TA with pcDNA3 or with saline injection had no effect on miR-142 expression. #Normalized to untreated WT SN (n = 5). **p < 0.01, Mann–Whitney U test. B, miR-124 and miR-142 expression levels in SN in WT, nNOS KO, and nNOS KO mice treated by electroporation of nNOS in TA 5 d previously. #Normalized to untreated WT SN. Expression levels of both miR-124 and miR-142 in SN were significantly lower in nNOS KO mouse than in WT mouse SN. Expression of nNOS in nNOS KO TA muscle significantly increased the levels of miR-124 and miR-142 expression (n = 3). *p < 0.05, Mann–Whitney U test. C, Rab3a 3′-UTR-directed luciferase activity reveals modulation by miR-124 and miRNA-142, but not their mutant variants. A luciferase expression construct bearing Rab3a 3′-UTR was coexpressed with expression plasmids for miRNAs. Coexpression of both miR-124 and miR-142 caused significant reduction of luciferase activity. Coexpression of miR-124mut or miR-142mut or the controls, miR-206 and miR-LacZ, had no effect on luciferase activity (n = 10). #Normalized to luciferase activity of Rab3a 3′-UTR only transfection. ***p < 0.001, Student's t test. D, miRNA modulation of endogenous expression of Rab3a. miR-124, miR-124mut, miR-142, and miR-142mut were transfected separately in the neuroblastoma cell line Neuro-2a, which expresses Rab3a. Expression levels of Rab3a protein in each culture were compared by immunoblot (top) after 5 d. Rab3a protein levels (bottom) were significantly reduced in Neuro-2a cells transfected with miR-124 or miR-142 compared with untreated Rab3a protein level. Mutant miRNAs, miR-124mut, or miR-142mut had no effect. #Normalized to GAPDH (n = 3). *p < 0.05, Mann–Whitney U test.
Finally, to test for a direct interaction of miR-124, miR-142, and miR-206 with the Rab3a 3′-UTR, we cotransfected each miRNA separately with a luciferase-based miRNA reporter containing 3′-UTR-Rab3a in HEK293 and measured luciferase activity (Greco and Rameshwar, 2007). Both miR-124 and miR-142 significantly reduced the relative luciferase activity to 0.47 ± 0.11 and 0.52 ± 0.06 of the original value, respectively, in cells expressing miRNA reporter bearing a 3′-UTR-Rab3a (n = 10, p < 0.001; Fig. 8C). Conversely, when cells were cotransfected with reporter constructs bearing mutant miR-124 (miR-124mut) and mutant miR-142 (miR-142mut) (see Materials and Methods for sequences) in place of WT reporters, luciferase levels were not changed (1.04 ± 0.09 and 0.95 ± 0.10 of control, respectively, p > 0.98; Fig. 8C). miR-206 did not influence luciferase activity of 3′-UTR-Rab3a (0.96 ± 0.08, p > 0.98; Fig. 8C). miRNA targeted to the β-galactosidase 3′-UTR had no effect on luciferase activity of 3′-UTR-Rab3a (1.04 ± 0.13, p > 0.98).
To investigate the role of miRNA in the modulation of endogenous levels of Rab3a, we used the neuroblastoma cell line neuro-2a (Nishimura et al., 1995). After a 5 d transfection of neuro-2a with the plasmids that express miR-124 and miR-142, Rab3a protein was significantly reduced relative to GAPDH (0.56 ± 0.12, untransfected, vs 0.15 ± 0.08, miR-142, and 0.22 ± 0.06, miR-124, p < 0.05; Fig. 8D). However, expression of the mutant forms of miR-124 and miR-142 in neuro-2a cells had no effect on Rab3a protein expression (0.53 ± 0.13, miR-124mut and 0.56 ± 0.1, miR-142mut, p > 0.97; Fig. 8D). These results suggest that muscle activities of calpain, Cdk5, and nNOS influence presynaptic quantal release through miRNA-based modulation of Rab3a mRNA stability that controls Rab3a expression levels.
Discussion
In this study, we have shown that muscle calpain activity plays a prominent role as an upstream trigger to couple increases in junctional sarcoplasmic Ca2+ levels to modulation of muscle nNOS activity and presynaptic neurotransmitter release through an miRNA mechanism. This feedback mechanism, which proceeds through a protein kinase cascade, appears to be present both in normal and pathological states. Figure 9 summarizes the steps underlying this nNOS feedback pathway in the NMJ.

Summary schema of the calpain-Cdk5-nNOS retrograde pathway in NMJ illustrating that in SCS diseased muscle, mutant AChR-mediated Ca2+ overload activate calpain and amplify Cdk5 activity via calpain-dependent cleavage of p35 to p25. Increased Cdk5 activity reduces CaMKII activity by decreasing CaMKII phosphorylation at Thr286. Decreased CaMKII activity reduces the ratio of nNOS phosphorylation at Ser 847 to Ser 1412, which finally amplifies NO production in SCS muscle. More NO arrival at motor neurons increases the cGMP level, which is likely responsible for increased miRNA 124 and miRNA 142 expression that directly reduces the Rab3a mRNA level and protein production in motor neuron terminals. Decreased Rab3a expression influences QC release at the endplate; however, inhibition of calpain-Cdk5-nNOS pathway by CPST, Cdk5DN, or 7-NI recoveres Rab3a expression and improves quantal release at motor neuron terminals.
Calpain is a cysteine protease whose substrates include several cytoskeletal proteins and signaling molecules, such as Cdk5 p35 coactivator (Lee et al., 2000). Calpain plays roles in cell cycle, cell migration and adhesion, axon guidance, NMJ formation, and synaptic plasticity (Robles et al., 2003; Jánossy et al., 2004; Perrin et al., 2006; Chen et al., 2007; Zadran et al., 2010) and has been strongly implicated in numerous cell death pathways. However, until this study, its perceived role in synaptic plasticity has been limited to influencing synaptic strength directly within the cell in which is activated. It has not been shown previously to act through CaMKII and nNOS to exert a retrograde effect on synaptic transmission potentially underlying homeostatic synaptic plasticity, nor in contributing to presynaptic depression in neuromuscular disease. Its role in degradation in the NMJ appears to be of secondary importance (Groshong et al., 2007). These observations may provide insights into new avenues for therapy for these disorders. If this pathway can be identified in CNS synapses, it could provide new insights into synaptic plasticity, learning, and memory.
Cdk5 is a proline-directed serine/threonine kinase that has numerous identified protein substrates in the CNS and in non-neuronal cells. It plays a central role in CNS development, function and disease, and is involved in a wide array of physiological processes, including neuronal migration, axon guidance, synaptic transmission, endocytosis, and synaptic plasticity (Nguyen and Bibb, 2003; Hirasawa et al., 2004; Hawasli et al., 2007; Poore et al., 2010). Until now, Cdk5 was only known to influence cellular behavior within its cell of origin (Nguyen and Bibb, 2003; Evans and Cousin, 2007). Beyond coupling Cdk5 activation to synaptic activity through calpain, this study broadens the scope of influence of Cdk5 to include a retrograde effect on the presynaptic neuron through its effect on NO production. This finding may have significant physiological and therapeutic implications, particularly if a comparable role can be identified in CNS synapses.
The role of NO generated by nNOS in neuromuscular transmission has been controversial, with some studies demonstrating enhancement and others suggesting suppression of transmitter release (Etherington and Everett, 2004; Nickels et al., 2007). Several studies have already suggested that NO, mediates a form of retrograde feedback in synaptic transmission (Lindgren and Laird, 1994; Wang et al., 1995; Etherington and Everett, 2004; Graves et al., 2004). The present study characterizes a pathway by which synaptic nNOS can be activated through excessive cholinergic activity after calpain activation and further implicates NO as a source of negative feedback. This study also suggests that nNOS activity plays a significant role in modulating synaptic strength in clinical disease. Excessive NO is also a source of oxidative damage in pathological states (Martin et al., 2005; Bayir et al., 2007). However, in this model, NO appears to play its primary role as a signaling molecule in both healthy and diseased NMJ. Localized targeting of nNOS in the NMJ may be a reasonable therapeutic strategy to improve synaptic transmission in disorders of cholinergic excess.
The role of miRNA in synapse development and function is well established (Manakov et al., 2009; Schratt, 2009; Smalheiser and Lugli, 2009; Impey et al., 2010). There is also a growing realization that miRNA plays a prominent role in neurological and muscular disease (Bicker and Schratt, 2008; Chen et al., 2009; Christensen and Schratt, 2009; Williams et al., 2009; Huang et al., 2011). However, until now, the pathways through which NO affects presynaptic function have not been shown to occur through changes in expression of presynaptic proteins via a miRNA mechanism. Yet to be determined is the mechanism by which changes in cGMP or other NO targets lead to changes in specific miRNA levels in the presynaptic motor neuron. The characterization of this cascade provides new insights into transsynaptic communication and provides a wide array of potential targets for therapeutic intervention.
Footnotes
This work was supported by the National Institutes of Health (Grant #RO1 NS33202 to C.M.G.). We thank Dr. Andrew G. Engel, Dr. Ricardo Maselli, and Dr. Rosana Scola for providing SCS and CEAD patient muscle samples and Dr. Engel and Dr. Maselli for helpful comments on the manuscript.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Christopher M. Gomez, Department of Neurology, AMB S237, MC2030, University of Chicago Medical Center, 5841 S. Maryland, Chicago, IL 60637. gomez001{at}uchicago.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}