

Abstract
Activation of the nerve growth factor (NGF) receptor trkA and tissue acidosis are critically linked to inflammation-associated nociceptor sensitization. This study explored how increased acidity is linked to sensory neuron sensitization to NGF. Adult Wistar rat primary sensory neurons grown at physiological pH 7.4, then either kept at pH 7.4 or challenged for 30 min in pH 6.5 medium, provided a model of acidosis. Nonpermeabilizing trkA immunofluorescence revealed a significant increase in trkA mobilization to the plasma membrane from intracellular stores in response to proton challenge. This was confirmed using a surface protein biotinylation assay and Brefeldin A disruption of the rough endoplasmic reticulum-Golgi-trans-Golgi network. Mobilization of trkA to the membrane at pH 6.5 was abolished in neurons treated with the acid-sensitive ion channel blocker, amiloride. While elevated levels of NGF-independent trkA phosphorylation occurred at pH 6.5 alone, the level of activation was significantly increased in response to NGF challenge. Exposure of sensory neurons to pH 6.5 medium also resulted in strong calcium (Ca2+) transients that were reversible upon reintroduction to physiological pH. The pH 6.5-induced mobilization of trkA to the membrane was Ca2+ dependent, as BAPTA-AM Ca2+ chelation abrogated the response. Interestingly, KCl-induced depolarization was sufficient to induce mobilization of trkA to the cell surface at pH 7.4, but did not augment the response to pH 6.5. In conclusion, increased mobilization of trkA to neuronal membranes in response to either acidosis or neuronal depolarization provides two novel mechanisms by which sensory neurons can rapidly sensitize to NGF and has important implications for inflammatory pain states.
Introduction
Alterations in the extracellular environment due to inflammation or tissue injury, have tremendous affects on sensory neuron peripheral sensitization (Nicol and Vasko, 2007). Key drivers of inflammatory pain states are increased expression of nerve growth factor (NGF) in regions of inflammation (Lewin et al., 1993, 1994; McMahon et al., 2006) and signaling via the NGF receptor trkA (Ji et al., 2002; Zhang et al., 2005), expressed by ∼40–45% of adult rat lumbar primary sensory neurons (Karchewski et al., 1999). Indeed, therapeutically targeting this axis has shown to be beneficial to resolution of pain states (McMahon et al., 1995; Koltzenburg et al., 1999; Winston et al., 2003; Jimenez-Andrade et al., 2007; Ugolini et al., 2007).
Tissue acidosis is another prominent and early contributor to pain states caused by the release of protons in response to many conditions including inflammation, injury, and ischemia (Reeh and Steen, 1996). Decreased extracellular pH initiates discharges in some nociceptors (Steen et al., 1995, 1996) due to activation of ion channels directly gated by protons and that include a family of acid sensing ion channels known as ASICs (Waldmann et al., 1997; Ugawa et al., 2002; Jones et al., 2004; Deval et al., 2008). ASICs can be activated by modest drops in pH, exhibiting a sustained current that does not inactivate while the pH remains acidic (Yagi et al., 2006). Further, the restriction of ASIC3 expression to a predominantly trkA-positive subpopulation (Molliver et al., 2005) and the ability of NGF to regulate its expression (Mamet et al., 2003) provides a longer term mechanism to increase the sensitivity of these neurons in inflammation-driven pathologies.
While NGF can regulate expression of ASICs, the impact acidosis has on rapidly altering responsiveness of neurons to NGF as part of the sensitization process has not been studied. Two investigations support that an acidic environment may alter trkA presentation on neuronal membranes. The first by Ross et al. (2001) reported that exposure of PC12 cells to a pH of 6.5 for 15 min increased the specific binding of NGF to PC12 cell membranes to 150% of that observed at control pH 7.4. The second exploits the potential for acidosis to alter neuronal activity and effect calcium transients in sensory neurons. The study by Du et al. (2000) links electrical stimulation and calcium influx to increased presentation of trkB on the surface of hippocampal neurons. Collectively, this led us to speculate that the increased proton-mediated activation of ASICs might alter calcium and membrane dynamics such that more trkA is rapidly mobilized to the membrane.
Thus, we hypothesized that modest drops in extracellular pH leading to calcium fluxes acts like a dynamic switch to rapidly mobilize trkA to the cell membrane surface of adult sensory neurons, which in turn serves to increase the sensitivity of these neurons to NGF. Our findings reveal a cellular mechanism whereby even small changes in pH can rapidly shift sensitivity to a critical driver of the inflammatory pain state—NGF.
Materials and Methods
Animals.
Male Wistar rats 2–3 months of age and weighing 200–300 g were used in all experiments (University of Saskatchewan ARC or Charles River Laboratories). All animal procedures were conducted in accordance with the policies of the Canadian Council on Animal Care and approved by the University of Saskatchewan Animal Care Committee.
Primary neuronal culture and treatments.
All experimental conditions examined were performed at minimum in triplicate from three separate experiments. Animals were killed by CO2 inhalation, followed by rapid dissection of dorsal root ganglia (DRGs) from all spinal levels and collection into L15 media (Invitrogen). The DRG were cleaned of connective tissue and dissociated by incubation at 37°C in 1 mg/ml collagenase (Sigma-Aldrich) for 90 min with trituration, followed by 0.1% trypsin (Sigma-Aldrich) for 30 min. Horse serum was added to quench the trypsin digest. Cells were spun down, resuspended in DMEM, and filtered through a 100 μm mesh filter. The cell suspension was further purified by placing on the top of a 15% bovine serum albumin (BSA) solution, and then spun at 850 rpm, 20 min, at 4°C. The neurons were counted and plated at a density of 5000–7000 neurons per 22 × 22 mm coverslip coated with poly-l-lysine (25 μg/ml) (Sigma-Aldrich) and laminin (10 μg/ml) (Sigma-Aldrich). The cells were cultured for 2 d in DMEM containing 10 ng/ml 2.5S NGF (Cedarlane Laboratories).
Isolation of trkA-positive neurons.
The CELLection Dynabead technique was used to isolate the trkA-positive subpopulation of sensory neurons. Briefly, magnetic Dynabeads (Dynal Biotech) were incubated with monoclonal trkA antibody (Alexis Biochemicals) at a concentration of 0.4 μg of antibody per 100 μl Dynabead suspension containing 1 × 107 beads for 30 min at 4°C in a microtube. The tube was placed in a Dynal MPC magnet (Dynal Biotech) for 2 min and the supernatant was discarded. The anti-trkA-coated beads were further washed and resuspended to 4× the original volume with 10 mm PBS. The dissociated DRG neurons were incubated with the anti-trkA-coated Dynal beads for 1 h with gentle gyroscopic mixing. To isolate trkA-expressing neurons, the tube was placed in the Dynal MPC magnet for 2 min and the supernatant, containing the trkA-negative cells, was removed. The isolated trkA-positive cells and magnetic beads were dissociated from one another using the DNase releasing buffer (Dynal Biotech). Isolated trkA-positive cells were then resuspended in DMEM and seeded onto coated coverslips as previously described.
In vitro acidosis.
Physiological control medium, pH 7.4, was prepared with DMEM with HEPES buffer as needed. Acidic media of pH 5.5 or 6.5 were prepared with 25 mm HEPES and MES buffer as needed. Before pH shift, cells were changed to DMEM medium without NGF, washed with 37°C DMEM containing 1.5 μg/ml sheep-anti-NGF (Cedarlane Laboratories) 2× 15 min, then once with DMEM alone to block biological responses due to residual exogenous NGF. Cells were then exposed to either an acidic pH of 5.5, 6.5, or a control pH of 7.4 with or without a variety of treatments as described below. Cells were incubated at 37°C in the selected pH media for 15 min, 30 min, 1 h, or 2 h. Initial experiments revealed that exposure to medium at pH 5.5 resulted in highly compromised cells showing evidence of leaky membranes that were not evident at pH 6.5, therefore all subsequent experiments were performed with the latter pH drop. Initially the pH of the medium was tested at the end of the experiment to ensure that the pH of the medium was not altered; no change was observed.
Treatments.
To inhibit the trafficking of trkA from the Golgi to the cell membrane, 2 d cultured DRG neurons were washed with DMEM twice, incubated with DMEM containing anti-NGF (1.5 μg/ml) for 30 min, and changed to DMEM with or without Brefeldin A (BFA; Calbiochem; 5 μg/ml) 2 × 25 min. Then the media were replaced with a pH 6.5 DMEM or pH 7.4 DMEM with or without BFA for 30 min, followed by immediate processing for trkA immunohistochemistry under nonpermeabilizing conditions.
To examine whether BFA treatment was inducing cell membrane internalization at pH 6.5 (which would confound interpretation of the results), 2 d DRG neuronal cultures were washed with DMEM containing function-blocking sheep anti-NGF (Cedarlane Laboratories; 1.5 μg/ml) 2 × 15 min, followed by incubation with the membrane FM1–43FX (Invitrogen) for 5 min on ice. Cells were then subjected to one of the following four experimental conditions: (1) DMEM pH 7.4, (2) DMEM pH 6.5, (3) DMEM pH 6.5 + BFA (5 μg/ml), or (4) DMEM pH 6.5 + NGF (50 ng/ml; a condition known to induce trkA receptor internalization; Beattie et al., 2000) for 15 min at 37°C. Following the 30 min exposure to acidic or control pH as above, cells were rinsed once with cold PBS and fixed with cold 2% paraformaldehyde (PFA) for 30 min. The FM 1–43FX signals were immediately captured using a Zeiss Axioskop fluorescence microscope and quantified using Northern Eclipse software. FM 1–43FX is nontoxic to cells and virtually nonfluorescent in aqueous medium, but when internalized and trapped inside endocytotic vesicles they fluoresce intensely (Griesinger et al., 2002; Life Technologies, Invitrogen).
To antagonize proton-activation of ASICs, 2 d cultured DRG neurons were washed with DMEM twice, incubated with DMEM containing anti-NGF (1.5 μg/ml) for 30 min, and then changed to DMEM. The cells were switched to pH 6.5- or pH 7.4-specific media with or without 250 μm amiloride (Sigma-Aldrich) for the duration of the 30 min pH challenge. Cells were then processed immediately for trkA immunohistochemistry under nonpermeabilizing conditions.
To chelate available Ca2+, 2 d cultured DRG neurons were washed 2× with DMEM and incubated in DMEM containing anti-NGF (1.5 μg/ml) and BAPTA-AM (50 μm in 0.2% dimethylsulfoxide; DMSO) or DMSO (0.2%) for 30 min. Then the media were changed to pH 6.5 and 7.4 with either BAPTA-AM (50 μm) or DMSO (0.2%). After 30 min., the cells were processed for trkA immunohistochemistry under nonpermeabilizing conditions.
KCl was used to depolarize the sensory neurons. Two day cultured DRG neurons were washed twice with DMEM and incubated in DMEM containing anti-NGF (1.5 μg/ml) and switched to DMEM. The cells were then switched to pH 6.5- or pH 7.4-specific media with or without KCl of 15 or 45 mm for the 30 min pH challenge, then processed for trkA immunohistochemistry under nonpermeabilizing conditions.
NGF challenge.
The responsiveness of the increased pool of trkA receptors mobilized to the plasma membrane, to NGF challenge (trkAY490 phosphorylation), was assessed by exposing the cells to DMEM with or without 50 ng/ml 2.5S NGF (Cedarlane Laboratories) for 15 min following the 30 min exposure to acidic or control pH as above. To eliminate potential contributions of brain-derived neurotrophic factor (BDNF; expressed by trkA neurons) on this response to NGF challenge, either via modifying the affinity of NGF for trkA (MacPhee and Barker, 1997) or acting on the 8% of sensory neurons that also express trkB (Karchewski et al., 1999), as Y490 is common to all trk receptors, function-blocking anti-BDNF (Millipore Bioscience Research Reagentsl 1.5 μg/ml) was added to the media for the 30 min pH shift before and during the 15 min NGF challenge.
The level of constitutive trkAY490 phosphorylation is altered with pH shift alone in the absence of NGF challenge. To eliminate possible contributions by endogenous BDNF or NGF to this response, acidosis experiments were repeated with function-blocking sheep anti-NGF (Cedarlane Laboratories; 1.5 μg/ml) and the function-blocking sheep anti-BDNF (1.5 μg/ml) in the media used in the 30 min acidosis experiment.
At the end of the experiment, the cultures were fixed and processed for phosphoY490-trkA immunohistochemistry under permeabilizing conditions as described below.
Surface protein biotinylation.
Cell-surface proteins were biotinylated and then isolated using the Pierce Cell Surface Protein Isolation Kit (Thermo Fisher Scientific). DRGs from 12 rats (representing three separate experiments) were harvested and dissociated as above and grown for 2 d on laminin (10 μg/ml) and poly-l-lysine (25 μg/ml)-coated 60 mm Petri dishes in DMEM containing 10 ng/ml 2.5S NGF.
The 2 d in vitro (DIV) cells were subjected to a shift in pH for 30 min (as described previously), washed with cold 10 mm PBS, and incubated with 0.5 mg/ml Sulfo-NHS-SS-Biotin in PBS with gentle rocking for 30 min. Quenching Solution was added to the cells to stop the reaction, then cells were centrifuged at 500 × g at 4°C for 3 min and resuspended in 25 mm Tris-buffered saline. After centrifugation, the cells were lysed using a combination of Lysis Buffer (Pierce Biotechnology) containing 1× protease inhibitor mixture (Sigma-Aldrich), trituration by repeated pipetting and sonication (Vibra-Cell; Sonics and Materials) five times for 1 s. After 30 min with rotation the cell lysate was then centrifuged at 10,000 × g for 3 min at 4°C and the supernatant collected for the experiment.
Proteins were loaded into 100 μl of NeutrAvidin gel resin included in the protein isolation kit and incubated for 1 h at room temperature with end over end mixing followed by centrifugation at 1000 × g with the elutant representing cytoplasmic proteins. The gel slurry and attached membrane proteins were washed three times with 100 μl of wash buffer containing 1× protease inhibitor mixture. Bound proteins were released by incubating the resin with 100 μl SDS sample buffer containing 50 mm dithiothreitol for 1 h at room temperature. The column was centrifuged 1000 × g for 3 min. Total protein was measured using the Bradford method (Bradford, 1976).
SDS-PAGE and Western blot analysis.
Equal volumes of biotinylated cell-surface proteins were loaded onto an 8% SDS-PAGE gel and run at 100 V. Similarly, equal volumes of cytoplasmic proteins were also loaded onto an 8% SDS-PAGE gel and run at 100 V. Electrophoresed proteins were transferred onto a nitrocellulose membrane and blocked overnight at 4°C with 5% milk (Bio-Rad) in 10 mm PBS containing 0.05% Tween 20 (PBST). The primary antibodies (anti-rabbit trkA; 1:2000 Alexis Biochemicals; or anti-mouse GAPDH 1:2000, Novus Biologicals) were then applied at room temperature for 1 h in 5% milk in 10 mm PBST. After 1 h, the blot was washed 3 × 5 min with PBST and secondary antibodies goat anti-mouse IRDye 680 and sheep anti-rabbit IRDye 800 (1:10,000; LI-COR) applied in the dark for 30 min at room temperature. The blot was washed again 3 × 5 min in PBST and then visualized with a LI-COR Odyssey infrared imager (LI-COR).
Immunohistochemistry.
Before processing for immunohistochemistry, cells were fixed with 1% PFA in 10 mm PBS for 30 min at room temperature (nonpermeabilizing conditions) or with methanol for 20 min at −20°C (permeabilizing conditions). Cells were washed with PBS (nonpermeabilizing) or PBST (0.05% Tween 20) (permeabilizing) for 15 min., then blocked in a solution containing 5% horse serum and 0.1% BSA in 10 mm PBS with (permeabilizing) or without (nonpermeabilizing) 0.05% Tween 20 at 4°C overnight. Primary antibodies included a rabbit polyclonal anti-trkA (1:2000; Alexis Biochemicals), a rabbit polyclonal anti-phospho-p38MAPK (1:50; Cell Signaling Technology), and a rabbit polyclonal anti-phospho-trkA (1:100; Neuromics) diluted in blocking solution, and incubated at room temperature (trkA) or 4°C (phospho-trkA and phospho p38MAPK) for 24 h. The anti-trkA antibody recognizes the extracellular fragment of the trkA protein at amino acids 1–416, the phospho-trkA antibody recognizes the intracellular phosphorylated Y490 residue, while the phospho-p38MAPK recognizes p38MAPK phosphorylated at T180 and Y182 residues. Other control experiments ascertained the effectiveness of the permeabilizing versus nonpermeabilizing procedures by processing sample coverslips from each procedure with an antibody directed against the cytoskeletal protein βIII-tubulin (1:1000; Millipore) diluted in PBS. After exposure to the primary antibodies, cells were washed in PBS with (permeabilizing) or without (nonpermeabilizing) 0.05% Tween 20. The secondary antibody, a donkey anti rabbit Cy3 (1:500; Jackson ImmunoResearch), recognizing the Fab2 fragment, diluted in 10 mm PBS, 5% horse serum, and 0.1% BSA was applied for 30 min at room temperature, followed by washes in 10 mm PBS. Immunostained coverslips were mounted onto slides using ProLong Gold with DAPI nuclear stain (Invitrogen Inc).
Preparation of NGF and 125I-NGF binding.
β-NGF (2.5S NGF) was purified from mouse submandibular glands by cation-exchange chromatography (Mobley et al., 1976; Chapman et al., 1981) and radio-iodinated by the lactoperoxidase method (Sutter et al., 1979) with final separation of radioligand from free iodine and lactoperoxidase (Richardson et al., 1988) on a cartridge (Accell CM; Waters). 125I-NGF with a specific activity of 80–200 μCi/μg was used within 24 h of preparation.
Radioautographic studies.
Naive rats were perfused via the aorta with cold 0.1 m PBS, pH 7.4, and the fifth lumbar (L5) DRG were removed, placed in a cryomold, and quickly frozen in cooled isopentane. Sections 6–10 μm thick were thaw mounted on gelatin-coated slides and stored l–3 d at −80°C. They were then incubated for 90 min at 20°C with 20 pm 125I-NGF in 0.1 m PBS, pH 7.4, with magnesium chloride (0.5 mm), cytochrome C (1 mg/ml), leupeptin (4 μg/ml), and phenylmethylsulfonyl fluoride (0.5 mm) (Richardson et al., 1986). Following incubation with 125I-NGF, the sections were rinsed for 3 min in several changes of cold PBS, fixed by immersion for 10 min in 2% PFA and 2% glutaraldehyde, rinsed briefly in distilled water, and dried with cool filtered air. Sections were defatted, dipped in radiosensitive emulsion (Kodak NTB2), exposed at 4°C in lightproof boxes for l–6 d, developed, and counterstained with thionin.
Analysis and quantification of immunohistochemical signal.
Images of the immunostained cells were captured under identical parameters for control and experimental conditions processed at the same time, using a 40× oil-immersion lens on a Zeiss Axioskop fluorescence microscope with a Qimaging Retiga EXI Fast 1394 camera. Neuronal images were quantified for cell diameter and average fluorescence signal using Northern Eclipse software (Empix Imaging). For each experiment, the exposure, gain, and offset were kept constant when capturing images. For each coverslip, cells with detectable immunohistochemical signal above the background levels observed when the primary antibody was omitted, regardless of cell size, were photographed and analyzed. Five coverslips were analyzed per condition per trial and ∼200 cells/trial with immunofluorescence signal above background levels per condition were captured from slides that had coverslips from both pH conditions on them. The exposure threshold was set as the level where the no primary control slide no longer had detectable immunofluorescence signal. Care was taken to alternate which coverslip the analysis was begun on each slide so as to minimize bias associated with potential fading of the signal due to exposure when capturing the digital images. Slides were also analyzed qualitatively in a blinded fashion by V.M.K. and the qualitative and quantitative results compared. There was excellent and consistent agreement between the two forms of analyzes. The data were normalized to the mean signal intensity of the pH 7.4 neurons of the respective trial and then pooled from a minimum of three trials.
To determine whether changes in the immunofluorescence signal were significant, a nonparametric U test was used between groups (Mann–Whitney) since it could not be assumed that our data followed a Gaussian distribution or in the case where multiple datasets were compared a one-way ANOVA with post hoc nonparametric Tukey's tests was performed. All statistical calculations were performed using Prism 5.0 (GraphPad Software).
Ca2+ imaging.
Before Ca2+ imaging and pH shift, cells cultured for 2 DIV as above were changed to DMEM medium without NGF, washed twice with 37°C DMEM containing 1.5 μg/ml sheep-anti-NGF (Cedarlane Laboratories), and then once with DMEM alone to block biological responses due to residual exogenous NGF. Sensory neurons were then incubated in the membrane-permeant high-affinity Ca2+ indicator Rhod-2 AM (20 μm; Invitrogen) for 20 min. The solution was made fresh daily in DMEM containing 0.5% DMSO. DRG neurons were removed from the Ca2+ indicator solution and placed in a microscope chamber and perfused with DMEM, gravity fed at a flow rate of 3 ml/min, and maintained at 35 ± 0.5°C with an inline heater (Warner Instruments). DRG neurons were left to rinse in the microscope chamber for 20 min to remove residual Ca2+ indicator before being exposed to pH-specific culture medium (pH 7.4 or pH 6.5).
Ca2+ fluxes were visualized using an X-Cite 120PC system (EXFO Electro-Optical Engineering) light source (12% intensity) directly coupled to an Olympus BX51WIF upright research microscope by a 3 m LLG. The light intensity was attenuated with two neutral density filters in series (Olympus N.D.25 and N.D.6) and shutter controlled with a ProScan II Controller (Prior Scientific). The illumination and fluorescence light was filtered using a TRITC filter set (41002 Olympus BX2 mounted–HQ535/50× HQ610/75 m Q565LP) and detected with two high-sensitivity cooled CCD cameras. A 16-bit, 512 × 512 ImagEM EM CCD Camera (C9100–13; Hamamatsu) cooled to −65°C and a 16-bit, 1344 × 1024 ORCA-R2 CCD camera (C10600–10B; Hamamatsu) cooled to −35°C. Images were acquired using variable frame rates, binning, and defined regions of interest. A total of 15 fields were analyzed over the course of four separate experiments.
Results
Intracellular pools of NGF receptors and impact of extracellular pH on membrane integrity
To assess the mobilization of the NGF receptor trkA to the membrane of sensory neurons in response to extracellular stimuli, certain criteria need to be satisfied. First, there must exist an intracellular pool of receptors ready to be inserted into the plasma membrane. High-affinity NGF receptor binding radioautography or trkA immunohistochemistry on tissue sections of lumbar DRG revealed a large intracellular pool of NGF receptors (Fig. 1), consistent with previous findings (Verge et al., 1989, 1992; Averill et al., 1995). Second, if the impact of extracellular acidosis on the mobilization of trkA to the neuronal membrane is to be examined, it must be ascertained that the cell membrane will not be compromised by exposure to the acidic conditions used. This is to assure that the trkA immunofluorescence signal being quantified is derived only from cell surface-associated trkA protein and not internal sources of trkA. Thus, control experiments examining the impact of acidosis on neuronal membrane integrity determined whether the intracellular neuronal cytoskeletal protein βIII-tubulin (Avwenagha et al., 2003) could be detected when a nonpermeabilizing immunofluorescence approach was used following a shift in medium from neutral to acidic pH for 30 min. Results revealed that when the pH was shifted from 7.4 to 6.5, βIII-tubulin was not detectable using the nonpermeabilizing immunohistochemical approach used in these studies (data not shown). Indeed, βIII-tubulin could only be visualized immunohistochemically when the membrane had been deliberately permeabilized by methanol fixation and the addition of Tween 20 in the wash buffer. However, because pH levels as low as 5.3–5.7 have been observed in response to an inflamed environment (Jacobus et al., 1977; Steen et al., 1995; 1996; Andersson et al., 1999), a 30 min pH shift from 7.4 to 5.5 was also examined. Exposing the cells to pH 5.5 did compromise the integrity of the cell membrane (i.e., βIII-tubulin immunofluorescence signal was detected using nonpermeabilizing immunohistochemistry; data not shown) and thus this pH was eliminated from the experimental design.

NGF-responsive adult sensory neurons have a large cytoplasmic pool of high-affinity NGF receptors. Six micrometer cryostat sections of L5 DRG processed for high-affinity NGF binding (left) or trkA immunohistochemistry (right) reveal a large cytoplasmic pool of proteins that are able to bind 20 pm radio-iodinated NGF with high affinity or that are immunoreactive to trkA-selective antibodies. Scale bar, 20 μm.
Decreased pH increases mobilization of trkA to the neuronal surface
To assess whether moderate drops in extracellular pH (such as with nerve injury or inflammation) directly alter presentation of trkA on the membrane, adult sensory neurons were first cultured for 2 d in the presence of low levels of NGF (to maintain expression of trkA; Verge et al., 1992). Then, just before pH shift, cells were changed to medium without NGF, washed twice with medium containing 1.5 μg/ml sheep-anti-NGF, and then once with DMEM alone, to block biological responses due to residual exogenous NGF, which can promote trkA insertion by transcytosis into membranes in sympathetic neurons (Ascaño et al., 2009). Cells were then exposed to media at either an acidic pH of 6.5 or a control pH of 7.4. Neurons exposed to the acidic medium displayed an elevated level of trkA immunofluorescence on the extracellular surface across all size ranges of neurons, when compared with the control neurons incubated in the medium at a pH of 7.4 (Fig. 2). The increased mobilization of trkA to the cell surface was visible over both the cell bodies and the neurites. Quantitative analysis revealed a statistically significant difference in the detectable levels of cell-surface trkA immunofluorescence signal between the pH 7.4 and 6.5 experimental groups (p < 0.001; Fig. 2). To assess whether removal of NGF by anti-NGF treatment altered the level of trkA presentation on neuronal membranes, neurons at pH 7.4 were exposed to anti-NGF treatment for 0 min, 30 min, or 60 min followed by assessment of cell-surface trkA expression using immunofluorescence approaches. The absence of NGF and presence of anti-NGF did not significantly alter the mean level of trkA detected on the cell surface (as normalized to the 0 min control group) at pH 7.4 (0 min, 1 ± 0.041; 30 min, 0.9760 ± 0.03741; 60 min, 0.9908 ± 0.036; from three separate experiments with 120 neurons analyzed per condition). Thus, the removal of NGF from the medium before pH shift does not appear to alter the presentation of trkA on the neuronal membrane.

Exposure of sensory neurons to an acidic environment results in mobilization of trkA to the membrane. A, B, Fluorescence photomicrographs depicting the relative level of membrane-associated trkA expression in representative adult DRG neurons exposed to control (pH 7.4, A) or acidic (pH 6.5, B) media for 30 min, then processed to detect trkA under nonpermeabilizing conditions. Scale bar, 20 μm. C, Scatter plot depicts the relationship between trkA immunofluorescence signal labeling intensity (y-axis; normalized to the mean signal intensity from the pH 7.4 neurons; open circles, pH 6.5; closed circles, pH 7.4) and perikaryal diameter (x-axis) for all neurons analyzed from three separate experiments. D, Summary bar graph of data (from C) depicts relative mean levels of membrane-associated trkA on adult DRG neurons exposed to acidic (pH 6.5) or control (pH 7.4) media for 30 min. Note the elevated level of trkA detected under acidic conditions, most notable in small to medium size neurons. Asterisks indicate significant differences between experimental groups (Mann–Whitney U test; ***p < 0.001; pH 7.4, n = 213; pH 6.5, n = 239).
The incidence of trkA expression was also analyzed to assess how closely the culture situation mimicked that observed in vivo and also to determine whether exposure to an acidic environment altered the incidence of neurons with detectable membrane-associated trkA expression. More than 12,000 neurons were analyzed for incidence of detectable trkA immunofluorescence signal in the heterogeneous population. TrkA-positive neurons represented 40–45% of the population per pH condition examined, with an average of 43.7% for neurons at pH 7.4 and 43.8% for those challenged with media at pH 6.5 for 30 min (Table 1). This percentage also corresponds to that previously reported for sensory neurons that are responsive to NGF as indicated by the presence of high-affinity binding sites for NGF (Verge et al., 1989) or the expression of detectable trkA mRNA (Verge et al., 1992; Karchewski et al., 1999). Further, there was no shift in the perikaryal size range of the neuronal subpopulation expressing trkA when the pH was reduced to pH 6.5 from pH 7.4, collectively suggesting that exposing the neurons to reduced pH for 30 min does not reveal a novel population of NGF-responsive neurons. Finally, to assess whether removal of NGF by anti-NGF treatment altered the level of trkA presentation on neuronal membranes, neurons at pH 7.4 were exposed to anti-NGF treatment for 0 min, 30 min, or 60 min followed by assessment of cell-surface trkA expression using immunofluorescence approaches. The presence of anti-NGF did not significantly alter the mean level of trkA detected on the cell surface (0 min, 1 ± 0.041; 30 min, 0.9760 ± 0.03741; 60 min, 0.9908 ± 0.036). Thus, the removal of NGF from the medium before pH shift does not appear to alter the presentation of trkA on the neuronal membrane.
Effects of acidosis on incidence of sensory neurons expressing cell surface-associated trkA
Temporal analysis revealed that at the earliest time point examined, 15 min, a maximal level of trkA was already mobilized to the membrane of primary sensory neurons in response to the pH shift from 7.4 to 6.5. Surface trkA levels remained maximally elevated at the 30 min time point, with still significantly elevated, albeit lesser amounts of cell surface-associated trkA still observed following 1 and 2 h exposure to the pH 6.5 condition (Fig. 3A).

pH challenge rapidly mobilizes trkA to the membrane. A, Temporal quantitative analysis of relative levels of membrane-associated trkA immunofluorescence signal in adult DRG neurons exposed to control (pH 7.4) or acidic (pH 6.5) media for times as indicated and normalized to the mean signal intensity from the control pH group. B, Quantitative analysis of trkA immunofluorescence signal levels on sensory neurons exposed to acidosis and then reintroduced to control pH as normalized to the mean signal intensity from the control pH group. Summary bar graph reveals that trkA mobilized to the membrane in response to acidosis is not likely internalized when exposed back to pH 7.4 medium for 30 min, a condition required for biotinylation of cell-surface membrane-associated proteins in C. C, Representative Western blot where equivalent amounts of eluted biotinylated surface proteins (Memb trkA) or cytoplasmic proteins (Cyto trkA) from the same sensory neurons exposed to physiological pH 7.4 or acidic pH 6.5 media were loaded and then probed to detect levels of trkA protein. GAPDH served as a loading control for the cytoplasmic proteins (Cyto GAPDH). D, Summary bar graphs of densitometry performed on membrane and cytoplasmic eluents from the biotinylation experiments with conditions as indicated reveal a significant increase in the amount of cell-surface trkA protein detected following exposure to acidic conditions (one-way ANOVA with post hoc Tukey's; **p < 0.01; ***p < 0.001).
Cell-surface protein biotinylation was used as an alternative approach of assessing alterations in cell-surface trkA levels in response to reduced pH. However, before doing this procedure, it had to be ascertained that switching the cells that had been exposed to the pH 6.5 medium back to the neutral medium of pH 7.4 for 30 min (condition required to biotinylate cell-surface proteins), did not impact the mobilization of trkA to the cell surface that had been induced by the prior 30 min exposure of the cells to pH 6.5. Quantitative analysis of the trkA immunofluorescence signal over neurons exposed to pH 6.5 for 30 min then reintroduced to pH 7.4 for the 30 min biotinylation step revealed that there was no discernible impact on the degree of trkA that had been mobilized to the membrane in response to acidosis (Fig. 3B). This suggests that reintroduction of the pH 6.5 neurons to pH 7.4 did not significantly induce internalization or further mobilization. The “pH 6.5” sample revealed an increased amount of cell-surface trkA protein compared with “pH 7.4” (Fig. 3C), confirming the observations made using nonpermeabilizing immunohistochemical approaches. Quantification of the three biotinylation assays and normalization to the pH 7.4 state revealed that there was a significant increase in membrane-associated trkA at a molecular weight of just over 140 kDa detected in response to reduced pH (pH 7.4, 100 ± 3.59 vs pH 6.5, 142.37 ± 6.07; p < 0.01), while the decrease in cytoplasmic levels was not significant (pH 7.4, 100 ± 8.86 vs pH 6.5, 86.93 ± 3.69; p < 0.01).
pH challenge rapidly mobilizes trkA to the membrane from internal stores
We next investigated whether the increased surface expression of trkA in response to reduced extracellular pH was due to the trafficking of trkA to the membrane from a pre-existing pool in sensory neurons. Blocking the vesicular trafficking system with BFA, which destroys the rough endoplasmic reticulum (ER) Golgi-trans-Golgi network (TGN) pathway (Lippincott-Schwartz et al., 1991; Klausner et al., 1992) prevented trkA from being mobilized to the membrane from intracellular stores in response to the pH challenge (Fig. 4A). It should be noted that in other studies BFA increased endocytosis selectively from the apical surface of the MDCK kidney epithelial cell line (Prydz et al., 1992). However, because the data were normalized to the control pH 7.4 group, it could be ascertained that BFA did not have any discernible affect on the control pH group (Fig. 4A), suggesting that there was no notable endocytotic recycling of the receptor in the time frame analyzed. To further test whether BFA treatment impacted endocytosis, cells were briefly labeled with the fixable membrane stain FM 1–43FX, which is nontoxic to cells and virtually nonfluorescent in aqueous medium, but when internalized and trapped inside endocytotic vesicles fluoresces intensely (Griesinger et al., 2002). While there was no indication that cells exposed to pH 6.5 with or without BFA treatment had any discernible levels of internalization beyond that observed at neutral pH, when challenged with NGF, a condition that induces trkA internalization (Beattie et al., 2000), a significant increase in internalized membrane was observed (Fig. 4B,C; p < 0.01). Finally, while easiest to quantify at the level of the cell body, the mobilization of trkA to the cell surface in response to acidosis was not only restricted to this compartment. As Figure 2 shows, there was also a robust mobilization to the neurites. Qualitatively BFA treatment also attenuated the heightened immunofluorescence signal that was normally observed over the neurites in response to pH 6.5 exposure (data not shown).

Acidic pH challenge rapidly mobilizes trkA to the membrane from internal stores. A, Bar graphs summarize relative changes in neuronal cell-surface trkA expression from three separate experiments (detected with immunofluorescence) over neurons exposed to control (pH 7.4) or acidic (pH 6.5) media for 30 min with or without exposure to Golgi collapsing compound BFA and as normalized to the mean signal intensity from the control pH group. Note: A significant increase is observed in the mean levels of trkA mobilized to the neuronal membranes of sensory neurons in response to acidosis when compared with the control pH, a response that is blocked with BFA treatment. B, Immunofluorescence photomicrographs and summary histograms (C) depict degree of FM 1–43FX-stained neuronal membrane internalization in response to conditions as indicated. Note: Significant membrane internalization was only observed in the NGF challenge control group and not in response to acidic pH challenge with or without BFA treatment. (Data normalized to the control pH of each experimental condition and pooled from three separate experiments; A, C, one-way ANOVA with post hoc Tukey's, **p < 0.01; ***p < 0.001). Scale bar, 20 μm.
Increased mobilization of trkA to the cell membrane augments neuronal sensitivity to NGF
To investigate the functional implications of the increased trkA surface expression induced by exposure to a reduced extracellular pH of 6.5, we assessed whether the degree of trkA receptor activation/Y490 phosphorylation (Stephens et al., 1994) in response to NGF challenge was altered in neurons previously exposed to acidic versus control conditions. Sensory neurons were therefore exposed to control media supplemented with or without 50 ng/ml of the mature form of NGF (2.5S NGF) for 15 min following the 30 min exposure to acidic (pH 6.5) or control (pH 7.4) DMEM. Function-blocking anti-BDNF (1.5 μg/ml) was added to the media as a control for the duration of the pH shift and during NGF challenge to block potential activation of trkB by released endogenous BDNF and to prevent BDNF-induced dampening of trkA signaling (MacPhee and Barker, 1997). This was done because some trkA-positive sensory neurons express and release the neurotrophin BDNF in an activity-dependent manner (Balkowiec and Katz, 2000; Karchewski et al., 2002) and thus, have the potential to activate trkB receptors. Further, NGF-evoked BDNF release, while not established for sensory neurons, has been shown in PC12 cells (Krüttgen et al., 1998).
Sensory neurons cultured under control physiological conditions without NGF challenge had extremely low levels of constitutively active trkA, as visualized by immunohistochemical detection of phospho-trkA-Y490 under permeabilizing conditions (Fig. 5A). Neurons exposed to either an acidic pH of 6.5 or control pH of 7.4 displayed increased levels of activated/phosphorylated trkA in response to the 15 min NGF challenge (Fig. 5A). Quantification of the immunofluorescence signal for individual neurons cultured at physiological pH (7.4) revealed that NGF challenge resulted in elevated phospho-trkA signal most evident over medium to large size neurons. Interestingly, exposure to acidic conditions (pH 6.5) alone for 30 min before NGF challenge appeared sufficient to activate trkA as evidenced by elevated levels of phospho-trkA immunofluorescence signal over neurons of all size ranges, a response that was significantly augmented by NGF challenge (Fig. 5B,C). In addition, the average phospho-trkA labeling intensity after NGF challenge was both statistically significant between experimental groups before NGF challenge and within experimental groups following NGF challenge (Fig. 5D). To ascertain whether residual NGF in the culture dish might contribute to the observed trkA activation on the absence of NGF challenge (Fig. 5C,D), the experiments were repeated in triplicate with both anti-NGF and anti-BDNF in the pH challenge media before the NGF challenge phase where only anti-BDNF was included for reasons stated above. The results still showed a consistent low level of trk activation in response to pH 6.5 challenge, which did not differ from that obtained without the anti-NGF (data not shown), suggesting that this activation was NGF independent. Further, the impact of including the anti-NGF and anti-BDNF on trkA mobilization to the membrane in response to reduced pH was also assessed and revealed no impact of the antibodies on this response (data not shown).

Increased mobilization of trkA to neuronal membrane enhances response to NGF challenge. A, Fluorescence photomicrographs depicting relative levels of activated/phosphorylated trkA (phospho-trkA) detected under permeabilizing conditions in representative adult DRG neurons exposed to acidic (pH 6.5) or control (pH 7.4) media (in the presence of anti-BDNF) for 30 min, followed by a 15 min challenge with pH-specific medium + anti-BDNF alone or with anti-BDNF + 50 ng/ml NGF. Scale bar, 10 μm. Scatter plots illustrate the relationship between phospho-trkA-like labeling intensity (y-axis; normalized to the mean signal intensity from the control pH group) and perikaryal diameter (x-axis) for all sensory neurons analyzed from three separate experiments exposed to control (pH 7.4, B) or acidic (pH 6.5, C) media as above and then challenged with 50 ng/ml NGF (open circles) or not (filled circles). D, Summary bar graph depicting relative levels of activated trkA (phospho-trkA; normalized to pH 7.4 control) in representative adult DRG neurons exposed to acidic (pH 6.5) or control (pH 7.4) media for 30 min and then challenged with 50 ng/ml NGF and as normalized to the mean signal intensity from the control pH group. E, Fluorescence photomicrographs of sensory neurons exposed to identical conditions as in A and processed to detect activated/phosphorylated p38MAPK Note: A significant increase is observed in the levels of phospho-trkA detected in sensory neurons in response to acidosis with a parallel response observed for phospho-p38MAPK (E) (one-way ANOVA with post hoc Tukey's; ***p < 0.001; n = 131–210 neurons analyzed per experimental condition). Scale bar, 20 μm.
Finally, a downstream response to trkA activation was also examined, namely p38MAPK phosphorylation, as it is linked to NGF-mediated increases in ASIC3 expression during inflammation (Mamet et al., 2003). Qualitatively, the phosphorylation of p38MAPK paralleled the response observed with respect to trkA activation (Fig. 5A,D) with increased immunofluorescence signal localized to both cytoplasm and nuclei, suggesting that there were cellular consequences to the increased mobilization of trkA to the neuronal cell surface.
Brief exposure to extracellular acidic conditions activates trkA in an NGF-independent manner
Even though the cultures grown in 10 ng/ml NGF for 2 d were washed with media containing activity-blocking sheep anti-NGF to quench activation of trkA and inactivate any residual NGF before exposure of these sensory neurons to an acidic environment, a higher basal level of trkA activation/phosphorylation was still detected for the pH 6.5 group relative to those kept at physiological pH before NGF challenge (Fig. 5C,D). The most prevalent form of trkA activation and phosphorylation is induced by the binding of NGF to key residues on the extracellular domain (Kaplan et al., 1991; Ultsch et al., 1999). To control for the possibility that the shift to an acidic environment might trigger release of small amounts of endogenous NGF from the few non-neuronal cells in the culture (Bandtlow et al., 1987; Heumann, 1987), the experiments were repeated three times, adding activity-blocking sheep anti-NGF in addition to the activity-blocking sheep anti-BDNF in the 7.4 and 6.5 pH-specific media used for the pH shift. These conditions would inactivate any endogenous BDNF or NGF that is released by the neurons. Under these new control conditions, a 30 min exposure to an acidic pH of 6.5 still resulted in a significant increase in the level of presumably NGF-independent activated/phosphorylated trkA being detected (data not shown). Additionally, whether the experiments were conducted in the presence or absence of anti-NGF, no statistical differences were observed, supporting that the increased level of trkA phosphorylation observed in response to acidosis is by a means other than by neurotrophin-mediated trkA activation. Notably, a similar response with respect to p38MAPK phosphorylation was also observed in pH 6.5-exposed neurons in the absence of NGF challenge, similar to that observed with respect to trkA activation (Fig. 5A,D,E), suggesting that this form of trkA activation is functionally relevant.
Increased neuronal activity is linked to mobilization of trkA to the membrane
Depolarization of hippocampal neuron resulted in rapid recruitment of trkB to the neuronal membrane (Meyer-Franke et al., 1998). Thus, we examined whether a similar mechanism might contribute to the rapid mobilization of trkA to the cell surface of sensory neurons that we observe with reduced pH. Elevated levels of protons associated with acidosis can result in increased activation of ASICs in sensory neurons that serve as highly sensitive receptors for extracellular protons (tissue acidosis). ASICs are expressed by the NGF-responsive subpopulation of neurons (Molliver et al., 2005) and are readily activated by moderate acidosis, resulting in the generation of a sustained depolarizing current (Immke and McCleskey, 2001; Smith et al., 2007; Deval et al., 2008).
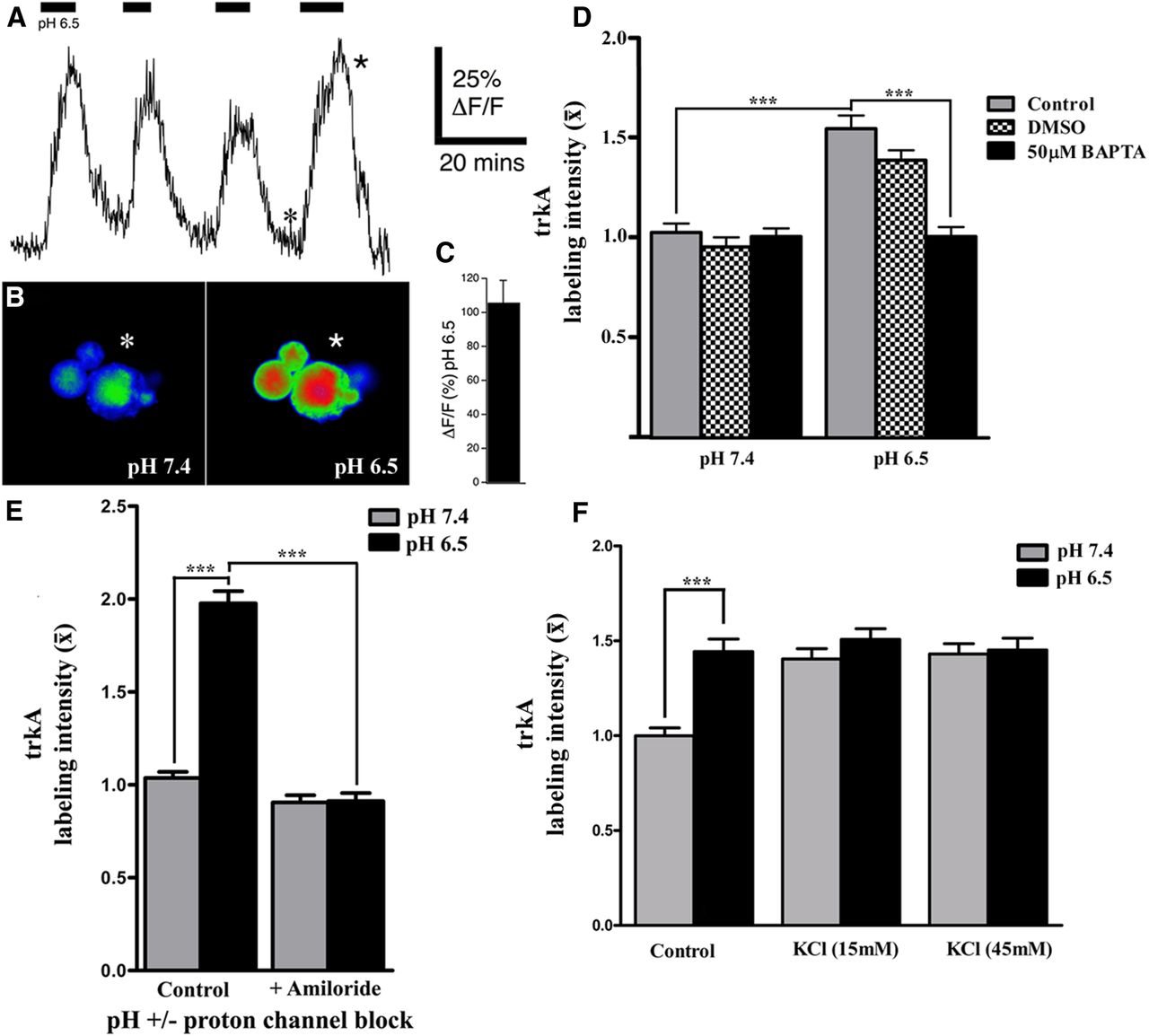
Increased neuronal activity triggers large and rapid changes in cytosolic free Ca2+ and imaging with fluorescent Ca2+ indicators has been widely used to monitor neuronal spiking across populations of CNS neurons in vitro (MacLean and Yuste, 2009) and in vivo (Stosiek et al., 2003). To gain insight into whether calcium responses are altered in response to pH 6.5, we examined if switching the pH of the bathing solution from 7.4 to 6.5 could induce Ca2+ transients in small to medium size rat DRG neurons (consistent with the size range of the majority of trkA-positive neurons; Verge et al., 1992). Dissociated neurons were first loaded by bath-applied Rhod-2 AM. An increase in intracellular Ca2+ concentration ([Ca2+]i) was elicited by exposure to the acidic medium (Fig. 6A), which was reversed by switching the neurons back to pH 7.4, with each of the pH 6.5 challenges producing a comparable Ca2+ transient (Fig. 6A–C). These data support that acidosis is capable of evoking calcium transients, and the reproducible nature of the response serves as further proof that the neuronal membranes remain intact throughout the pH challenge. To assess whether alterations in intracellular Ca2+ signaling contribute to the mobilization of trkA to the membrane in response to acidosis, the impact of chelating available Ca2+ was examined. Ca2+ was chelated by pre-incubation of the neurons with cell-permeant BAPTA-AM (50 μm in 0.2% DMSO) or DMSO (0.2%) for 30 min. Media were changed to pH 6.5 or pH 7.4 with either BAPTA-AM (50 μm) or DMSO (0.2%) for 30 min, followed by processing the cells for cell-surface trkA immunohistochemistry under nonpermeabilizing conditions. Ca2+ chelation abolished the pH 6.5-induced trkA mobilization to the cell membrane, while exposure to the DMSO control condition did not have any discernible impact on this response (Fig. 6D).

Neuronal activity is linked to mobilization of trkA to membrane. A, Acid-induced [Ca2+]i increases in DRG cells. Typical sample trace exhibiting reversible repeatable Ca2+ transients upon exposure of sensory neurons to a lowered pH of 6.5. The six-point asterisk in the trace refers to the pseudocolor image seen in B (pH 7.4) exhibiting baseline fluorescence. The five-point asterisk highlights a typical peak Ca2+ response seen in the corresponding pseudocolor image seen in B (pH 6.5). C, Bar graph representing the mean Ca2+ increase observed upon repeated exposure to a lowered pH of 6.5 (n = 6 neurons; ±SEM). D, E, Summary bar graphs of data (normalized to pH 7.4 controls), depicting relative changes in the level of membrane-associated trkA expression in adult DRG neurons exposed to acidic (pH 6.5) or control (pH 7.4) media for 30 min in the presence or absence of Ca2+ chelator BAPTA-AM (50 μm), DMSO, or the acid-sensing ion channel blocker amiloride (250 μm). Note: Blocking proton-sensitive channels or chelating Ca2+ greatly attenuates the amount of trkA detected on the cell surface in response to acidosis when compared with pH 6.5 alone. F, Summary bar graphs from experiments (normalized to pH 7.4 controls), depicting relative changes in membrane-associated trkA expression in adult DRG neurons exposed for 30 min to acidic (pH 6.5) or control (pH 7.4) media for 30 min with or without KCl depolarization (15 mm or 45 mm) as indicated. Note: KCl-induced depolarization of neurons exposed to pH 7.4 results in mobilization of trkA to the membrane in a manner equivalent to that observed in response to a lowered pH of 6.5; but KCl does not induce further mobilization in neurons already exposed to pH 6.5. (one-way ANOVA with post hoc Tukey's; ***p < 0.001; n = 101–140 neurons analyzed per experimental condition).
We next examined the contributions of ASIC channel activity to pH-induced cell-surface mobilization of trkA. To antagonize proton activation of ASICs, cells were exposed to pH 6.5- or pH 7.4-specific media with or without the general ASIC antagonist, amiloride (250 μm; Tang et al., 1988; Ugawa et al., 2002) for the duration of the 30 min pH challenge, followed by immunohistochemical detection of cell-surface trkA expression. Antagonizing ASIC activation also prevented the mobilization of additional trkA to the neuronal cell surface (Fig. 6E).
Finally, calcium transients are not direct proof of increased neuronal activity. Thus, to determine whether neuronal depolarization is sufficient to induce to the same degree of mobilization of trkA to the membrane as a moderate drop in extracellular pH, the cultures were exposed to KCl at concentrations shown to depolarize neuronal membranes (Di Virgilio et al., 1987). Prepared cells were washed in DMEM with anti-NGF, then switched to pH 6.5- or pH 7.4-specific media with or without [KCl] of 15 mm or 45 mm for the 30 min pH challenge, followed by processing for trkA cell-surface expression. For sensory neurons cultured under physiological conditions (pH 7.4), a 30 min exposure to either 15 mm or 45 mm KCl triggered mobilization of trkA to the neuronal membranes in a manner equivalent to that observed following exposure to pH 6.5 medium alone (Fig. 6F). Notably, KCl treatment did not augment the degree of trkA mobilization to the cell membrane for the neurons exposed to pH 6.5 DMEM, suggesting that the cells were effectively activated with respect to this response by the acidic challenge alone.
Discussion
While individual molecules such as NGF have been shown to contribute to the process of peripheral sensitization (Woolf et al., 1994), we focused on whether the acidosis that accompanies inflammation impacts how sensory afferents sensitize to NGF. Our novel findings reveal that even a modest drop in pH can rapidly shift neuronal sensitivity to NGF, a critical driver of the inflammatory pain state. Switching from an extracellular environment of pH 7.4 to 6.5 is sufficient for us to visualize ∼50% more immunofluorescence signal for trkA on the cell surface in as little as 15 min through the activation of ASICs channels and in a Ca2+-dependent manner. We also demonstrated that depolarization of neurons at physiological pH alone is sufficient for the response and that this mobilization resulted in both ligand-independent and ligand-dependent increases in receptor activation. Collectively, these findings have numerous implications for NGF-mediated pathological and biological processes.
Decreased pH can rapidly sensitize sensory fibers as seen with inflammatory states such as acid injections in muscle to mimic muscle pain (Issberner et al., 1996) or acidosis due to cardiac ischemia (Yagi et al., 2006). Although the time frame we chose to examine the mobilization of trkA may seem long in terms of cell-signaling events, acidosis can persist for an extended period of time, lasting in some cases for hours. The observation that significantly increased levels of trkA are still mobilized to the membrane after 2 h exposure to pH 6.5 (our latest time point analyzed) is consistent with extracellular acidosis having a protracted affect with respect to potentially increased sensitivity to NGF.
trkA mobilizes to the membrane from an internal pool in response to acidosis
The rapid insertion of additional trkA receptors on the cell surface in response to extracellular pH 6.5 implies that the receptors are either newly synthesized or from an existing intracellular pool. In the case of the former, axons, which lack trkA receptor mRNA (Vogelaar et al., 2009; Gumy et al., 2011), would require both synthesis and transport to the axonal compartment. The 15 min time frame observed for the trkA mobilization to the cell surface make de novo synthesis unlikely. Rapid mobilization of additional trkA protein to the membrane surface over the neurites suggests that a pre-existing pool of receptors must exist in the axons as well. This pool is likely associated with the recently described Golgi-like structures in peripheral axons (Merianda et al., 2009), as we found BFA treatment also attenuated acid-induced trkA surface mobilization to the neurite membrane in addition to over the cell body. The receptor that was translocated to the membrane was of a molecular weight consistent with the fully glycosylated/mature form of trkA (Schecterson and Bothwell, 2010), suggesting that the immature form, which is capable of being transactivated in an NGF-independent manner, is either retained in the Golgi or undergoes maturation before translocating. Although the exact nature of the signal that causes release of the fully glycosylated trkA from the Golgi is not known, recent work supports that the protein muc18–1-interacting protein 2 (Mint2) may serve to retain trkA in the Golgi, inhibiting its cell-surface sorting until signaled to do so (Zhang et al., 2009).
Typically, receptors are delivered to the membrane via de novo synthesis, followed by packaging and transport through the ER and Golgi apparatus. Even though trkA localizes to the ER and Golgi (Sorkin, 2005), the 15 min time point for maximally observed insertion of trkA on the cell surface is likely too brief to be a consequence of described protein insertion in the membrane via traditional secretory pathways. Previous work in sympathetic neurons has shown that it takes up to an hour for proteins to be synthesized, packaged, transported, and inserted into the membrane (Carbonetto and Fambrough, 1979). Further, the time frame required for Golgi-trans-network to cell membrane trafficking has been shown in other cell types to be within the 15–30 min time frame examined in this study (Wacker et al., 1997; Hirschberg and Lippincott-Schwartz, 1999). The nature of the signal that affects the rapid insertion of additional mature trkA receptors into the plasma membrane in response to acidosis is not known. Perhaps extracellular acidosis results in cellular responses that accelerate the maturation process of trkA through the Golgi, because we only observed trkA of a molecular weight consistent with the fully glycosylated form present on the membrane surface. Additionally, the strong calcium transients observed in response to the switch to pH 6.5 may serve to remove the inhibitory influence that calneurons impose on the TGN to plasma membrane trafficking, as shown in hippocampal neurons (Mikhaylova et al., 2009), thereby facilitating insertion of trkA into the neuronal membrane.
Functional implications for increased cell-surface trkA expression
We found that brief exposure of the cells to acidic conditions, followed by NGF challenge, resulted in a significantly enhanced level of trkA activation. While the trkA that mobilized to the membrane in response to pH 6.5 exposure was of a molecular weight consistent with fully glycosylated/mature trkA, we also evidenced NGF-independent trkA activation. This form of trkA activation can occur merely as a result of trkA overexpression on the cell surface (Hempstead et al., 1992) or as a result of Golgi-localized trkA proteins that are transactivated in response to depolarization-induced release of molecules such as pituitary adenylate cyclase-activating polypeptide (PACAP) or adenosine (Rajagopal et al., 2004; Gwak and Hulsebosch, 2011). The latter however, is less likely given that it is estimated to take at least 1 h to transactivate trkA receptors (Lee and Chao, 2001).
The heightened level of trkA activation likely has ramifications on both short- and long-term sensitization processes as it regulates the activity and expression of a wide variety of receptors, ion channels, and signaling molecules (Mantyh et al., 2011). In the short term, it modulates the activity of receptors and ion channels. For example, NGF decreases the threshold of activation of the nociceptive transient receptor potential vanilloid 1 (TRPV1) receptors (Chuang et al., 2001), mediates TRPV1 trafficking to the membrane (Stein et al., 2006), increases purinergic receptor P2X3-mediated currents and Ca2+ transients (D'Arco et al., 2007) and for sympathetic neurons, rapidly modulates the activity of at least four voltage-gated currents (Luther and Birren, 2009). In the long term, increased NGF can lead to increased transcription of many nociception-associated genes such as its receptors trkA and p75 (Verge et al., 1989, 1992); the neuropeptides Substance P, calcitonin gene-related peptide (Lindsay and Harmar, 1989; Verge et al., 1995), and PACAP; Jongsma Wallin et al., 2001, 2003); sodium channels (Dib-Hajj et al., 1998; Fjell et al., 1999; Kerr et al., 2001); and P2X3 (Ramer et al., 2001; Simonetti et al., 2006). It is also interesting to note that activation of trkA was linked in our study to a parallel activation of p38MAPK, which has been shown to be linked to NGF-mediated increases in ASIC3 expression during inflammation (Mamet et al., 2003). The duration of our studies unfortunately was too brief to examine this latter aspect.
Neuronal depolarization induces trafficking of trkA to the cell surface
We observed strong Ca2+ transients upon switching sensory neurons to a pH 6.5 environment and a Ca2+ dependency of acid-induced trkA mobilization to plasma membrane. This, coupled with our ability to prevent trkA trafficking to the membrane by exposure to the nonselective ASIC antagonist, amiloride, made us speculate that neuronal depolarization resulting from proton-mediated activation of ASIC receptors might be sufficient to induce the mobilization of trkA to the cell surface at physiological pH. Depolarization is one mechanism by which trafficking of protein to the neuronal membrane (not involving de novo synthesis) occurs (Kiss et al., 1994; Meyer-Franke et al., 1998; Bao et al., 2003; Kingsbury et al., 2003; Bouchard et al., 2008). Kingsbury et al. (2003) demonstrated that depolarization of cultured mouse cortical neurons resulted in an increase in both membrane bound functional trkB protein and in trkB mRNA production. Other studies have shown that depolarization, through a Ca2+-mediated event, not only induced the trafficking of trkB to the neuronal plasma membrane, but also altered the localization of trkB mRNA within neurons in a Ca2+-dependent manner, with more trkB message appearing in the neurites of depolarized neurons (Tongiorgi et al., 1997, Meyer-Franke et al., 1998). Depolarization has also been shown to induce trkA mRNA in MAH cells (Birren et al., 1992). Thus, a similar mechanism with respect to trkA trafficking and perhaps even expression is likely occurring in trkA neurons in response to the increased sensitization and depolarization that often accompanies inflammatory states and that acidosis contributes to.
In conclusion, our finding that even small changes in pH may rapidly shift the sensitivity of sensory neurons to NGF provides a novel mechanism for rapid modulation of cell-surface interactions with NGF. These data also establish a relation between neuronal activity and membrane-associated trkA expression that may serve to rapidly sensitize neurons to NGF under a variety of pathological conditions.
Footnotes
This work was supported by a grant from the Canadian Institutes of Health Research (V.M.K.V.) MOP74747. G.E.B. was supported by a University of Saskatchewan Graduate Scholarship and Z.Y. by a University of Saskatchewan/China Doctoral Scholarship. We are indebted to J. Johnston for excellent technical and editorial assistance.
- Correspondence should be addressed to Valerie M.K. Verge, Cameco MS/Neuroscience Research Center, Saskatoon City Hospital, Room 5800, 701 Queen Street, Saskatoon, Saskatchewan, Canada, S7K 0M7. valerie.verge{at}usask.ca





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}