

Abstract
CXCR4, a receptor for the chemokine CXCL12 (stromal-cell derived factor-1α), is a G-protein-coupled receptor (GPCR), expressed in the immune and CNS and integrally involved in various neurological disorders. The GABAB receptor is also a GPCR that mediates metabotropic action of the inhibitory neurotransmitter GABA and is located on neurons and immune cells as well. Using diverse approaches, we report novel interaction between GABAB receptor agents and CXCR4 and demonstrate allosteric binding of these agents to CXCR4. First, both GABAB antagonists and agonists block CXCL12-elicited chemotaxis in human breast cancer cells. Second, a GABAB antagonist blocks the potentiation by CXCL12 of high-threshold Ca2+ channels in rat neurons. Third, electrophysiology in Xenopus oocytes and human embryonic kidney cell line 293 cells in which we coexpressed rat CXCR4 and the G-protein inward rectifier K+ (GIRK) channel showed that GABAB antagonist and agonist modified CXCL12-evoked activation of GIRK channels. To investigate whether GABAB ligands bind to CXCR4, we expressed this receptor in heterologous systems lacking GABAB receptors and performed competition binding experiments. Our fluorescent resonance energy transfer experiments suggest that GABAB ligands do not bind CXCR4 at the CXCL12 binding pocket suggesting allosteric modulation, in accordance with our electrophysiology experiments. Finally, using backscattering interferometry and lipoparticles containing only the CXCR4 receptor, we quantified the binding affinity for the GABAB ligands, confirming a direct interaction with the CXCR4 receptor. The effect of GABAergic agents on CXCR4 suggests new therapeutic potentials for neurological and immune diseases.
Introduction
CXCR4 is a G-protein-coupled receptor (GPCR) that selectively binds CXCL12 (stromal-cell derived factor; SDF-1α). This chemokine and its receptor have been found to play important roles in several processes involved in ischemic stroke and its subsequent repair (Wang et al., 2012), brain tumor pathogenesis (Rempel et al., 2000), human immunodeficiency virus (HIV) encephalopathy (Li and Ransohoff, 2008), multiple sclerosis, and stem cell migration (Carbajal et al., 2010). CXCR4 is widely expressed in a variety of cell types including leukocytes, where it promotes migration, recruitment, and activation (Bonavia et al., 2003; Salcedo and Oppenheim, 2003; Juarez et al., 2004; Choi and An, 2011; Comerford and McColl, 2011); neurons, where it modulates electrical activity (Banisadr et al., 2002; Guyon and Nahon, 2007; Rostène et al., 2011); and various cancers and metastases (Wang et al., 2006), where it is involved in tumor progression (Liu et al., 2006; Gao et al., 2010; Zhao et al., 2010). CXCR4 also binds the HIV-1 viral envelope glycoprotein, gp120 (Doranz et al., 1997; Gabuzda and Wang, 2000). Thus CXCR4 is an important therapeutic target for stroke, inflammation, neuromodulation, cancer, and in the prevention of HIV infection.
CXCR4 couples to the Gi family of proteins activating multiple G-protein-dependent and -independent pathways (Lazarini et al., 2003; Busillo and Benovic, 2007). In neurons, CXCR4 stimulation has been shown to activate a G-protein inward rectifier K+ (GIRK) and a voltage-gated K channel Kv2.1 associated to neuronal survival, and to increase high voltage-activated (HVA) Ca2+ currents (Guyon and Nahon, 2007; Shepherd et al., 2012).
The GABA type B (GABAB) receptor is also a GPCR activated by GABA, the chief neuro-inhibitory neurotransmitter in mammalian systems. GABAB receptors are obligatory heterodimers with two homologous subunits (GB1 and GB2) required for functioning, are widely expressed and distributed in the CNS (Kaupmann et al., 1998), and can activate diverse intracellular pathways (Guyon and Leresche, 1995; Laviv et al., 2011). GABAB receptors are also expressed on cells of the immune system with a possible link to the inflammatory response (Tian et al., 2004; Rane et al., 2005). As a consequence, there is a rich pharmacology aimed at targeting GABAB receptors, with numerous compounds currently being used with the presumption that they are highly selective for these receptors (Bowery, 1993; Froestl, 2010). Given that CXCR4 and GABAB have coexpression on immune cells and neurons, and the evidence for possible cross talk between these receptors, we hypothesized that ligands binding GABAB are involved in allosteric or direct interaction with CXCR4. Here we describe the experiments that test our hypothesis. First we show inhibition of CXCL12-induced migration of cancer cells by GABAB ligands. We tested these same ligands in electrophysiology experiments using dopaminergic neurons in acute slices of substantia nigra and oocytes that lead us to binding experiments using GABAB agents at the CXCR4 receptor. While somewhat unexpected, we observed that GABAB agents such as baclofen, the antagonists CGP 55845 and CGP 54626, and GABA can directly bind the chemokine receptor CXCR4.
Materials and Methods
Molecular biology.
Rat CXCR4 gene containing pcDNA 3.1 (−) plasmid was obtained from Richard Miller lab, University of Michigan. CXCR4 was then either subcloned in pGEMHE vector using XmaI and XbaI sites (for electrophysiology) or pGEMHE-EGFP-X vector (X being the linker sequence SRGTSGGSGGSRGSGGSGG) using XhoI and XbaI sites. cRNA was then prepared from the above clones using mMessage T7 RNA kit. Eitan Reuveny of the Weizmann Institute of Science provided GIRK1/2 cDNA for experiments.
Oocyte expression.
Stage V–VI oocytes were collected from anesthetized Xenopus laevis and defolliculated with collagenase (Boehringer Mannheim). Oocytes were incubated at 16°C in external medium (N96) of the following composition (in mm): 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, 5 pyruvate, and 100 mg/L gentamycin, pH 7.2. Fifty nanoliters of RNA was injected. The concentration of RNA injected was 0.003 μg/μl for GIRK1 and GIRK2, 0.005–0.1 μg/μl for CXCR4. Expression protein was allowed 24–72 h before the start of the experiments at 12°C or 18°C.
Cell culture and transfection.
The human embryonic kidney cell line 293 (HEK293) was maintained in DMEM with 5% fetal bovine serum (FBS) on poly-lysine-coated glass coverslips at ∼6 × 106 cells per 25 mm coverslip and transiently cotransfected using Lipofectamine 2000 (Invitrogen) with CXCR4 in PCDNA 3 (−), GIRK1-F137S (homotetramerization mutant), and eYFP at a ratio of 7.5:7.5:0.5 with 1.6 μg of DNA total per 18 mm diameter coverslip. Patch clamp was performed 24–36 h after transfection.
The human neuroblastoma cell line SH-SY5Y was obtained from ATCC by the MCB Tissue Culture Facility and maintained as monolayers in DMEM with 5% FBS. The human breast adenocarcinoma cell line, MDA-MB-231, was obtained from ATCC by the MCB Tissue Culture Facility and maintained as monolayers in RPMI 1640 supplemented with 10% FBS, 1 mm pyruvate, and 2 mm glutamine. All cells lines were used at a low passage.
Agarose spot assay.
Agarose spot assay was performed as described previously (Vinader et al., 2011). Briefly, low melting point agarose (Ultrapure, LMP agarose, Invitrogen) was diluted into sterile PBS to make a 0.5% agarose solution. The agarose solution was autoclaved, removed from the heat, and cooled down to 40°C.
To prepare the CXCL12/agarose solution, lyophilized CXCL12 (R&D Systems) was reconstituted to a final stock concentration of 12.5 μm in sterile water. CXCL12 solution was then added to the molten 0.5% agarose solution at 40°C to produce a final concentration of 10 nm (+) or 100 nm CXCL12 (++). Control agarose solutions were prepared by substituting same volumes of water for CXCL12 (Solutions − and −−). Four independent drops (10 μl, ∼2 mm diameter) of the four solutions (maintained at 40°C) were pipetted onto the base of a sterile 20 mm diameter glass-bottomed cell culture dish (MatTek Corporation; see Fig. 1C). The dishes were then cooled for 5 min at 4°C to allow the agarose spot to solidify. MDA-MB-231 cells were trypsinized and resuspended to a final concentration of 105 cells/ml in RPMI 1640 supplemented with 10% FBS, 1 mm sodium pyruvate, and 2 mm l-glutamine. Cells were then exposed to AMD3100 (2 μm), gp120 and CD4 (300 nm each), CGP 55845 (5 μm), CGP 54626 (5 μm), GABA (100 μm), baclofen (100 μm), or control PBS with no drug. Following this incubation, cells were added to the dishes containing the agarose spots (1 ml per plate at a final concentration of 105 cells.ml−1) and then incubated for 4 h at 37°C to allow the cells to adhere. The culture media was replaced with RPMI medium containing 0.1% FBS and the drugs or control, and the dish incubated at 37°C in 5% CO2 overnight. The degree of cells invading underneath the agarose spot was analyzed by collecting pictures under an inverted microscope Olympus IX91 using a ×10 objective equipped with an Andor Camera and Software, and then counting the number of cells using ImageJ software.
Six representative fields of view of equal size sectors were individually counted per agarose spot, and the mean number of invading cells per field of view calculated. The values reported herein are the average of three independent dishes. The experiment was reproduced twice and gave similar results.
Reverse transcription-PCR.
The content of a monolayer of confluent cells in a 25 ml flask was trypsinized and washed twice in cold PBS and the cell pellet was used for extraction of total RNA using TRIzol. cDNA was then made from total RNA using SuperScript II Reverse Transcriptase kit (Invitrogen). CXCR4 and GABAB genes were then amplified using CXCR4 Forward 20 AGGTAGCAAAGTGACGCCG, CXCR4 Reverse 220 GATGGTGGGCAGGAAGATT (Guyon et al., 2006) and GAGABR2 Forward GGACCTGGATTCTCACCGTGGGCTA, GABABR2 Reverse TGCTGGGTCCGGCTCCATGCTGTA (Osawa et al., 2006) primers, respectively, and products were analyzed on agarose gel.
Immunoblot analysis.
Both floating and attached cells were collected, washed twice with cold PBS, and processed for plasma membrane protein fraction using Qproteome Plasma Membrane Protein Kit (Qiagen). Equal amounts of membrane protein (15–20 μg) from cells were separated on 12% SDS-PAGE and transferred electrophoretically to a polyvinylidene difluoride membrane. Immunoblot analyses were performed with 1:1000 dilutions of CXCR4 and GABABR2 antibodies using standard procedure. The blots were visualized using the enhanced chemiluminescence detection reagents and the manufacturer's protocol.
Electrophysiology.
Electrophysiological recordings in brain slices from the rat substantia nigra were performed as described previously (Guyon et al., 2008). Briefly, male Wistar rats were bred in the local animal facilities and maintained on a 12 h dark/light cycle (07:00/19:00) with food and water ad libitum. All of the protocols were performed in accordance with French standard ethical guidelines for laboratory animals (agreement no. 75–178, 05/16/2000). Brain slices were obtained from 12- to 23-d-old rats anesthetized with 1% halothane. Following decapitation, brains were rapidly removed and placed in cold phosphate/bicarbonate-buffered solution (PBBS) composed of the following (in mm): 125 NaCl, 2.5 KCl, 0.4 CaCl2, 1 MgCl2, 25 glucose, 1.25 NaH2PO4, and 26 NaHCO3, pH 7.4 when bubbled with 95% O2/5% CO2. Transversal substantia nigra slices (250 μm) were then transferred to an incubating chamber maintained at 34°C in oxygenated PBBS. After 1 h, slices were transferred to another incubating chamber at room temperature (22–25°C) filled with PBBS containing additional CaCl2 (final concentration 2 mm) and used for electrophysiological recordings. Slices were placed under a Nomarski microscope (Zeiss) equipped with infrared video camera (AxioCam; Zeiss) in a recording chamber superfused at a flow rate of 1 ml/min with oxygenated PBBS. Pictures were taken by using a digital camera (AxioCam; Zeiss) and connected image-acquisition software (AxioVision). Dopaminergic neurons were recorded in the whole-cell mode in voltage clamp and characterized as described previously by the presence of the hyperpolarization-activated cation current and low-threshold Ca current (IT), and using single-cell reverse transcription (RT)-PCR to reveal the presence of mRNA for the tyrosine hydroxylase as described previously (Guyon et al., 2006). Patch-clamp pipettes had a resistance of 3–6 MΩ when filled with the internal solution containing the following (in mm): 120 CsCl, 5 MgCl2, 1 CaCl2, 10 EGTA, 4 Na ATP, 0.4 Na GTP, and 10 HEPES supplemented with 15 mm phosphocreatine and 50 U/ml creatine phosphokinase (pH adjusted to 7.3 with CsOH). Statistical significance between groups (average data expressed as mean ± SEM, n = number of neurons) was tested using either the one-way ANOVA or repeated-measures one-way ANOVA test followed by a Newman–Keuls post hoc test with a threshold of significance of *p < 0.05, **p < 0.01, and ***p < 0.001 using a statistical software package (SigmaStat 2.03 from Jandel Scientific or Origin from MicroCal). Changes of extracellular solution were obtained by a fast multibarrel delivery system positioned close to the cell tested.
Electrophysiological recording in Xenopus oocytes was done 2–5 d after cRNA injection. Two-electrode voltage-clamp was performed with a GeneClamp 500 amplifier interfaced to a Digidata 1200 A/D (Molecular Devices). The interface was controlled with a PC computer (Dell) running pClamp version 10.2 (Molecular Devices). Microelectrodes were filled with 3 m KCl and had a tip resistance of 0.15–1.5MΩ. The oocytes were placed in a small chamber continually perfused with high K+ Ringer's solution (100 mm KCl, 2 mm NaCl, 1.8 mm CaCl2, 1 mm MgCl, 25 mm HEPES, pH 7.5). Agonists and blockers were applied in the bath perfusion. The holding potential was set at −30 mV. Current–voltage records were obtained during 500 ms voltage jumps to potentials between −120 and +50 mV. All data were produced for more than one oocyte batch and analyzed with Clampfit 10.2 (Molecular Devices).
Whole-cell patch-clamp recordings in transfected HEK293 cells used an Axopatch 200A amplifier. Cells were voltage clamped at −60 mV. Pipettes had membrane resistance of 2–5 MΩ and were filled with a solution containing the following (in mm): 145 KCl, 10 NaCl, 1 MgCl2, 3 Na ATP, 0.4 Na GTP, and HEPES 10, pH 7.3, for HEK293 cells. The extracellular recording was the following (in mm): 145 NaCl, 4 KCl, 1 MgCl2, 2 CaCl2, and 16 HEPES, pH 7.4 (theoretical EK+ = −90 mV). HEK293 cells that had been successfully transfected with the Kir3.1 and Kir3.2 subunits were identified by their YFP fluorescence. To enhance the amplitude of the CXCL12-evoked GIRK currents, we recorded inward current through these inwardly rectifying channels (see Fig. 6). The K+ concentration of the extracellular bathing solution was raised to 60 mm and the NaCl concentration reduced to 90 mm. Furthermore, to ensure that currents evoked by prolonged exposure to agonist did not decline because of the inward current raising the K+ concentration, thereby reducing the electrochemical drive for further entry of K+, we used a protocol that minimized the amount of K+ entry into the cell during the perfusion of the drug as described previously (Johnson et al., 2006). Cells were initially held at −60 mV and when the buffer was changed from low to high [K+], the membrane potential was stepped to −25 mV, which is the reversal potential for GIRK channel activation under these conditions. To measure GIRK channel activation in response to CXCL12, the membrane potential was then stepped from −25 to −60 mV for only 100 ms every 2 s. In this way, the current response to the agonist could be measured whereas the amount of K+ entering the cell during a recording was minimized. All drugs were applied in known concentrations in the superfusing solution that flowed at ∼1 ml.min−1.
Lipoparticle binding assays with backscattering interferometry.
Binding assays with backscattering Interferometry (BSI) were performed as described previously (Baksh et al., 2011). BSI is a unique form of interferometry consisting of a channel in a microfluidic chip, a laser-based source, and a detection camera. Briefly, lipoparticles containing CXCR4 (Integral Molecular), stored at 4°C, were used and binding was measured in the typical endpoint (Baksh et al., 2011). Ligand binding to the lipoparticle was accomplished by incubating a fixed amount of lipoparticle solution with varying concentrations of ligands (CXCL12 and GABAB modulators) for 1 h at room temperature. Solutions containing the same concentration of ligand and a null lipoparticle (lipoparticles without a receptor expressed) were used as reference samples to account for any nonspecific binding to the particle. Dopamine was used as a nonbinding negative control ligand and did not show any binding signal. For each sample, a solution of ligand and the null lipoparticle was introduced into the channel and the BSI signal was measured. The channel was rinsed and the solution containing the ligand and the CXCR4 lipoparticle was introduced into the channel and the signal measured. This procedure was repeated iteratively for increasing ligand concentrations. Data were collected with a program written in-house using LabView (National Instruments). The binding signal was calculated as the difference in phase between the null lipoparticle-ligand solution and the CXCR4 lipoparticle–ligand complex. The background signal due to the presence of the receptor was subtracted from all measurements. This corrected binding signal was plotted versus concentration to form a saturation binding isotherm and the affinity was calculated by fitting to a square hyperbolic function using GraphPad Prism software.
Tag-lite receptor-ligand binding assay.
To perform the binding assay at the CXCR4 ligand binding site (see Fig. 8), Cisbio Bioassays provided CXCR4 terbium cryptate-labeled HEK cells and CXCR4 and GABAB d2-labeled ligands. The assay was run in 384 low-volume white plates and readings were performed on PHERAstar FS flash lamp. It was performed twice with identical results.
For the competition assay with the CXCR4 ligand d2-labeled and unlabeled GABAB ligands, we used 10 μl of transfected terbium-labeled CXCR4 expressing HEK cells (resuspended in Tag-lite buffer), 5 μl of unlabeled GABAB ligands (baclofen, GABA, CGP 55 845, and CGP 54626) at different concentrations, and 5 μl of labeled CXCL12 at 12.5 nm. Cells were incubated 2h00 at room temperature.
To test the GABAB d2-labeled ligand (CGP 54626 derivative) on CXCR4 receptors, we used 10 μl of transfected terbium-labeled CXCR4 expressing cells (resuspended in Tag-lite buffer), 5 μl of Tag-lite buffer (or unlabeled CGP 54626 at 50 μm), and 5 μl of d2-labeled CGP 54626 derivative at different concentrations. Cells were incubated 2h00 at room temperature.
Drugs.
Murine recombinant CXCL12 was obtained from R&D Systems and baclofen, CGP 55845, CGP 54626, and AMD3100 were from Tocris Bioscience. The following drugs were from Sigma: tetrodotoxin (TTX), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), tetraethylammonium, 4-aminopyridine, CsCl, d-APV, GABA, and gabazine. gp120 and soluble hCD4 were obtained through the National Institutes of Health (NIH) AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases, NIH. Drugs were applied in the bath.
Statistics.
Statistical significance between groups (average data expressed as mean ± SEM, n = number) was tested using either the Student's t test or nonparametric Mann–Whitney test or ANOVA followed by t test. Statistical analysis was done using SigmaPlot (Jandel Science) and Origin (MicroCal) software. For BSI, data analysis and curve fitting was done with GraphPad Prism. Values of p < 0.05 were considered significant. This study was approved by Centre National de la Recherche Scientifique and University of Nice Sophia Antipolis.
Results
CXCR4 and GABAB often coexpress in the same cell type (Banisadr et al., 2002), have complementary functionality, and may be involved in cross talk (Duthey et al., 2010). We describe two new examples of such interaction, one involving chemotaxis the other involving ionic current modulations, in which agents historically believed to act selectively on GABAB receptors also affect the CXCR4 system. Furthermore, we provide evidence that these ligands specifically bind to CXCR4 and quantify the binding affinity for these interactions.
Chemotactic assays
There is a great deal of interest in stem cell therapy for the treatment of neurological disease (Peterson, 2004). It has previously been reported that baclofen inhibited CXCL12-induced migration of human peripheral blood mononuclear cells and suggested that the mechanism for this was heterologous desensitization of several chemokine receptors (Duthey et al., 2010). In another report, CXCL12-induced migration of hematopoietic stem and progenitor cells was also blocked by GABA and baclofen, through a suppression of the CXCL12-induced calcium (Seidel et al., 2007). GABA has been shown to inhibit the migration of cancer cells, although the mechanisms underlying these effects remain elusive (Ortega, 2003; Entschladen et al., 2004). To explore further the action of GABA and baclofen on tumoral cell migration induced by CXCL12, we used the human breast adenocarcinoma cell line MDA-MB-231, which has previously been characterized to express the CXCR4 receptor and to migrate toward CXCL12 in a migration assay (Vinader et al., 2011). Our RT-PCR and Western blots results are illustrated here (Fig. 1A,B) showing that both CXCR4 RNA and protein and GABAB receptors are endogenously expressed in these cells. An agarose gel chemotaxis assay was used to quantify cell migration as outlined in the Materials and Methods section (Fig. 1C). As proof of concept, photographic snapshots of the migration of cells under the agarose spots as controls (minus CXCL12), plus CXCL12, and plus CXCL12 and pre-incubation with the GABA antagonist CGP 55845 are shown in Figure 1D. It should be noted that at the concentration of antagonists used, the cells remained viable for the duration of the experiment (as attested by their morphology), except for CGP 54626 (5 μm), which was thus excluded from the study. Preliminary experiments to optimize the assay found that a concentration of CXCL12 (100 nm) gave a 10-fold increase in migration over basal, thus further experiments were performed with this concentration (Fig. 1E). Pre-incubation with CXCR4 antagonist AMD3100 and gp120/CD4 significantly inhibited the CXCL12-induced migration by, respectively, 48 and 51% but not back to basal levels. A similar inhibition of cell migration was measured when cells were pre-incubated with GABAB receptor antagonist (CGP 55845) and agonists (baclofen and GABA). These results suggest that there may be interaction between CXCR4 and GABAB agents.

GABAB agents reduce the migration of the breast cancer cells MDA-MB-231 in a chemotactic assay. A, RT-PCR reveals the presence of endogenous mRNAs for CXCR4 and GABABR2 in both HEK and MDA-MB-231 cells. B, Western blot reveals the presence of endogenous CXCR4 and GABABR in both HEK and MDA-MB-231 cells. HELA cells were used as a positive control. C, Schematic of the agarose spots on the 35 mm Petri dish. −, −−, +, and ++ are markings on the back of the Petri dish: + and ++, agarose drops made with the 10 and 100 nm CXCL12 agarose solution; − and −−, control drops in which equivalent amount of water was added to the agarose solution, respectively, to + and ++. The red inserts correspond to typical fields that were analyzed. D, Images of the agarose spots showing the cells crawling under the spot: Control (no chemoattractant), 100 nm CXCL12, and 100 nm CXCL12 and 5 μm CGP 55845. Scale bar, 100 μm. The dashed line represents the upper limit of the drop. E, Histogram showing the number of MDA-MB-231 cells under the agarose spots. Error bars indicate the mean of n = 18 fields (6 in 3 independent drops). Notice the dose-dependent increase in the migration with CXCL12 concentration and the reduction in migration in the presence of CGP 55845 (5 μm), GABA (100 μm), baclofen (100 μm), AMD3100 (2 μm), and gp120/CD4 (300 nm each). *p < 0.05, ***p < 0.001, t tests after ANOVA against control group. ++p < 0.02, +++p < 0.01, t test against 100 nm CXCL12 group.
Brain slices experiments
We previously reported patch-clamp whole-cell experiments showing that CXCR4 stimulation by CXCL12 chemokine positively modulates HVA calcium currents in dopaminergic neurons in acute brain slices from rat substantia nigra (Guyon et al., 2008). HVA calcium currents (Fig. 2A) were prevented by 200 nm AMD3100 (Fig. 2B), as previously described (Guyon et al., 2008). We investigated whether the CXCL12 HVA calcium currents (Fig. 2A) were also prevented by 500 nm 55845, which was significantly reducing the chemotactism induced by CXCL12 on MDA-MB-231 cells. In whole-cell patch-clamped dopamine neurons, 500 nm CGP 55845 significantly blocked the potentiating CXCL12 effect on HVA calcium currents (Fig. 2B), suggesting that the effect of CGP 55845 also interfered with CXCR4 in mammalian neurons, and that the use of GABAB pharmacological agents could interfere with CXCR4 receptors in the brain. GABAB agonists could not be tested here because they depress HVA Ca currents by stimulating GABAB receptors endogenously expressed on neurons (Guyon and Leresche, 1995).

The facilitator effect of 10 nm CXCL12 on HVA Ca currents recorded in dopaminergic neurons of the substantia nigra in rat brain slices is prevented by 500 nm CGP 55845. A, HVA calcium current recorded in a dopaminergic neuron in response to a voltage step from −60 to −10 mV in the absence (control) or the presence of 10 nm CXCL12. Calcium currents were isolated by the continuous perfusion of 1 μm TTX, 4-aminopyridine, 10 mm tetraethylammonium, 2 mm CsCl, 50 μm APV, 10 μm CNQX, and 1 μm gabazine. B, The effect of 10 nm CXCL12 on HVA Ca currents was significantly blocked by the CXCR4 antagonist AMD3100 (AMD, 200 nm) and by 500 nm CGP 55845, the GABAB receptor antagonist (applied in a different set of neurons).
Therefore, as shown in the two examples above, agents supposed to act on GABAB receptor also affect the CXCR4 system. We investigated the possibility that GABAB agents could also interact with CXCR4 receptor. To test this hypothesis, we first used a heterologous model of expression of wild-type CXCR4 in Xenopus oocytes that lack GABAB receptors (Uezono et al., 1998) or express them at low levels in a small subpopulation of cells (Yang et al., 2001).
Heterologous expression of CXCR4 on Xenopus oocytes
We performed electrophysiology experiments on Xenopus oocytes expressing wild-type CXCR4 chemokine receptor together with the GIRK channels (GIRK1 and GIRK2) as a reporter of CXCR4 activity. Activation by the CXCR4 ligand CXCL12 in high K+ Ringer's solution induced an inward current at holding potentials of −30 mV (Fig. 3A). The amplitude of the current response depended on the concentration of CXCL12, saturating at ∼1 nm (n = 11), with half-maximal stimulation at 0.49 ± 0.02 nm and Hill coefficient of 1.87 (Fig. 3B).

Electrophysiological recordings of oocytes expressing CXCR4, GIRK1, and GIRK2. A, Inward currents recorded in response to various concentrations of CXCL12 as indicated by the bars. B, Concentration–response curve of CXCL12. Currents were measured at the peak and plotted against log[CXCL12]. Values are the mean of several experiments as indicated by the numbers in parenthesis. The curve was fitted to a sigmoid curve using MicroCal Origin software, with EC50 of 0.49 ± 0.02 nm and a Hill number of 1.87. C, Current–voltage relationship obtained in control, in the presence of 1 nm CXCL12 and in the presence of 1 nm CXCL12 + 100 μm Ba2+. D, Current–voltage relationship of CXCL12-evoked current (current recorded in 1 nm CXCL12 minus control current). E, The inward current activated by 1 nm CXCL12 was blocked by 100 nm AMD3100, which induced a small inward current by itself. The response to 1 nm CXCL12 partially recovered after washout of AMD3100. F, The inward current activated by 1 nm CXCL12 was blocked by 30 nm gp120 applied together with 30 nm CD4, which induced a small inward current by itself. The response to 1 nm CXCL12 partially recovered after washout of gp120/CD4.
To verify that the currents induced by chemokine perfusion resulted from the activation of GIRK channels, we characterized the properties of these currents. The voltage dependence of the current showed a strong inward rectification, as expected for GIRK (Fig. 3 C,D), and the current showed a strong inhibition by Ba2+, also consistent with GIRK (Fig. 3C). The selective CXCR4 antagonist AMD3100 antagonizes the CXCL12-elicited GIRK current (Fig. 3E). AMD3100 (200 nm) induced a 76.67 ± 3.12% decrease (n = 4, from 2.15 ± 0.49 to 0.52 ± 0.16 μA, p < 0.02) in the current elicited by 1 nm CXCL12, confirming that the chemokine activates GIRK through the CXCR4 receptor. It should be noted that AMD3100 sometimes induced an inward current by itself (see Fig. 5E), but always prevented the effect of CXCL12. We also tested another CXCR4 antagonist, the recombinant protein HIV-1 IIIB gp120 (coapplied with recombinant CD4 as described previously; Tran et al., 2005). As illustrated in Figure 3 F, the gp120/CD4 complex antagonized the effects of CXCL12 by 80.5 ± 11.4% (n = 10, from 1.86 ± 0.28 to 0.59 ± 0.28 μA, p < 0.02), accompanied by a small direct induction of inward current (15.8 ± 5.2%, n = 6, of the current induced by 1 nm CXCL12), as previously described (Madani et al., 1998). No response to CXCL12, AMD3100, or gp120/CD4 was observed in water-injected oocytes (n = 5) or in oocytes expressing only the GIRK channels (GIRK1 and GIRK2) (n = 5; data not shown). Thus, we conclude that the genuine CXCR4 functionally activates the GIRK channel in oocytes.
Compounds acting on GABAB receptor act directly on CXCR4
We then tested the hypothesis that compounds acting on GABAB receptor could act directly on CXCR4. Both GABAB antagonists CGP 55845 and CGP 54626, baclofen, and GABA were inactive on oocytes injected only with GIRK1 and GIRK2 (n = 3 each; data not shown), suggesting the lack of functional expression of endogenous GABAB receptors on the batches of oocytes that we used (Yang et al., 2001). Surprisingly, in oocytes coexpressing CXCR4 and the G-protein-coupled GIRK channels (GIRK1 and GIRK2), both GABAB receptor antagonists CGP 55845 and CGP 54626 induced an inward current similar to the one induced by CXCL12 (n = 30 and 16, respectively for CGP 55845 and 54626; Figs. 4, 5A1,B1). The effect of both GABAB receptor antagonists was dose dependent (Fig. 4). The EC50s (see Table 1) were, respectively, 1.87 and 61.02 nm with Hill numbers of 1. Similarly, the GABAB receptor agonists baclofen and GABA were both able to reversibly activate inward currents in a dose-dependent manner (Fig. 4). They both acted in a similar concentration range (EC50s of 6.9 and 7.5 μm, respectively, and Hill numbers of 1). Notice that the Hill number of the concentration–response curve for CXCL12 (1.92) was higher than the Hill number of the concentration–response curve of the GABAB receptor agonists/antagonists, which suggests that GABAB receptor agonists/antagonists use different mechanisms of activation of CXCR4 than CXCL12.

Dose-dependent effect on CXCR4 receptor of several pharmacological agents acting on GABAB receptor. A, Inward currents recorded in oocytes expressing CXCR4, GIRK1, and GIRK2 in response to the drugs indicated on the left, at various concentrations as indicated by the bars. B, Concentration–response curves of several pharmacological agents acting on GABAB receptor as compared with CXCL12. Currents were measured at the peak, normalized to the maximum response, and plotted against log[CXCL12]. Values are the mean of several experiments, which number (n) is indicated in Table 1. The curves were fitted to sigmoid curves using MicroCal Origin software. EC50 and a Hill number are also indicated in Table 1 (mean ± SEM).

Effects of various pharmacological agents acting on GABAB receptors on oocytes coinjected with CXCR4 and GIRK1 and GIRK2. A1, B1, CGP 55845 (500 nm) and CGP 54626 (500 nm) both induced an inward current. A2, B2, These currents were reversibly blocked by 200 nm AMD3100 recordings obtained in other oocytes than A1 and B1. C, The inward current elicited by 1 nm CXCL12 was reversibly blocked by 500 nm CGP 55845, which could itself induce an inward current as in the oocyte or have a very small effect by itself. D, The inward current elicited by 1 nm CXCL12 was reversibly blocked by 500 nm CGP 54626, which itself induced an inward current in the oocyte presented in this example. E, The effects of 10 μm baclofen and 10 μm GABA were blocked by perfusion of 200 nm AMD3100. F1, F2, The inward current elicited by 1 nm CXCL12 as blocked by perfusion of both 10 μm baclofen (F1) and 10 μm GABA (F2). G1, G2, The inward currents elicited by 10 μm baclofen (G1) and 10 μm GABA (G2) were blocked by 30 nm gp120. The current even became outward in the presence of 30 nm gp120 suggesting that a proportion of CXCR4 receptors was constitutively activating GIRK in these oocytes, which was blocked by the gp120.
EC50s and Hill numbers (mean ± SEM) of dose-response curves obtained by recording inward currents on oocytes expressing CXCR4, GIRK1, and GIRK2 in response to the different drugs indicated on the left as illustrated in Figure 4
To investigate the mechanism of action of GABAB receptor agonists and antagonists on CXCR4, we did additional experiments of coapplication of GABAB receptor antagonists/antagonists together with compounds known to interact at the binding site of CXCL12 on CXCR4 (CXCL12 itself, AMD3100, gp120). Of interest, in the presence of the CXCR4 antagonist AMD3100 (200 nm), the current induced by GABAB receptor antagonists switched from inward to outward, and this effect was reversible upon washout of AMD3100 (Fig. 5A2,B2; n = 3 each). In the presence of both GABAB receptor antagonists CGP 55485 and CGP 54626, the current induced by CXCL12 was blocked or became outward (Fig. 5C,D, respectively, n = 8 and n = 4). The CXCR4 antagonist, AMD3100, switched the effect of baclofen and GABA from an inward current to a small outward current (Fig. 5E; n = 3 each). As the GABAB receptor antagonists, the GABAB receptor agonists baclofen and GABA switched the effect of CXCL12 from an inward to a small outward current (Fig. 5 F1,F2, respectively; n = 5 each). Finally, in the presence of baclofen (n = 3) and GABA (n = 6), gp120 induced an outward current (Fig. 5 G1,G2).
To summarize, in Xenopus oocytes expressing CXCR4 together with GIRK, GABAB receptor antagonists/agonists are able to activate GIRK. However, once the CXCR4-CXCL12 binding site is occupied (by CXCL12, AMD, or gp120), they have an opposite effect. Similarly, once GABAB receptor antagonists/agonists interact with the CXCR4, the occupation of the CXCL12 ligand-binding domain induces an inactivation of GIRK instead of activation. Therefore, GABAB agents act on CXCR4 in an allosteric manner at a site separate from the CXCL12-CXCR4 binding domain. Indeed, if it was simple competition at this site, one would expect a partial or full block of the effect of the ligand rather than a switch from activation to inactivation states.
As the CXCR4 antagonist AMD3100 unexpectedly behaved as an agonist in Xenopus oocytes, we thus decided to use another expression system to validate our results with agents acting on GABAB receptors in a mammalian cell line. HEK293 cells were further chosen as a model because they express native CXCR4 although at very low levels (Busillo et al., 2010) and because a ligand-binding assay is available in this cell line.
GABAB agents also interact with CXCR4 expressed in HEK293 cells
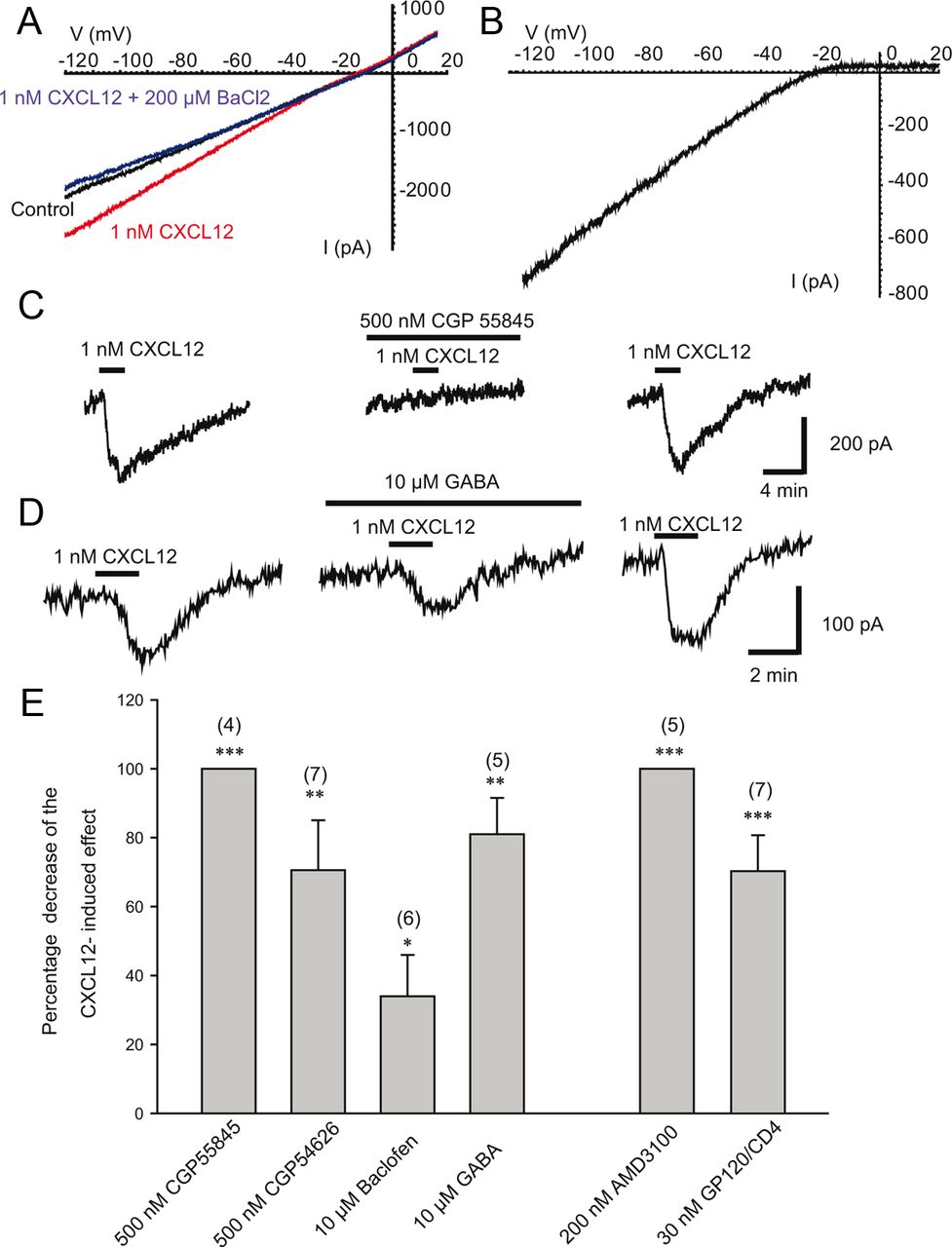
Whole-cell patch-clamp recordings were then performed in HEK293 cells transiently expressing CXCR4, GIRK1-F137S, and YFP and superfused with an extracellular solution containing raised [K+]. In cells transfected with CXCR4 and GIRK1-F137S, CXCL12 (1 nm) evoked currents that displayed a high degree of inward rectification (Fig. 6A,B). Barium (200 μm), a blocker of Kir channels, abolished the CXCL12-evoked current (Figure 6A; n = 5).

Effects of various pharmacological agents acting on GABAB receptors on patch-clamp recordings from HEK293 cells transiently expressing CXCR4 and GIRK channels. A, Current–voltage relationship in control conditions (leak current in the presence of the extracellular solution with elevated KCl), in the presence of CXCL12, or in the presence of CXCL12 + 200 μm of the GIRK channel blocker BaCl2, as indicated. B, CXCL12-induced current. Leak current has been subtracted. CXCL12 evoked an inwardly rectifying current that had a reversal potential of −24.6 mV. C, Representative traces showing the effect of 1 nm CXCL12 before, during, and after the washout of 500 nm CGP 55845, as indicated by the bars. Cells were held at −25 mV and then stepped to −60 mV for 100 ms every 2 s (see Materials and Methods). D, Representative traces showing the effect of 1 nm CXCL12 before, during, and after the washout of 10 mm GABA, as indicated by the bars. E, Histogram showing the percentage of decrease induced by the various compounds indicated on the GIRK current induced by 1 nm CXCL12 in HEK293 cells transiently expressing the CXCR4 and GIRK channel.
The reversal potential for the CXCL12-evoked current was −23.4 ± 4.6 mV (n = 5), which agrees closely with the calculated equilibrium potential for K+ of −25 mV under these recording conditions. At a holding potential of −60 mV, the maximum current evoked by 1 nm CXCL12 was −383.8 ± 63.9 pA (n = 42). This current was fully and reversibly blocked by 200 nm AMD3100 (n = 5), which induced an outward current of +230 ± 86 pA by itself (n = 5), suggesting that some CXCR4 was constitutively active. gp120/CD4 (3 nm each) also blocked 70.0 ± 10.4% (n = 7) of the current induced by 1 nm CXCL12. Interestingly, CXCL12-induced current was also significantly blocked by 500 nm CGP 55 845 and CGP 54 626 (Fig. 6 C,E). These GABAB antagonists had no effect when applied alone. Similarly, GABA and to a lesser extent baclofen also significantly blocked the CXCL12-induced current (Fig. 6D,E) while these GABAB receptor agonists had no effect when applied alone.
When transfected with GIRK+ and YFP without CXCR4, some transfected HEK293 cells (6/17 tested) exhibited a small inward current of −79.2 ± 24.5 pA (n = 6) at −60 mV in response to CXCL12 (1 nm). No current was detected in response to AMD, gp120/CD4, CGP 55845, or CGP 54626 (n = 9 each). Only a small amount of HEK293 cells transfected with GIRK only (2/7 cells) presented a small inward current in response to baclofen (10 μm) or GABA (10 μm; between −25 and −75 pA) suggesting that low amounts of GABAB receptors were present endogenously only in a subpopulation of HEK293 cells, as confirmed by the RT-PCR or Western blot experiments that exhibited a small band for both human GABABR2 and CXCR4 receptor (Fig. 1A,B).
Baclofen and other GABAB agents directly interact with CXCR4 expressed in lipoparticles
To substantiate the data above that GABAB agents are directly binding the CXCR4 receptor, we used BSI, a molecular interaction photometer, to quantify ligand/receptor binding. We previously reported binding specificity of the native pairs CXCR4/CXCL12 binding using red blood cell “ghosts” with human SUP-T1 lymphoma T-cells expressing CXCR4 and BSI (Baksh et al., 2011). Here, we expressed the CXCR4 receptor using lipoparticle technology: protein directly incorporated into virus-like particles with a lipid bilayer surface that provides concentrated protein (50–200 pm/mg) in the native conformation (Jones et al., 2008). Equilibrium dissociation constants were calculated using saturation analysis. To demonstrate that the refractive index (RI) change is not due simply to the introduction of the pairs, we used dopamine as a control in each experiment (at five concentrations that span the concentration range in all the other curves), and measured no RI change. In Figure 7A the saturation isotherm measured binding of CXCL12 with a KD of 0.49 nm (± 0.16 nm), in agreement with a previously published report (Jones et al., 2008). Next, we measured binding of the two GABAB antagonists CGP 55845 and CGP 54626 (Fig. 7B,C) calculating KD values of 11 nm (±0.5 nm) and 35 nm (±6.2 nm), respectively. Finally, we measured binding of two GABAB agonists to CXCR4, baclofen, and GABA (Fig. 7D,E) calculating KD values of 10.3 ± 2.8 μm and 0.57 ± 0.24 μm. To confirm that this binding is at a different site from that of CXCL12, we pre-incubated the lipoparticles with CXCL12 and performed a binding assay using baclofen. Figure 7F shows that baclofen is still binding CXCR4 even when CXCL12 is bound and the affinity and signal magnitude for baclofen with the receptor is enhanced by CXCL12 binding. To demonstrate that the observed binding was not due to CXCL12 interacting with baclofen, the assay was performed in the absence of CXCR4, with no observed binding signal (Fig. 7F). These results are proof in principle that GABAB ligands directly bind CXCR4, and at a site distinct from the active site.

Representative plots of BSI signal versus ligand concentration for the determination of binding constants for CXCR4 to the following ligands: CXCL12 (A), CGP 55845 (B), CGP 54626 (C), baclofen (D), and GABA (E). Dopamine (tested at 5 different concentrations: 0.02, 2, 200, 2, and 200 nm) was used as a nonbinding control ligand for all five plots. The binding of baclofen to the CXCR4–CXCL12 complex (0.2 nm CXCR4 + 20 nm CXCL12) is represented in F; also shown is the negative control of the interaction of baclofen with CXCL12. For all plots, error bars indicate SDs of the measurements from three independent trials.
GABAB agents do not interact with CXCR4 at the CXCL12 ligand binding site
To check whether GABAB antagonists and agonists do not bind competitively with CXCL12, we used the Tag-lite receptor ligand technology, which is designed to show a binding between the ligand and the receptor by a homogeneous and nonradioactive assay on living cells (Fig. 8A). To check the ability of unlabeled GABAB ligands to bind to CXCR4 receptor, a competition assay was first performed with the CXCR4 ligand d2-labeled and unlabeled GABAB ligands, testing a wide range of concentrations of unlabeled compounds against a fixed concentration of CXCL12-d2 ligand. None of the GABAB ligand was able to compete with the d2-labeled ligand while the d2-labeled CXCL12 was displaced by the unlabeled CXCL12 with KI in accordance with expected values (Fig. 8B). These data suggest that these ligands do not bind to the CXCL12-CXCR4 binding site. We also tested whether the GABAB d2-labeled ligand (CGP 54626 derivative) could interact at the CXCL12 binding site of CXCR4 receptors. The specific binding curve, obtained by subtracting the nonspecific signal from the total binding signal, revealed that the d2-labeled CGP 54626 derivative (used as a GABAB fluorescent ligand) does not bind to the CXCR4 receptor at the CXCL12 binding pocket (Fig. 8C). Therefore, as hypothesized, GABAB agents do not interact at the binding site of CXCL12 on CXCR4 but modulate the activation of the receptor by allosteric interaction.

Baclofen and other ligands of GABAB receptor do not bind at the CXCL12/SDF1 binding site on CXCR4. A, Principle of the Tag-lite receptor-ligand technology. The receptor is expressed at the cell surface with a SNAP tag and then labeled with a fluorescent donor dye (terbium cryptate) through an appropriate substrate. On the other side, the ligand is labeled with a red acceptor dye (d2). If the fluorescent donor dye labeled on the receptor is excited by a nitrogen laser or flash lamp (∼340 nm) when the ligand binds to the receptor, there is a transfer of energy between the donor dye to the acceptor dye, resulting in the latter dye emitting light, in a time-resolved manner, at 665 nm. The receptor-ligand binding can hence be monitored at 665 nm on a time-resolved mode. B, Competition assay run with d2-labeled CXCL12 at KD (12.5 nm). None of the unlabeled GABAB ligand was able to compete with the d2-labeled ligand while the d2-labeled CXCL12 was displaced by the unlabeled CXCL12 as expected. C, The specific binding curve was obtained by subtracting the nonspecific signal from the total binding signal. It revealed that the d2-labeled CGP 54626 derivative (used as a GABAB fluorescent ligand) does not bind to the CXCR4 receptor at the CXCL12 binding pocket.
Discussion
Our study demonstrates that GABAB agonists and antagonists directly bind CXCR4 by allosteric action. This finding has important physiological implications for the immune and nervous systems and is also highly relevant in pointing a novel molecular target to baclofen, a drug widely used in human health.
Direct effect of GABAB receptor agents on CXCR4
The human breast cancer cell line MDA-MB-231 expressing endogenously CXCR4 allowed us to demonstrate the effects of both GABAB receptor agonists and antagonists on native CXCR4 by using the chemotactic properties of the chemokine CXCL12. In neurons, the GABAB receptor antagonist blocked the effects of CXCL12 on HVA calcium currents. However, in neurons, it is hard to decipher the relative effect of GABAB pharmacological agents on CXCR4 or GABAB receptor because neurons express both CXCR4 and GABAB in similar amounts. To resolve whether these agents have a direct effect at CXCR4, we used X. laevis oocytes or HEK293 cells expressing CXCR4, which have relatively no endogenous GABAB receptor expression compared with the overexpressed CXCR4. These cells allow for pharmacological studies although we acknowledge that the receptor might behave differently in vivo. For example, we observed in TIRF experiments that CXCR4 does not form dimers when expressed in X. laevis oocytes (A. Guyon, unpublished data) contrary to what is described in mammalian cells. In oocytes expressing CXCR4 receptors, GABAB agonists and antagonists both behaved as agonists as they were able to activate GIRK current. This was not a direct effect on GIRK as the effect was absent in oocytes injected with GIRK only. These compounds acted on CXCR4 since they interfered with CXCL12 and AMD3100 for the activation of GIRK. In HEK293 cells, however, GABAB agonists and antagonists both blocked the CXCL12-induced current without inducing any currents by themselves. Therefore, it is likely that in mammalian cells (neurons and immune cells), GABAB receptor agents behave as antagonists on CXCR4, and thereby block the effects of CXCL12.
Similarly, AMD3100 behaved as an agonist in the Xenopus oocyte experiments. AMD3100 has previously been described to behave as a partial agonist and has shown some agonistic effects in other models (Zhang et al., 2002; Tran et al., 2005). Such partial agonist activity precludes the use of this agent as an antagonist of metastatic activity in mammalian carcinoma because stimulation of CXCR4 at the surface of tumor cells could theoretically increase dissemination. However, AMD3100 did not behave as an agonist in HEK293 cells transiently transfected with CXCR4 and GIRK. Contradictorily, it induced an outward current, likely due to blocking the activity of tonically activated CXCR4. Similarly, in the chemotactic assay, AMD3100 blocked the migration of MDA-MB-231 cells toward the agarose drop containing CXCL12. Thus, AMD3100 behaves differently depending on the cell type expressing CXCR4. This could be due to distinct intracellular pathways, to a different folding or conformation of the protein, or to heterologous dimerization depending on the tested model.
The BSI assays showed a direct interaction of GABAB receptor antagonists and agonists with CXCR4, with KD values that are in the range of our electrophysiological results. The Tag-lite competition ligand-binding assay demonstrated that GABAB receptor antagonists/agonists do not interact at the ligand-binding domain for CXCL12 on CXCR4. This result is corroborated by the difference in Hill numbers observed in the concentration–response curves between CXCL12 on one side (close to 2) and the GABAB agents on the other side (Hill number close to 1). This difference indicates a separate mode of action. It is hard to speculate on the site and mechanisms of interaction of GABAB agents on CXCR4 and these questions will be the topic of future studies. Using Tag-lite technology, we found no fluorescent resonance energy transfer interaction between the CXCR4 terbium cryptate, fixed at the CXCL12 binding site, and the GABAB receptor-labeled ligands. Forster's radius (R0, distance equivalent to 50% transfer efficiency) of 9 nm CXCR4 terbium cryptate-GABAB d2-labeled ligand pair, indicates that the allosteric site of GABAB ligands is thus far from the CXCL12 binding pocket on CXCR4. Another way to test this would be using the crystal structure of the receptor in the presence of the GABAB receptor antagonists/agonists to allow determining where these compounds interact on the chemokine receptor. Silencing CXCR4 expression through siRNA technology would also provide some clue to the relevance of GABA–CXCR4 interactions in mammalian cells and whether a heterologous dimerization may be occurring. Other mechanisms of interactions between GABAB and CXCR4 are currently under investigation, and will address both direct interaction and activation of second messenger cascade.
Physiological implication
This study shows that GABA is able to block the effect of CXCL12 on CXCR4. Thus, it is likely that when the GABAergic system is activated, GABA released in the brain will antagonize the effect of CXCL12 on its receptor CXCR4, and thus could influence the chemokine neurotransmission as well as the inflammatory response in the CNS. We now demonstrate that there is reciprocal cross talk between these two systems as it has previously been shown that CXCR4 stimulation by CXCL12 can increase presynaptic GABA release (Guyon and Nahon, 2007).
Indeed, in dopaminergic neurons of the rat substantia nigra, we have previously shown that CXCR4 stimulation by CXCL12 induces an increase of release of presynaptic neurotransmitter, particularly of GABA (Guyon et al., 2006). We have also shown that CGP 55845 (500 nm) blocks the outward GIRK current induced by CXCL12 (recorded in the presence of glutamate receptor blockers). At that time, we interpreted this result as an effect mediated through GABAB receptor stimulation by GABA spilling over following CXCL12 presynaptic stimulation and increase in GABAB release. However, in view of the present results, we can re-interpret our data. Indeed, the GIRK currents might have been activated by the stimulation of postsynaptic CXCR4 by CXCL12, which was then blocked by CGP 55484.
CXCR4 activation by CXCL12 has been shown to increase presynaptic neurotransmitter release and particularly GABA release in several neuronal populations (Guyon and Nahon, 2007; Bhattacharyya et al., 2008; Qu et al., 2008). If GABA can in turn block the effects of CXCL12, this could represent a negative feedback loop for presynaptic chemokine release. Indeed, when applying CXCL12 for several minutes, a transient increase in the frequency of spontaneous postsynaptic currents is frequently observed, followed by a reduced activity (Guyon et al., 2006, their Figure 3). This reduction could be due to an antagonistic effect of GABA, although desensitization of CXCR4 itself cannot be excluded. Similarly, it has been shown that elevated concentrations of CXCL12 exert more opposite effect than lower concentrations on the electrical activity of some neuronal populations that receive GABA inputs (Guyon and Nahon, 2007). The antagonistic effect of GABA released presynaptically in response to CXCL12 could contribute to these biphasic effects. In the future, it will be of interest to search for putative effects of GABAB receptor ligands on CXCR7, the other receptor for CXCL12 (Schönemeier et al., 2008), as well as on other chemokine receptors.
Putative applications in cancer treatment and inflammation
Baclofen treatment was demonstrated to reduce the incidence of some carcinogen-induced gastrointestinal cancers in rats (Tatsuta et al., 1990) as well as human hepatocarcinoma cell growth (Wang et al., 2008). In contrast, baclofen promotes human prostate cancer cell migration (Azuma et al., 2003). As CXCR4 is highly expressed in cancer cells, baclofen may have been acting through CXCR4 in these examples.
Similarly, it has been shown that GABA can affect cell proliferation and have anti-inflammatory properties on fibroblasts, although the mechanism of action of GABA was not elucidated (Han et al., 2007). We suggest that GABA may have acted through the CXCR4 receptor, as CXCR4 is expressed on fibroblasts (Qu et al., 2008). Baclofen is currently used for the treatment of spasticity in patients with spinal cord injury, cerebral palsy, traumatic brain injury, multiple sclerosis, and other disorders (Plassat et al., 2004; Guglani and Lodha, 2007; Kolaski and Logan, 2008; Rekand and Grønning, 2011). Recently, it has been used in the treatment of alcohol dependence and withdrawal (Addolorato et al., 2006). The allosteric effects of such agents at CXCR4 likely contribute to these beneficial effects as CXCR4 often colocalizes with GABAB receptors.
As a conclusion, this study opens new perspectives on the putative use of baclofen and other GABAB agents acting at CXCR4 for their therapeutic potential to treat quite a broad range of diseases, such as ischemic stroke, brain tumors, HIV encephalopathy, and multiple sclerosis, as well as affecting stem cell migration (Duthey et al., 2010) and other cancers.
Footnotes
This work was supported by Centre National de la Recherche Scientifique (CNRS) (INSB), ANR-MNPs-018-01, Fondation France Parkinson, Fondation de la Recherche Médicale, and the National Science Foundation (Grant CHE-0848788). G.S. was supported by the Atip-Avenir fund from the CNRS and INSERM. We are grateful to Richard Miller for generously providing us with the rat CXCR4 clone and for all his advice. We thank Ehud Isacoff for his generous welcome. We greatly appreciated the assistance of Dr. Shashank Bharill, Dr. Ryan Arant, and Dr. Hitomi Okada. We thank Dr. Josh Levitz for providing us with GIRK1-F137S and Drs. Thomas Berger, Zhu Fu, Benji Gaub, Grant Kauwe, Susy Kohout, Andreas Reiner, Sasha Shekhar, Sandra Wiese, and all the Isacoff lab for fruitful discussions and technical support. We thank Dr. Piotr Bregestovski, Dr. Gregory Conductier, and Dr. Brice Flammang for their advice on experiments. We thank Audrey Recouly, Fabienne Chevallier, and Florence Servent from Cisbio Bioassays, Codolet, France, for the receptor-ligand binding assay using the Tag-lite technology. We thank Dr. Joseph Rucker from Integral Molecular for generously providing the lipoparticles. We acknowledge Ann Fischer and Michelle Richner from the University of California Berkeley MCB Tissue Culture Facility for cell culture support; Dr. Franck Bihl from IPMC, France; and Drs. Victoria Vinader and Kamyar Afarinkia from the University of Bradford, UK, for their advice on chemotactic assay. We acknowledge Michelle Sexton for help in preparation of this manuscript and Franck Aguila for artwork. The following reagents were obtained through the National Institute of Health (NIH) AIDS Research and Reference Reagent Program, Division of AIDS, National Institute of Allergy and Infectious Diseases (NIAID), NIH: Recombinant HIV-1 IIIB gp120 (CHO), cat#11784 from DIAIDS and NIAID (Immunodiagnostics) and Recombinant Soluble Human CD4, cat#4615, from Progenics Pharmaceuticals.
- Correspondence should be addressed to Alice Guyon, Centre National de la Recherche Scientifique, Institut de Pharmacologie Moléculaire et Cellulaire, 06560 Valbonne, France. alice.guyon{at}ipmc.cnrs.fr





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}