

Abstract
Synaptic scaling is a form of synaptic plasticity that contributes to the homeostatic regulation of neuronal activity both in vitro and in vivo, by bidirectionally and proportionally adjusting postsynaptic AMPA receptor (AMPAR) abundance to compensate for chronic perturbations in activity. This proportional regulation of synaptic strength allows synaptic scaling to normalize activity without disrupting the synapse-specific differences in strength thought to underlie memory storage, but how such proportional scaling of synaptic strength is accomplished at the biophysical level is unknown. Here we addressed this question in cultured rat visual cortical pyramidal neurons. We used photoactivation and fluorescence recovery after photobleaching of fluorescently tagged AMPAR to show that scaling down, but not up, decreases the steady-state accumulation of synaptic AMPAR by increasing the rate at which they unbind from and exit the postsynaptic density (Koff). This increase in Koff was not diffusion limited, was independent of AMPAR endocytosis, and was prevented by a scaffold manipulation that specifically blocks scaling down, suggesting that it is accomplished through enhanced dissociation of AMPAR from synaptic scaffold tethers. Finally, simulations show that increasing Koff decreases synaptic strength multiplicatively, by reducing the fractional occupancy of available scaffold “slots.” These data demonstrate that scaling down is accomplished through a regulated increase in Koff, which in turn reduces the fractional occupancy of synaptic scaffolds to proportionally reduce synaptic strength.
Introduction
Efficient information storage during learning and memory is thought to depend upon the presence of homeostatic mechanisms such as synaptic scaling that normalize synaptic strengths (Abbott and Nelson, 2000; Turrigiano and Nelson, 2004). Synaptic scaling is a cell-autonomous process in which neurons detect changes in their own firing through a set of calcium-dependent sensors, and then slowly (over many hours) increase or decrease the accumulation of synaptic AMPA receptors (AMPARs) to compensate (Turrigiano et al., 1998; Ibata et al., 2008; Goold and Nicoll, 2010). A hallmark of synaptic scaling is the proportional regulation of postsynaptic strengths that preserves the relative differences in weights across synapses (Turrigiano et al., 1998). Despite great recent interest in the mechanisms of synaptic scaling, the biophysical mechanisms that underlie this proportional scaling of AMPAR accumulation remain unknown.
Synaptic AMPARs are highly dynamic; after insertion into the membrane they diffuse laterally and can be transiently captured at the postsynaptic density through binding to scaffold proteins (or “slots”), before unbinding and diffusing away again and/or being internalized at endocytic zones (Gerrow and Triller, 2010; Kennedy et al., 2010; Opazo and Choquet, 2011). Given these dynamics, the steady-state number of synaptic AMPAR (AMPARss) should depend on a number of factors, including the concentration of diffusing surface receptors (controlled by relative rates of exocytosis and endocytosis), the number of synaptic slots available to transiently bind and immobilize them, and the relative rates at which AMPAR bind to (Kon) and unbind from (Koff) these slots (Earnshaw and Bressloff, 2006; Gerrow and Triller, 2010; Opazo and Choquet, 2011; Opazo et al., 2012). Long-lasting forms of postsynaptic plasticity have generally been ascribed to enhanced AMPAR forward trafficking and exocytosis (synaptic strengthening, Malinow and Malenka, 2002), regulated endocytosis and degradation of AMPAR (synaptic weakening, Malenka and Bear, 2004), or changes in the number of AMPAR slots (Lisman and Raghavachari, 2006). It is unknown whether synaptic weakening can be achieved through an alternative mechanism: by targeting the rate at which AMPAR unbind from synaptic scaffolds and diffuse away (Koff).
Here we used photoactivatable green fluorescent protein (PAGFP)-tagged GluA2, as well as fluorescence recovery after photobleaching (FRAP) of superecliptic phluorin (SEP)-tagged GluA2 (Lippincott-Schwartz et al., 2003), to probe the dynamics of AMPAR turnover after scaling up or down. This approach allowed us to quantify independently the magnitude and dynamics of the synaptic receptor pool that is freely diffusing, and the pool that is reversibly bound to synaptic scaffold proteins. Both methods revealed that scaling down doubled the rate at which the bound fraction of AMPAR exited the synapse (Koff), whereas scaling up did not affect Koff. Scaling down and increased Koff did not rely on increased AMPAR endocytosis, but did depend on scaffold interactions. Finally, simulations indicate that the change in Koff we measure can account quantitatively for multiplicative scaling down. Together, these data show that scaling down is accomplished through a regulated increase in the rate at which AMPAR unbind from synaptic scaffolds.
Materials and Methods
Neuronal cultures and transfection.
Cortical cultures were prepared from visual cortex of postnatal day 1–2 Long–Evans rat pups of both sexes, and plated onto beds of confluent astrocytes as described previously (Wierenga et al., 2005). All experiments were performed on pyramidal neurons after 7–10 d in vitro. All experimental conditions were compared with age-matched controls from sister cultures and all data were obtained from at least three independent platings. Neurons were transfected 2–4 d before experimentation using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instruction. All experiments were performed 2–4 d after transfection unless otherwise noted. Scaling up was induced with tetrodotoxin (TTX; 2 μm) or in a few experiments with 6,7-dinitroquinoxaline-2,3-dione (DNQX; 20 μm) (Turrigiano et al., 1998; Blackman et al., 2012), and scaling down was induced by picrotoxin (PTX; 100 μm) (Turrigiano et al., 1998; Sun and Turrigiano, 2011); drugs were added 1 d before experimentation and refreshed 4–6 h before the experiment. FRAP measurements following DNQX and TTX treatment were not different so data were combined in the Figure 5A “scaling up” condition. EGFP-GluA2 was as described previously (Ibata et al., 2008); SEP-GluA2 and PAGFP-GluA2 constructs were generated by replacing EGFP with PAGFP or SEP. For imaging experiments mRFP or CFP was cotransfected with tagged GluA2. All FRAP experiments used the SEP tag except the data in Figures 5, A and B, and 8.
FRAP and photoactivation experiments.
FRAP and photoactivation experiments were performed using a Leica TCS SP2 or SP5 confocal/multiphoton microscope; the dynasore FRAP experiments in Figure 6 were performed on a Marianas Zeiss spinning disk confocal. Neurons transfected with EGFP-GluA2 or SEP-GluA2 were identified under epifluorescence and imaged using the 488/543 nm laser lines. GluA2 was used because synaptic scaling requires the GluA2 subunit (Gainey et al., 2009) and results in bidirectional changes in GluA2 accumulation (Ibata et al., 2008). Cultures were continuously perfused with imaging buffer throughout the experiment, as described previously (Ibata et al., 2008). The imaging buffer contained the following (in mm): 117 NaCl, 5.3 KCl, 1.8 CaCl2, 0.814 MgSO4, 1 NaH2PO4, 20 HEPES, 50 dextrose, and 100 mg bovine serum albumin, with a final osmolarity between 320 and 330 mOsm, pH 7.3. Experiments in Figure 5, A and B, were performed at 33°C. Experiments in Figures 2, 3, and 5, C and D, were performed at 25°C in the presence of acute 0.1 μm TTX to mimic conditions used to measure synaptic scaling of miniature EPSCs (mEPSCs). The control dataset from Figure 5C is also plotted in Figure 3C (1 μm bleach spot) to allow comparison of the FRAP kinetics when large and small bleach spots were used. For all measurements (here and below) laser power and image acquisition were adjusted to keep bleach rates negligible, and were then kept constant across experiments. Six to eight z-stack images were taken for each region of interest. After collecting the first two sets of baseline images laser power was increased to 100% and one to three synapses were bleached by five consecutive laser scans. For dendritic FRAP measurements and “large bleach spot” synaptic FRAP measurements an ∼10 μm dendritic segment was bleached. Fluorescence was quantified using in-house and commercial analysis programs (Igor Pro, ImageJ, and Leica LAS-AF lite). The data were normalized to fluorescence before bleaching (defined as 100%) and immediately after bleaching (0%). Because the synaptic fluorescence recovery curves reached a plateau by 60 min after photobleaching the data from the last two time points (60–80 min) were averaged to determine the recovery fraction (mobile fraction). For comparison of recovery time constants under different conditions curves were normalized to the recovery fraction (Normalized Recovery). Spines were defined as protrusions <2 μm in length that contained an AMPAR cluster, and were only included in analysis if they persisted throughout the imaging experiment.
PA-GFP experiments were conducted in imaging buffer at 33°C. A 780 nm two-photon laser was used to photoactivate PAGFP-GluR2 by consecutively scanning a dendritic region of interest (identified by expression of mRFP) of 10 μm in length three to five times. The imaging collection was performed as described for the FRAP experiment. At the end of each experiment, a second activation protocol was applied to reactivate the same region of interest to ensure that the punctum had not been lost; all included puncta had reactivation values of >60% of initial activation values; the averages across conditions were between 90 and 100% of initial values and were not significantly different. Data were normalized to PAGFP fluorescence before photoactivation (defined as 0%) and immediately after activation (100%).
NMDA-induced internalization of AMPAR.
Neurons were transfected with SEP-GluA2; pyramidal neurons were identified based on morphology and imaged on an Olympus IX70 inverted microscope, with a 60×, 1.25 NA oil-immersion objective. Cultures were perfused with modified imaging media (120 mm NaCl, 25 mm HEPES, 5 mm KCl, 2 mm CaCl2, 30 mm dextrose, 2 mm MgCl2, 1 μm TTX, pH 7.4) at 25°C. Baseline was established by imaging for 5 min before NMDA application. NMDA (20 μm) was applied to neurons in imaging media with 0.3 mm MgCl2 for 5 min to induce endocytosis of GluA2 receptors (Ashby et al., 2004). For the acute dynasore condition neurons were perfused with dynasore (80 μm[scap]; Tocris Bioscience) for 10 min before the experiment and during baseline images and NMDA application. Images were acquired with an Olympus IX80 microscope fitted with an ORCA-ER2 CCD camera, using a 6% neutral density filter to reduce bleaching; exposure times were kept constant across conditions. Background-subtracted SEP-GluA2 intensity at the cell body was calculated (excluding the nucleus) and normalized to the baseline. Intensity values at each time point were normalized to this average baseline to generate traces for each cell.
Measuring GluA2 endocytosis with CypHer5E.
Two days after transfection with SEP-GluA2 neuronal culture media was replaced with prewarmed live cell imaging media. Cells were placed on a Warner instrument DH35i stage heater and maintained at ∼33°C. Images were collected with an Olympus IX80 microscope fitted with an ORCA-ER2 CCD camera. Following acquisition of baseline images CypHer tagged rabbit anti-GFP antibody (Synaptic Systems; cat no. 132003 CPH-0.5 mg/ml) was added to the dish at a dilution of 1:4000 to 1:6000. Surface SEP fluorescence was monitored to make sure no clustering of the GluA2 receptor or change in surface receptor intensity was induced by the addition of CypHer tagged antibody. No CypHer fluorescence was observed in nontransfected cells. Images were collected at 5 min intervals for 1 h in the presence of the antibody. To verify that internal CypHer puncta remained attached to SEP-GluA2, in live neurons the internal pH was neutralized by permeabilizing with imaging buffer containing 0.01% Triton X-100 on the microscope stage while images were collected. To immunostain endogenous GluA2 receptors neurons were treated with 1:6000 CypHer-tagged rabbit anti-GFP antibody for 30 min in the incubator to allow internalization, then washed, fixed, and permeabilized with 0.1% Triton X-100 before labeling overnight with an anti-mouse GluA2 antibody (Millipore) and a mouse Alexa 546-conjugated secondary antibody.
Simulations.
The FRAP simulation was performed in Igor Pro. Baseline conditions consisted of 100 binding sites (N); empty sites filled with a rate Kon and filled sites emptied with a rate Koff. Then, after bleaching, the number of fluorescent receptors (Nfl) at time t + 1will be the following:
 where Nss is the steady-state number of bound receptors. At steady-state the receptor removal and addition rates are equal, so Nss = N * Kon/(Kon + Koff). The parameters Kon, Koff, and N were modified as described in text to determine the effect on Nss and on FRAP Tau.
where Nss is the steady-state number of bound receptors. At steady-state the receptor removal and addition rates are equal, so Nss = N * Kon/(Kon + Koff). The parameters Kon, Koff, and N were modified as described in text to determine the effect on Nss and on FRAP Tau.
Statistics.
All data were expressed as mean ± SEM for the number of repetitions (either puncta or neurons) indicated. Unpaired two-tailed Student's t tests or (for multiple comparisons) ANOVA followed by t tests with a Bonferroni correction were used, as appropriate. p ≤ 0.05 was considered statistically significant.
Results
Synaptic scaling in cultured neocortical pyramidal neurons was induced by blocking firing (scaling up) or increasing firing (scaling down) for 24 h, as previously described (Turrigiano et al., 1998; Sun and Turrigiano, 2011). To monitor synaptic AMPARs we transfected neurons with N-terminal-tagged GluA2; this tag is extracellular when receptors are in the plasma membrane. As shown previously by us and others (Wierenga et al., 2005; Ashby et al., 2006; Ibata et al., 2008; Kerr and Blanpied, 2012), N-terminal-tagged GluA2 accumulates at surface puncta that colocalize with the synaptic marker PSD-95 (Fig. 1A); there is also considerable diffuse dendritic fluorescence that corresponds to nonsynaptic surface AMPAR (Fig. 1A, arrow). GluA2 is obligatory for synaptic scaling (Gainey et al., 2009; Goold and Nicoll, 2010), and scaling protocols induced bidirectional changes in the intensity of the punctate GluA2 fluorescence (Fig. 1B; n = 59 control, 35 scaled up, 20 scaled down, and 22 scaled up + down puncta), as described previously for both endogenous and recombinant receptors (Wierenga et al., 2005; Ibata et al., 2008).

Measuring synaptic AMPAR removal with PAGFP-GluA2. A, Transfection with N-terminal-tagged GluA2 and mFRP-tagged PSD-95 reveals punctate accumulation at sites that colocalize with the synaptic marker PSD-95. Arrow highlights diffuse dendritic fluorescence corresponding to extrasynaptic GluA2. Scale bar, 10 μm. B, Synaptic GluA2 accumulation is bidirectionally regulated by chronic changes in activity (24 h of TTX or PTX). C, pH sensitivity of PAGFP-GluA2 signal. D, Example of dendritic PAGFP-GluA2 fluorescence before and after photoactivation, and (E) average dendritic intensity difference before and after photoactivation. Scale bar, 1 μm. F, Example of photoactivation of a 10 μm length of dendrite encompassing a synapse in a PAGFP-GluA2 transfected neuron; photoactivation reveals punctate fluorescence (0 min) that decays over time (10–60 min) and mRFP signal was used to identify neurons cotransfected with PAGFP-GluA2 (0 min, bottom). Reactivation of the same region reveals that the synapse persists. Scale bar, 5 μm. G, Time course of fluorescence decay after chronic TTX or PTX treatment; PTX is different from control from 30 min on. For all data here and below Ns are indicated in the Results section; *p < 0.05, **p < 0.01 compared with control.
Measuring AMPAR synaptic removal rate using PAGFP-GluA2
As a first means of assessing whether synaptic scaling protocols modulate the rate at which AMPAR exit the synapse we used PAGFP-GluA2 to activate receptors at synaptic sites, and then measured the rate of fluorescence decay as activated receptors unbind and diffuse away, or are internalized. PAGFP is pH sensitive and upon photoactivation ∼80% of the GluA2 signal was quenched by rapidly acidifying the medium, indicating that ∼80% of the signal is extracellular (Fig. 1C). Before photoconversion there was some basal PAGFP fluorescence (∼30% of the maximum dendritic signal after photoconversion; Fig. 1D,E), indicative of some degree of spontaneous photoconversion. Photoswitching PAGFP-GluA2 along a 10 μm length of dendrite encompassing a synapse revealed punctate PAGFP-GluA2 fluorescence, which then decayed over time (Fig. 1F). Because of the basal pool of activated dendritic receptors that can cycle into the synapse, the rate of fluorescence decay at the photoconverted synapse will not perfectly track AMPAR removal, so this method can only give information about relative rates of decay between conditions. Scaling synapses up (24 h TTX) had no effect on the rate of fluorescence decay compared with control synapses (Fig. 1G; n = 43 puncta control, 20 TTX, and 20 PTX). In contrast, scaling synapses down (24 h PTX) significantly increased the rate of fluorescence decay, with 50% of the fluorescence gone by ∼50 min for downscaled synapses, compared with ∼100 min for the control and TTX conditions (Fig. 1G; Control different from PTX from 30′ on, p < 0.05). These data suggest that scaling down, but not up, approximately doubles the rate at which synaptic AMPAR exit the synapse.
Using FRAP to probe AMPA Koff
As a second more quantitative means of measuring Koff we used FRAP of synaptic SEP-tagged GluA2. SEP is pH sensitive and quenching experiments revealed that ∼90% of the GluA2 fluorescence measured with SEP is extracellular (data not shown). If lateral diffusion of AMPAR is rapid relative to binding and unbinding from synaptic scaffolds, then when synaptic AMPAR fluorescence is bleached the component of FRAP due to scaffold interactions can be temporally separated from that due to diffusion. Under these conditions the slow component of recovery will be exponential with a time constant (tau) of 1/Koff, and will be insensitive to Kon (Lele et al., 2004, 2006; Sprague and McNally, 2005; see also simulation in Fig. 9).
To determine whether lateral diffusion of AMPAR is rapid relative to synaptic FRAP we began by measuring dendritic FRAP, where recovery primarily reflects free lateral diffusion of AMPAR in the dendritic membrane (Ashby et al., 2006; Frischknecht et al., 2009). When dendritic SEP-GluA2 was bleached recovery was rapid, with a tau of ∼1.5 min (Fig. 2A–D; n = 7), and was often incomplete (Fig. 2B); these measurements match well other published data (Ashby et al., 2006; Frischknecht et al., 2009). We next asked whether synaptic scaling protocols had any impact on the concentration or diffusion of dendritic SEP-GluA2. Neurons were grown under conditions of activity blockade or enhancement for 24 h, and then dendritic FRAP was measured. There was no difference in dendritic tau between conditions (Fig. 2C,D; n = 10 neurons control, 7 TTX, and 9 PTX). Further, synaptic scaling did not affect the intensity of the dendritic GluA2 signal (Fig. 2D, right), as reported previously (Wierenga et al., 2005). These data show that although synaptic scaling strongly modulates the accumulation of synaptic receptors (Fig. 1B; Wierenga et al., 2005; Ibata et al., 2008), the concentration and diffusion rates of dendritic receptors are unaffected. Thus synaptic scaling modulates AMPAR accumulation through processes that are specific for the synapse, rather than through generalized changes in trafficking to and mobility at the cell surface.

Dendritic FRAP in SEP-GluA2 transfected neurons. A, Example image of a nonsynaptic region of dendrite showing diffuse GluA2 fluorescence before and after bleaching. Dashed region (here and in subsequent figures) marks the photobleached area. Scale bar, 5 μm. B, Example recovery curve following dendritic FRAP, fit with a single exponential. C, Dendritic FRAP was not affected by chronic changes in activity (24 h TTX or PTX). D, Left, Dendritic FRAP tau values for control, scaling down, and scaling up conditions. Right, Fluorescence intensity of the SEP-GluA2 signal in the dendrite for the same three conditions.
Next, we performed FRAP of synaptic SEP-GluA2 (Fig. 3A). In contrast to the rapid recovery at dendritic sites, when synaptic SEP-GuA2 was bleached recovery was much slower, and followed a double-exponential time course, with a fast tau of ∼2 min, and a slower tau of ∼25 min (Fig. 3A–D). Thus complete synaptic FRAP is ∼10× slower than dendritic FRAP, with a fast component on the timescale of dendritic diffusion (Fig. 3F), and a slower component that likely reflects receptor interactions with scaffolds or other slow processes unique to synapses. The correspondence between the fast component of recovery and free diffusion is strengthened by the observation that the contribution of the fast component to synaptic fluorescence (∼23%; Fig. 3E, Tau1) reflects well the ratio of dendritic to synaptic fluorescence (∼23%; Fig. 3E; dendritic signal expressed as percentage of synaptic signal); this estimate of the freely diffusing component of synaptic AMPAR fluorescence is similar to that obtained from single receptor tracking experiments (Bats et al., 2007).

Synaptic FRAP has a fast and slow component. A, Example FRAP of a synapse (bleach region indicated by dashed circle). Note rapid partial recovery (2–4′) followed by slower recovery that plateaus ∼40–60′. Scale bar, 5 μm. B, Example FRAP curve of a single synapse, fit with a double exponential. C, Average FRAP curves for synapses using small (1 μm, synapse only) or large (10 μm, synapse and region of dendrite) bleach spots. Inset shows values of fast and slow taus for the two conditions. D, Average FRAP curves of spine and shaft synapses, normalized to recovery at 60′and fit with a double exponential. Inset shows values of fast and slow taus for the two populations of synapse. E, The magnitude of the fast component of synaptic recovery (calculated from double exponential fits) is comparable to magnitude of the dendritic GluA2 signal; data are expressed as fast tau recovery as percentage of the total synaptic GluA2 signal (Tau1), and dendritic signal as percentage of total synaptic GluA2 signal (dendrite percentage synapse). F, Comparison of dendritic tau to fast synaptic tau (Tau1).
If the slow component of synaptic recovery reflects binding or other protein–protein interactions between GluA2 and synaptic scaffold proteins, then the slow component should not be diffusion limited. To probe this further we first compared the rates of recovery at synapses when the bleach spot encompassed only the synapse (1 μm bleach spot), or included a 10 μm diameter additional region of dendrite (10 μm bleach spot). The slow tau was not significantly different for large and small bleach spots (Fig. 3C; n = 35 small and 5 large). Next, because diffusion into spines is slowed by geometry (Ashby et al., 2006; Hugel et al., 2009; Jaskolski et al., 2009) we compared FRAP at shaft and spine synapses. The fast tau was slower at spine than shaft synapses (Fig. 3D, inset); however, the slow component of FRAP was not slower (Fig. 3D; n = 32 puncta spine and 22 shaft). Together, these two controls suggest that the slow component of synaptic FRAP of GluA2 is not diffusion limited. While our dendritic FRAP measurements match well other reports (Ashby et al., 2006; Frischknecht et al., 2009), previous studies have not measured synaptic AMPAR FRAP over the ∼1 h needed for full resolution of the slow time-constant of recovery (Sharma et al., 2006; Bats et al., 2007; Makino and Malinow, 2009; Kerr and Blanpied, 2012).
To verify that measurements of FRAP Tau were not compromised by internal AMPAR fluorescence we compared FRAP of synaptic SEP-GluA2 and synaptic EGFP-GluA2; because EGFP is less pH sensitive than SEP, ∼20% of the signal comes from internal receptors, as opposed to ∼10% of the SEP signal. Despite this difference, FRAP tau was indistinguishable for the two fluorophores (Fig. 4A,B). In contrast the fractional recovery (the apparent “mobile fraction”) was greater when measured with SEP (∼75%) than when measured with EGFP (∼60%; Fig. 4B). This difference suggests that a fraction of the immobile receptors during synaptic FRAP comes from internal fluorescence, and the contribution of this internal fraction is greater when using EGFP than SEP. Thus the “immobile fraction” measured during synaptic FRAP likely overestimates the fraction of surface synaptic receptors that are immobile. Synaptic scaling protocols had no impact on the mobile fraction (measured using SEP tag; Fig. 4C); this differentiates scaling up from some forms of long-term potentiation (LTP) where mobile fraction is affected (Sharma et al., 2006; Makino and Malinow, 2009).

Comparison of FRAP of SEP- and EGFP-tagged GluA2. A, Normalized FRAP recovery curves for the two fluorophores. B, Comparison of mobile fraction (left) and FRAP Tau (right) for the two fluorophores. C, Synaptic scaling protocols do not affect the mobile fraction, here measured with SEP tag and normalized to control.
Synaptic scaling down is accompanied by a decrease in synaptic FRAP Tau
We next chronically blocked or enhanced activity for 24 h to induce scaling, and measured FRAP Tau. In this set of experiments FRAP was measured at close to physiological temperature (33°C), and to avoid reversal of scaling during the 1–2 h needed to measure FRAP cultures were maintained in the same conditions of activity blockade (scaling up, TTX) and activity enhancement (scaling down, PTX) used to induce synaptic scaling. Consistent with our PAGFP-GluA2 experiments, FRAP Tau was unaffected by scaling up, but was reduced to ∼50% of control after scaling down (Fig. 5A,B; n = 22 control puncta, 21 PTX, 20 TTX; PTX different from control, p < 0.03).
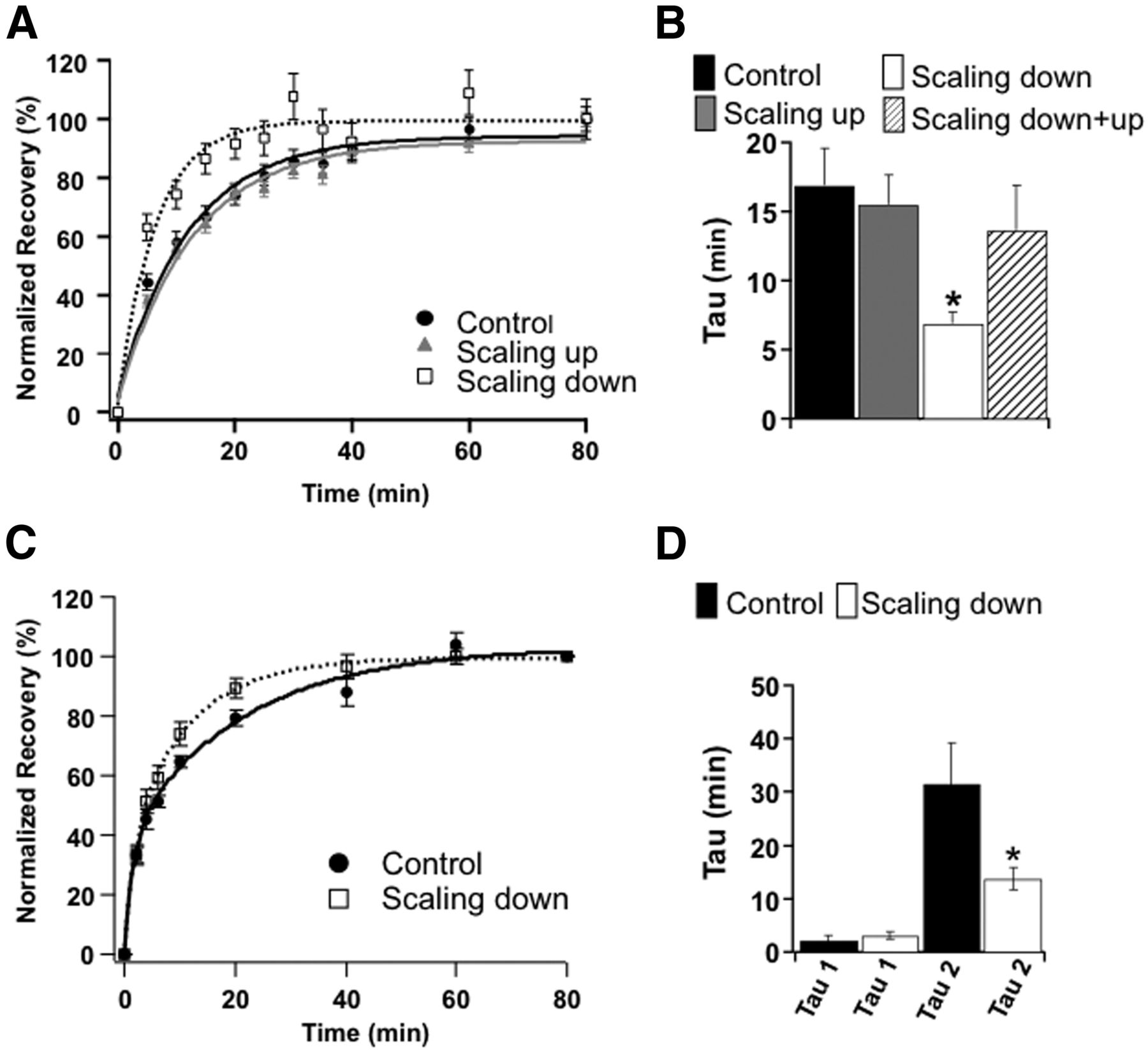
Scaling down selectively speeds up the slow component of synaptic GluA2 FRAP. A, Average synaptic FRAP curves for control synapses, or synapses that have been scaled up or down by chronic changes in activity (24 h TTX or PTX), measured at 33°C and in the continued presence of the respective drug (TTX or PTX). Curves are normalized to recovery at 60′. B, Average Taus for the indicated conditions; in this experiment curves were not sampled with high enough temporal resolution to resolve the fast component of recovery so were fit with a single exponential. C, FRAP for control or PTX-treated synapses, measured at 25°C and in the presence of acute TTX, and with greater initial temporal resolution. D, Fast and slow tau values from double-exponential fits of data in C. * indicates scaling down different from control.
Synaptic scaling of mEPSCs induced by chronic elevation or blockade of activity is typically measured at room temperature and in the presence of TTX; under these conditions the effects of chronic activity manipulations can be measured over the 1–2 h needed without significant reversal of scaling (Turrigiano et al., 1998; Wierenga et al., 2005). To rule out the possibility that the effect of PTX on FRAP Tau was due to an acute rather than chronic change in activity, we next measured FRAP under the conditions typically used to measure synaptic scaling: following 24 h of control or PTX treatment, both conditions were measured at room temperature in the presence of acute TTX (which does not affect the mobility of synaptic receptors, Groc et al., 2004; Ehlers et al., 2007). In addition we sampled the initial few minutes after bleaching at a faster rate to capture both the fast and slow components of recovery. Consistent with our earlier dataset (Fig. 5A,B), under these conditions as well the slow Tau was reduced by ∼50% at downscaled synapses (Fig. 5C,D; n = 35 puncta control, 33 PTX, p < 0.04). Further, there was no significant difference in the fast tau between control and downscaled synapses (Fig. 5D). These data show that chronic activity elevation does not affect the fast (diffusional) component of recovery at synapses, but specifically reduces the slow FRAP tau by ∼50%. Because the two time constants of recovery are well separated (Fig. 5D) and the slow component of recovery is not diffusion limited (Fig. 3), this halving of the slow FRAP Tau corresponds to a doubling of the dissociation constant Koff (Lele et al., 2004, 2006; Sprague and McNally, 2005).
The nonreciprocal regulation of Koff during scaling up and down is the first concrete evidence that these two processes target different aspects of the AMPAR cycling machinery. This observation raises the puzzling question of why, if scaling up does not reverse the change in Koff induced by scaling down, Koff does not eventually saturate and prevent further scaling down. We reasoned that, although scaling up does not itself target Koff, reducing activity might reverse or “reset” to control values the change in Koff induced by chronic activity elevation. To test this we first scaled synapses down for 24 h (with PTX), and then immediately scaled them up for 24 h (with TTX). Synaptic GluA2 abundance after scaling down + up was comparable to that after scaling up alone (Fig. 1B), and this manipulation reversed the change in Frap Tau induced by scaling down alone (Fig. 5B, scaling down + up). These data suggest that activity blockade simultaneously reverses any downscaling that has occurred previously, while also inducing scaling up via a distinct mechanism that does not target Koff.
Scaling down is not accomplished through enhanced AMPAR endocytosis
The increased AMPAR Koff during scaling down could result from enhanced endocytosis of synaptic receptors, or from an increase in the rate at which AMPAR dissociates from synaptic scaffolds. Which of these processes is the rate-limiting step for AMPAR synaptic exit is not known. To differentiate between these possibilities we first asked whether synaptic FRAP Tau was limited by the rate of AMPAR endocytosis, by using the soluble dynamin inhibitor dynasore to acutely block clathrin-mediated endocytosis (Jaskolski et al., 2009; Linden, 2012). We confirmed that acute application of dynasore (80 μm) prevented the internalization of SEP-GluA2 in response to brief NMDA receptor application (Fig. 6A,B; n = 4 neurons control and 3 neurons dynasore). We then performed FRAP of SEP-GluA2 in the presence or absence of acute dynasore (Fig. 6C–E). As reported previously for synaptic sites (Jaskolski et al., 2009), dynasore decreased the mobile fraction at both dendritic and synaptic locations (Fig. 6D; p < 0.01), suggesting that dynasore somehow “traps” or prevents free diffusion of a subset of receptors regardless of location; a similar effect of extracellular matrix on receptor diffusion has been observed (Frischknecht et al., 2009). Despite this effect on diffusion, dynasore had no impact on either dendritic (n = 6 control dendrites and 5 dynasore) or synaptic (n = 18 puncta control and 16 dynasore) FRAP Taus (Fig. 6E), indicating that the receptor turnover that drives recovery from FRAP is not limited by the rate of AMPAR endocytosis. These data suggest that the slow component of synaptic FRAP largely reflects AMPAR unbinding from scaffolds, rather than active receptor internalization.

FRAP tau is not affected by acute block of endocytosis. A, Acute dynasore blocks NMDA-induced GluA2 endocytosis. Examples of brief NMDA (20 μm) application (at t = 5 min) to control (top) and dynasore-treated (bottom) neurons. Scale bar, 10 μm. B, SEP-GluA2 fluorescence intensity after NMDA treatment (gray bar) for control and dynasore-treated neurons. C, Normalized synaptic FRAP curves from control and dynasore-treated SEP-GluA2 puncta. D, Average mobile fractions for control and dynasore treated SEP-GluA2 neurons; both synaptic (puncta) and dendritic mobile fractions were reduced by dynasore. E, Average taus for synaptic (puncta) and dendritic FRAP were not affected by an acute block of endocytosis.
To directly assess the role of AMPAR endocytosis in scaling down we wished to quantify the rate of GluA2 internalization in living neurons. To accomplish this we used a SEP antibody tagged with CypHer, a pH-sensitive cyanine dye that fluoresces at acidic and is quenched at physiological pH (Adie et al., 2003; Hua et al., 2011), to label surface SEP-GluA2 and then track the rate at which surface receptors were internalized (Fig. 7A–C). Because internalized surface proteins are rapidly trafficked to acidic intracellular compartments the appearance of CypHer fluorescence can be used to track the rate of internalization (Adie et al., 2003; Hua et al., 2011). We first verified that the CypHer signal remained colocalized with GluA2 after internalization using antibodies against GluA2 (Fig. 7A); further, neutralizing internal pH through permeabilization in living neurons converted the internal CypHer signal to a SEP signal, as expected if the antibody stays bound to SEP-GluA2 after internalization (Fig. 7B). To measure the rate of GluA2 internalization the CypHer antibody was added to the bath during imaging, and the rate of appearance of fluorescence in intracellular compartments tracked (Fig. 7C). Strikingly, the rate of GluA2 internalization was indistinguishable for control and downscaled neurons (Fig. 7D; n = 8 neurons each condition). Together these data indicate that, unlike some forms of long-term depression (LTD; Rial Verde et al., 2006; Waung et al., 2008; Linden, 2012), synaptic weakening during scaling down is not accomplished through increased AMPAR endocytosis.

Scaling down does not enhance the rate of GluA2 internalization. A, To determine whether the CypHer tag remains bound to GluA2 after internalization, neurons transfected with SEP-GluA2 were labeled with CypHer 5E (red), allowed to internalize receptor, then fixed, permeabilized, and labeled for total GluA2 (green). CypHer 5E puncta were colocalized with GluA2 puncta (example indicated by arrowhead); most GluA2 was not colocalized with CypHer (examples indicated by arrows). B, CypHer-SEP-GluA2-labeled neurons were permeabilized during live imaging to equalize internal and external pH; circles depict example puncta where CypHer signal (red, acidic pH) was converted to SEP signal (green, neutral pH), indicating that CypHer stays bound to SEP-GluA2 after internalization. C, Example time course of CypHer-GluA2 internalization in living dendrite. D, Rate of CypHer-GluA2 internalization at 32°C for control or PTX-treated neurons. Curves were not different at any time point. Scale bars, 5 μm.
Role of scaffold interactions in enhanced Koff
The data above suggest that scaling down occurs through changes in scaffold interactions that enhance GluA2 unbinding from synaptic tethers. We showed previously that scaling down is critically dependent on interactions with the synaptic scaffold PSD-95. Scaling down can be blocked by PSD-95 overexpression, which selectively blocks scaling down without impacting scaling up, or significantly affecting baseline mEPSC amplitude (Sun and Turrigiano, 2011). Consistent with a causal role for the change in FRAP tau in scaling down, PSD-95 overexpression had no significant effect on FRAP Tau under control conditions, but completely prevented the change in FRAP Tau normally induced by 24 h of PTX treatment (p < 0.03; Fig. 8A,B; n = 25 control, 20 PTX, 34 PSD-95 OE, and 17 PTX + PSD95 OE puncta). PSD-95 OE also blocked the reduction in synaptic GluA2 accumulation normally induced by 24 h of PTX (Fig. 8C). Thus a manipulation that blocks the change in FRAP tau also blocks scaling down. Further, these data show that the regulated change in FRAP tau during scaling down relies critically on a process that can be disrupted by excess PSD-95.
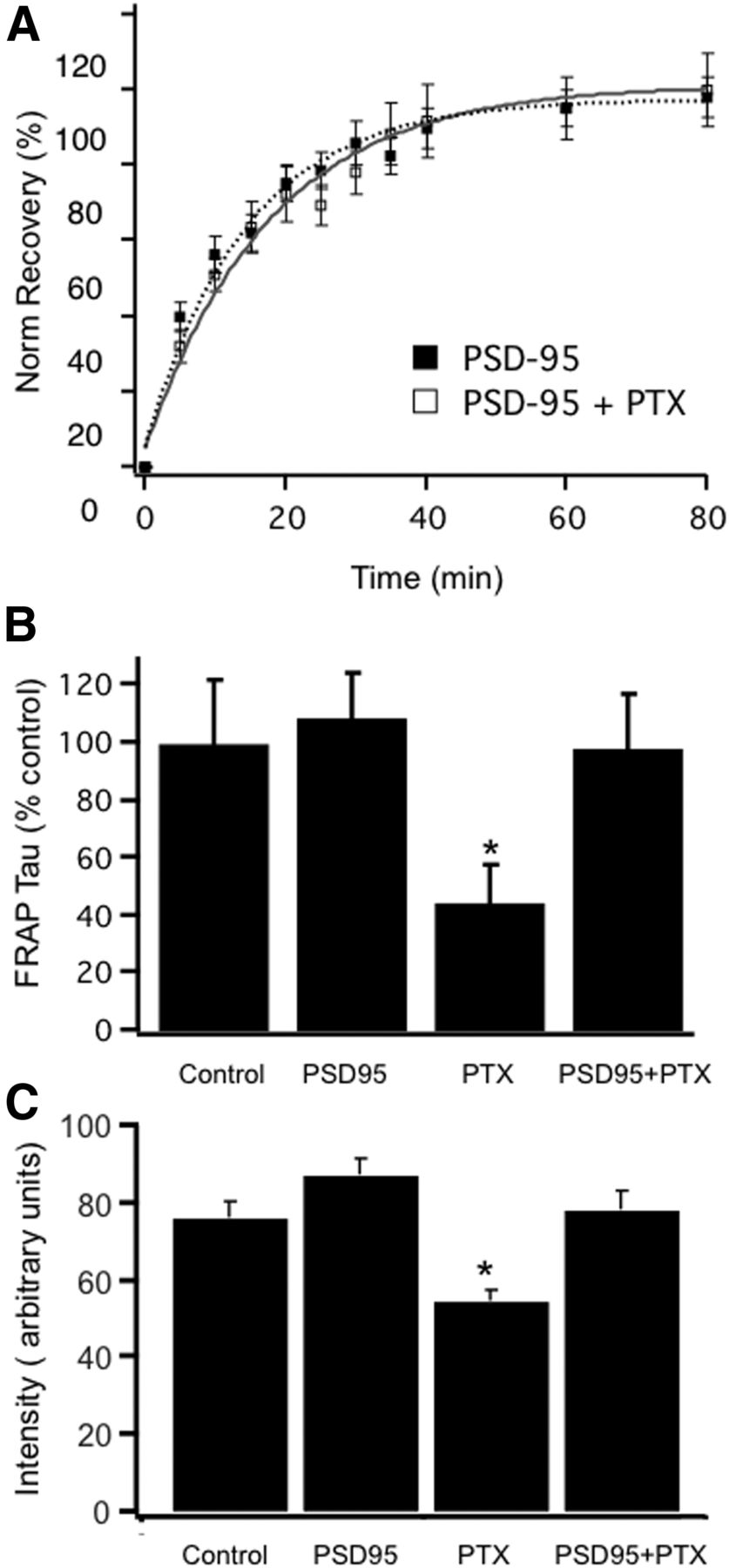
PSD-95 overexpression blocks the PTX-induced reduction in FRAP tau and prevents scaling down. A, Average synaptic FRAP curves for PSD-95 overexpression alone, or PSD-95 overexpression + 24 h PTX treatment. B, Average synaptic FRAP Taus for the indicated conditions, expressed as a percentage of control. C, Synaptic GluA2 intensity for the same conditions indicated in B. * indicates PTX different from PSD-95 + PTX.
Enhanced Koff scales synapses down
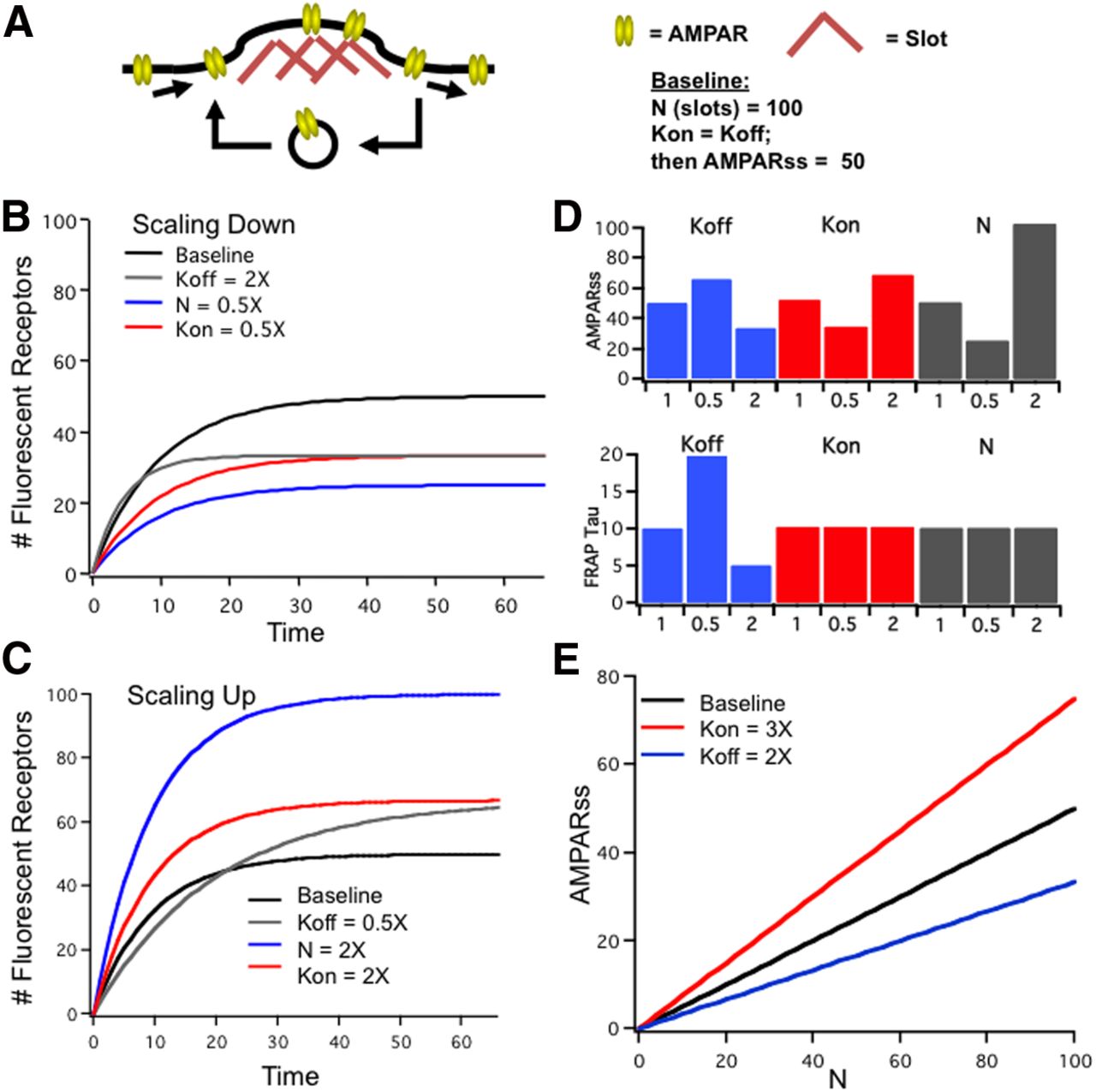
We next asked whether a change in the rate at which AMPARs unbind from synaptic scaffolds can account quantitatively for scaling down. Our data show that the concentration of diffusing receptors stays constant during scaling (Fig. 2D), that the fast (diffusional) fraction of synaptic recovery is not affected by scaling protocols (Fig. 5D), and that the slow component of recovery is not diffusion limited (Fig. 3C,D). We were therefore able to considered the binding and unbinding interactions independently of diffusion, and simulate the effects of changing Koff by considering a set of “slots” (N) where empty slots fill at a rate Kon, and filled slots empty at a rate Koff (Fig. 9A). We started with N = 100, and Kon = Koff = 0.1; then the steady-state number of bound AMPARss is N * Kon/(Kon + Koff), or 50, meaning that half of available slots are filled at any given time. We then simulated a FRAP experiment; as “bleached” receptors unbind and empty slots fill with unbleached receptors the fluorescence undergoes the expected exponential recovery until all bound receptors are fluorescent again; this steady-state value is AMPARss (Fig. 9B, Baseline). To simulate scaling down we doubled Koff, halved N, or halved Kon (Fig. 9B). While all three manipulations reduced AMPARss, only Koff affected tau; doubling Koff halved FRAP tau, and reduced AMPARss by 33% (Fig. 9B,D). To simulate scaling up we increased Kon, increased N, or reduced Koff; all three increased AMPARss, but only Koff affected Tau (Fig. 9C,D). As Tau does not change during scaling up, the increase in AMPARss likely occurs through an increase in N or Kon, but our data cannot distinguish between these possibilities.

A reduction in Koff reproduces synaptic scaling down. A, The bound fraction of receptors at the postsynaptic density modeled as a set of N slots; empty slots bind AMPAR with a rate Kon, and full slots lose AMPAR with a rate Koff. B, C, Simulated FRAP experiment illustrates the recovery to steady-state fluorescence after bleach for the indicated conditions during scaling down (B) or scaling up (C). D, Modifying N, Kon, or Koff all influence the steady-state number of bound receptors (top), but only changes in Koff affect the time constant of recovery (bottom). E, Reducing Koff scales down postsynaptic strength multiplicatively. Initial differences in postsynaptic strength across synapses were simulated as variations in the number of slots N; Kon and Koff were uniform across the population and were set to be equal, so for baseline occupancy of available slots at steady state was 50% across all synapses (black line). Doubling Koff (scaling down, blue line) decreased the slope of this relationship from 0.5 to 0.33, indicating that the fractional occupancy of available slots decreased and synaptic strength was reduced proportionally across synapses. Conversely, fractional occupancy was scaled up by an increase in Kon (red line).
A hallmark of synaptic scaling is that synaptic strength increases or decreases proportionally across synapses (Turrigiano et al., 1998), so we next asked whether changing Koff provides a mechanism for this proportional scaling. Synapse size is correlated with the number of postsynaptic receptors that accumulate (Kasai et al., 2010), so we modeled differences in postsynaptic strength across synapses as differences in the number of slots N; then for a given Kon and Koff, AMPARss will increase linearly as N increases (Fig. 9E, Baseline). When Koff was doubled, the slope of this line decreased, indicating that AMPARss was reduced proportionally across synapses (Fig. 9E, blue line); essentially, reducing Koff reduces the fractional occupancy of available slots. Conversely, increasing Kon scaled synaptic strengths up by increasing fractional occupancy (Fig. 9E, red line).
The simulation of scaling in Figure 9E does not consider the freely diffusing and immobile fractions of receptors. Interestingly, the immobile fraction of receptors stayed constant (at ∼25% of total synaptic fluorescence) during synaptic scaling (Fig. 4B,C), meaning that the immobile fraction and bound fraction both scale with synaptic strength. What the immobile fraction represents is not clear; some of these receptors are likely internal, while others may represent a population of receptors that are very tightly bound to scaffolds and turn over on a timescale not detectible in our FRAP experiments. Estimates from our fits of recovery curves give the freely diffusing fraction of total synaptic fluorescence at ∼23% (Fig. 3E), and the bound fraction (including the immobile fraction) at ∼77%, for control synapses. The magnitude of the freely diffusing AMPAR signal remains constant after scaling protocols, so if these receptors contribute to synaptic transmission this should add a small constant difference to postsynaptic strength during synaptic scaling (∼7%, or ∼1 pA for a typical mEPSC). This in turn would generate a very small deviation from perfect scaling, well within the error of the standard scaling analysis applied to mEPSC distributions (Turrigiano et al., 1998). Together, these results suggest that the change in Koff we measure makes a major quantitative contribution to scaling down of synaptic strength, with a small additional contribution from proportional changes in the immobile fraction.
Discussion
A hallmark of homeostatic synaptic scaling is the proportional adjustment of synaptic AMPAR abundance across synapses, but how this scaling of postsynaptic strength is accomplished at the biophysical level is unknown. Here we show that scaling down decreases the steady-state accumulation of synaptic AMPAR by increasing Koff, the rate at which AMPAR unbind from synaptic tethers and exit the synapse. Simulations show that this change in Koff generates a multiplicative decrease in postsynaptic strength, which can quantitatively reproduce synaptic scaling down. While many forms of synaptic weakening have been attributed to enhanced AMPAR endocytosis, this is (to our knowledge) the first demonstration that synaptic weakening can be accomplished by modulating the rate at which AMPAR dissociate from synaptic binding sites.
Our current understanding of AMPAR trafficking is that these receptors are inserted into the somatic, dendritic, or perisynaptic membrane, where they diffuse into the postsynaptic density and accumulate because they are reversibly trapped through interactions with synaptic scaffolds (Malinow and Malenka, 2002; Bats et al., 2007; Gerrow and Triller, 2010; Opazo and Choquet, 2011). In principle long-lasting changes in synaptic strength could be accomplished by targeting any (or all) of the factors that influence synaptic AMPAR abundance. In practice, although many molecular pathways have been identified that participate in regulated AMPAR trafficking during synaptic plasticity, for no form of plasticity do we fully understand which synaptic parameters are targeted to modify synaptic strength. For example, although LTP can transiently increase the rate of insertion of AMPAR into the plasma membrane (Malinow and Malenka, 2002; Kessels et al., 2009; Makino and Malinow, 2009; Patterson et al., 2010), it is still unclear how this contributes to the long-lasting and stable increase in synaptic AMPAR abundance. Similarly, the consensus is that synaptic weakening during LTD is due to enhanced AMPAR endocytosis from the plasma membrane (Lüscher et al., 1999; Malenka and Bear, 2004; Waung et al., 2008; Linden, 2012), but how this stably and locally reduces synaptic strength is still unclear.
The approach we took here, using photoactivation and FRAP of synaptic AMPAR, allowed us to differentiate and quantify three distinct pools of receptors regulated by distinct biophysical processes: a freely diffusing fraction (fast tau, ∼23%), a transiently bound fraction (slow tau, ∼52%), and a tightly bound fraction (the fraction that does not recover from FRAP, ∼25%). That the fast FRAP tau is driven by diffusion is supported by the observation that the recovery time constant and magnitude of this fraction of synaptic fluorescence correspond well to the recovery time constant and magnitude of the freely diffusing dendritic pool of AMPAR. In contrast, the slow tau was an order of magnitude slower and was not diffusion limited, so it must reflect slower protein–protein interactions such as binding to scaffolds or interaction with proteins that act as diffusional barriers (Opazo and Choquet, 2011; Santamaria et al., 2010; Renner et al., 2012). The transiently bound fraction of receptors turns over with a time constant of ∼20 min under basal conditions (at 33°C), indicating that the majority of receptors in the postsynaptic density are replaced fairly rapidly with receptors that diffuse in from lateral sites. Finally, there is a fraction of synaptic AMPAR that is immobile on the timescale of these experiments. What this immobile fraction of AMPAR fluorescence represents is not entirely clear. Some of this pool likely represents a very tightly bound fraction of surface receptors, but our data suggest there is also a contribution from internal immobile receptors. The fraction of immobile surface receptors is thus a fairly small fraction (perhaps 20%) of the total receptor pool.
This analysis allowed us to determine whether synaptic scaling modulates the synaptic accumulation of AMPAR through changes in the diffusional, transiently bound, or immobile fractions of synaptic AMPAR. We found that synaptic scaling does not affect the concentration of dendritic receptors or the rates of dendritic or synaptic diffusion, but instead scales the bound fraction of AMPAR up or down. In theory this could be accomplished by changing the number of binding sites within the postsynaptic density, or changing the association or dissociation constants between AMPAR and these binding sites (Lisman and Raghavachari, 2006; Opazo and Choquet, 2011). Koff does not change during scaling up, indicating that synaptic scaling operates via an increase in slots or an increase in the association constant Kon; our data cannot currently distinguish between these possibilities. In contrast, both our PAGFP and FRAP data show that during scaling down the transiently bound fraction is reduced through a regulated increase in Koff, which reduces the steady-state occupancy of available slots. The halving of Koff should reduce this fraction by 33% (Fig. 9C,E); when combined with the proportional decrease in the immobile fraction and lack of change in the diffusional component, total receptor fluorescence should decrease by ∼25%, close to the ∼28% reduction we observe in GluA2 fluorescence intensity following scaling down (Fig. 1B). Thus the change in Koff, as well the proportional change in the immobile fraction, together can account for the decrease in synaptic AMPAR accumulation observed during scaling down, with ∼2/3 of the contribution coming from the change in Koff.
How might Koff be regulated during scaling down? The molecular interactions that mediate scaling down are incompletely understood, but activity-dependent GluA2 dephosphorylation via Homer1A (Hu et al., 2010), along with the regulation of scaffold complexes that include SPAR and PSD-95 (Seeburg et al., 2008; Sun and Turrigiano, 2011), are known to be critical. Consistent with this, we show here that preventing synaptic scaling down by overexpression of PSD-95 (Sun and Turrigiano, 2011) prevents the enhancement of Koff by elevated activity. Interestingly, PSD-95 overexpression does not affect basal mEPSC amplitude (Sun and Turrigiano, 2011) or synaptic AMPAR accumulation, nor does it affect basal Koff, but specifically prevents the regulated increase in Koff and decrease in receptor accumulation induced by chronic activity elevation. This suggests that PSD-95 overexpression is acting as a dominant-negative that interacts with and sequesters proteins that are necessary for inducing the regulated change in Koff. Given the known AMPAR stabilization function of PSD-95-TARP interactions (Bats et al., 2007) this interactin is a potential target; however, because these neurons express a number of TARPS that may have redundant functions (Jackson and Nicoll, 2011), testing this idea will not be trivial. Together with these earlier studies, our data suggest that scaling down occurs via changes in GluA2-scaffold interactions that involve PSD-95-intracting proteins, and reduce the time that GluA2 stays bound to synaptic tethers.
The major mechanism that has been proposed to underlie postsynaptic weakening is enhanced AMPAR endocytosis, which likely contributes to many forms of LTD (Malenka and Bear, 2004; Linden, 2012). The molecular changes that drive synaptic scaling down have also been attributed to an enhanced internalization of synaptic AMPAR (Shepherd et al., 2006, Evers et al., 2010). Surprisingly, here we show that scaling down reduces synaptic AMPAR accumulation without enhancing AMPAR internalization, but instead uses the novel mechanism of enhancing Koff. This is in seeming contradiction to a proposed role for Arc-mediated AMPAR endocytosis in homeostatic plasticity (Chowdhury et al., 2006; Shepherd et al., 2006), but as scaling down of mEPSC amplitude was intact in Arc KO neurons (Shepherd et al., 2006) and can occur independently of Arc (Hu et al., 2010), Arc-mediated endocytosis appears to be dispensable for scaling down. Scaling down and LTD are thus fundamentally different processes: they use distinct scaffold interactions (Xu et al., 2008; Sun and Turrigiano, 2011) to target distinct AMPAR trafficking steps. These differences likely reflect the different functions, as well as temporal and spatial scales, of a synapse-specific and global form of plasticity. While LTD might be best accomplished by rapidly and locally internalizing a bolus of receptors (perhaps along with their associated scaffolds), the homeostatic function of scaling down requires a slower means of globally and proportionally regulated synaptic AMPAR abundance. A process that modifies GluA2-scaffold interactions to increase Koff seems ideally suited to achieve a global scaling down of postsynaptic strengths.
A striking finding of the present study is that scaling up and scaling down do not reciprocally regulate Koff, and thus at the level of receptor trafficking do not represent inverse processes. Together with a number of studies showing that the molecular underpinnings of scaling up and down are also distinct (Turrigiano, 2012), these data strongly suggest that scaling up and scaling down represent two distinct molecular processes that act in a “push–pull” manner to regulate postsynaptic strength. Interestingly, our data also suggest for the first time that scaling down can be reversed in a process that is distinct from scaling up, allowing synapses to avoid saturating changes in Koff during repeated rounds of scaling up and down. At many synapses LTP and LTD are known to target distinct processes, sometimes on opposite sides of the synapse (Malenka and Bear, 2004; Jörntell and Hansel, 2006; Sjöström et al., 2007), so this “resetting” could represent a general solution to allow increases and decreases in synaptic strength to target distinct processes without locking synaptic parameters at saturated values.
Footnotes
This work was supported by National Institutes of Health Grant NS036853 (G.G.T.). We thank B. Baxter and L. Wang for assistance with cultures, and E. Marder for helpful discussions.
- Correspondence should be addressed to Gina G. Turrigiano, Department of Biology and Center for Behavioral Genomics, MS08 415 South Street, Waltham, MA 02453. turrigiano{at}brandeis.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}