

Abstract
Animal and clinical studies show that gestational exposure to nicotine increases the propensity of offspring to consume nicotine, but the precise mechanism mediating this behavioral phenomenon is unclear. The present study in Sprague Dawley rats examined the possibility that the orexigenic peptide systems, enkephalin (ENK) and orexin (OX), which are stimulated by nicotine in adult animals and promote consummatory behavior, may be similarly responsive to nicotine's stimulatory effect in utero while having long-term behavioral consequences. The results demonstrated that nicotine exposure during gestation at low doses (0.75 or 1.5 mg/kg/d) significantly increased mRNA levels and density of neurons that express ENK in the hypothalamic paraventricular nucleus and central nucleus of the amygdala, OX, and another orexigenic peptide, melanin-concentrating hormone, in the perifornical lateral hypothalamus in preweanling offspring. These effects persisted in the absence of nicotine, at least until puberty. Colabeling of the cell proliferation marker BrdU with the neuronal marker NeuN and peptides revealed a marked stimulatory effect of prenatal nicotine on neurogenesis, but not gliogenesis, and also on the number of newly generated neurons expressing ENK, OX, or melanin-concentrating hormone. During adolescence, offspring also exhibited significant behavioral changes, increased consumption of nicotine and other substances of abuse, ethanol and a fat-rich diet, with no changes in chow and water intake or body weight. These findings reveal a marked sensitivity during gestation of the orexigenic peptide neurons to low nicotine doses that may increase the offspring's propensity to overconsume substances of abuse during adolescence.
Introduction
Tobacco is the most commonly used drug during pregnancy (Substance Abuse and Mental Health Services Administration, 2012). Gestational nicotine alters normal growth and development and has long-term physiological and behavioral consequences in the offspring. It increases tobacco use and risk of developing nicotine dependence in humans and consumption of nicotine in mice (O'Callaghan et al., 2009; Chistyakov et al., 2010; Rydell et al., 2012; Schneider et al., 2012). However, the mechanisms mediating this behavioral effect are unclear. Nicotine during gestation, generally tested at moderate to high doses, has suppressive effects on neurodevelopment in the forebrain, causing a decrease in cell number and survival, a decrease in neuronal area and number, and an increase in apoptotic cell death (Roy et al., 2002; Abdel-Rahman et al., 2005; Chen et al., 2005; Wei et al., 2011; Santiago and Huffman, 2012). With these effects accompanied by reduced functioning of two closely related dopaminergic and cholinergic neurochemical systems that mediate reward (Fung and Lau, 1989; Kane et al., 2004; Picciotto and Kenny, 2013), the resulting state of reward deficiency is suggested to increase the offspring's propensity to overconsume nicotine, raising dopamine to normal levels (Kane et al., 2004).
The present study tested the possibility that the orexigenic peptides enkephalin (ENK), orexin (OX), and melanin-concentrating hormone (MCH), which in adult animals stimulate consumption of nicotine and other reinforcing substances (Morganstern et al., 2011), may also be altered by gestational exposure to nicotine and be involved in promoting the offspring's nicotine intake. Self-administration of nicotine is reduced by hypothalamic administration of an OX receptor 1 antagonist (LeSage et al., 2010) and peripheral opioid antagonist (Ismayilova and Shoaib, 2010), and although yet to be studied with nicotine, central injection of MCH increases the consumption of ethanol (Duncan et al., 2005; Morganstern et al., 2011). Conversely, administration of nicotine stimulates expression of OX in the perifornical lateral hypothalamus (PFLH) and ENK in the hypothalamic paraventricular nucleus (PVN) and central nucleus of the amygdala (CeA) (Houdi et al., 1998; Loughlin et al., 2006) and OX mRNA is increased by chronic nicotine (Houdi et al., 1998; Kane et al., 2000). If these peptides do in fact have a role in potentiating the offspring's consumption, the effect of prenatal exposure to nicotine on the in utero development of these neurochemical systems would be expected to be stimulatory, rather than suppressive, in nature. A hint at this possibility comes from one study showing that nicotine increased ENK expression in the adrenal medulla on embryonic day 21 (E21; Wong et al., 2003).
In this study, we used a model of low-dose nicotine exposure in pregnant rats to test the hypothesis that nicotine during gestation stimulates the genesis and postnatal expression of orexigenic peptides in the hypothalamus and amygdala, possibly with long-term behavioral consequences. We demonstrate that prenatal nicotine at low doses stimulates: (1) ENK expression in PVN and CeA and both OX and MCH in PFLH of the offspring, which persists into adolescence; (2) neurogenesis of these peptide-expressing neurons; and (3) consumption in adolescent offspring of nicotine and the other reinforcing substances ethanol and dietary fat.
Materials and Methods
Animals and surgeries
Time-pregnant Sprague Dawley rats (220–240 g) from Charles River Breeding Laboratories arrived on E5 and were individually housed in plastic cages in a fully accredited Association for Assessment and Accreditation of Laboratory Animal Care facility (22°C, 12:12 h light/dark cycle with lights off at 12:00 P.M.) according to institutionally approved protocols as specified in the National Institutes of Health Guide to the Care and Use of Laboratory Animals and with approval of The Rockefeller University Animal Care and Use Committee. Rats were acclimated to laboratory conditions until E8, when they were briefly anesthetized with isofluorane and subcutaneously implanted with osmotic minipumps (2 week duration; model 2002; Alzet; Slotkin et al., 2006; Wei et al., 2011) filled with sterile saline control solution (referred to as “control”) or a solution of nicotine bitartrate yielding 0.075 or 1.5 mg/kg/d (free base) nicotine. Minipumps were primed to deliver solutions at a constant rate (0.50 μl/h) from subcutaneous implantation. No dams displayed abnormal recuperation from anesthesia or postimplantation infection. Rodent chow (LabDiet Rodent Chow 5001) and water were available ad libitum. Food intake was measured two times per week and body weight was recorded weekly. There were no differences between control and nicotine-treated dams in body weight (255–270 g) or chow intake (65–75 kcal/d). At birth, on postnatal day 0 (P0), litters were culled to n = 8. Litters of the nicotine and control dams were similar in terms of size (8–14), body weight (5.2–6.8 g), and male/female ratio (1.2/1.0), with no spontaneous abortions observed in either group. Only male offspring were tested, with 1 male pup taken from each litter and the number of rats/group (n = 4–8) equal to the number of litters. Offspring were killed at P15 or P40 by rapid decapitation and whole brain was collected for further analyses, which were performed by an evaluator blind to the experimental conditions. A total of eight groups of dams were tested, as listed in Table 1 and described below.
Groups and procedures
Radiolabeled in situ hybridization histochemistry to measure peptide mRNA levels
In the offspring of Group 1, mRNA levels of ENK, OX, and MCH were measured as described previously (Morganstern et al., 2010c; Chang et al., 2012) using radiolabeled in situ hybridization histochemistry (ISH) in the P15 (n = 7–8/group) or P40 (n = 4/group) offspring exposed in utero to the control solution or a 0.75 or 1.5 mg/kg/d dose of nicotine. Immediately after killing, whole brains were removed, fixed in 4% paraformaldehyde phosphate buffer (0.1 m, pH 7.2) at 4°C for 2 d, and then cryoprotected in 20% sucrose at 4°C for 3 d. Next, 30 μm alternate, cryostat, free-floating coronal sections were prepared as described previously (Chang et al., 2010). Gene expression level, expressed as optical density, was determined with a computer-assisted microdensitometry of autoradiographic images on the MCID image analysis system (Imaging Research) as described previously (Reagan et al., 2004; Chang et al., 2008). Gray level/optical density calibrations were performed with a calibrated filmstrip ladder (Imaging Research) for optical density, which was plotted as a function of microscale calibration values. All subsequent optical density values of digitized autoradiographic images fell within the linear range of the function. The values obtained represent the average of measurements taken from 10 sections per animal. Within each section, the optical density for the nucleus was recorded, from which the background optical density from a same size area in the corpus callosum was subtracted. Mean value of the optical density for the PVN, PFLH, and CeA, in the control, 0.75 mg/kg/d nicotine, or 1.5 mg/kg/d nicotine groups in each experiment was compared and reported as percentage of the control group, as described previously (Chang et al., 2008; Chang et al., 2012).
Digoxigenin-labeled ISH to measure peptide neuronal density
In P15 offspring of Group 2, ISH with a digoxigenin-labeled probe that provides better visualization was performed to more clearly demonstrate the anatomical distribution of changes in peptide expression in the control and 1.5 mg/kg/d nicotine groups as described previously (Chang et al., 2008; Chang et al., 2010). Briefly, digoxigenin-labeled antisense RNA probes and 30 μm free-floating cryostat coronal sections were used for ISH. AP-conjugated sheep anti-digoxigenin Fab fragments (1:1000; Roche Diagnostics) and NBT/BCIP (Roche Diagnostics) were used to visualize the signal. Sections were viewed on a Leitz microscope (10× objective), and the images were captured with a Nikon DXM 1200 digital camera and analyzed using Image-Pro Plus software (Version 4.5; Media Cybernetics), as described below.
BrdU immunofluorescence histochemistry to label proliferating cells
To label proliferating cells in the brains of the embryos, the dams of Groups 3, 4, 5, and 7, which were administered saline, 0.75 mg/kg/d nicotine, or 1.5 mg/kg/d nicotine, were also given intraperitoneal injections every 8 h during 4 d of BrdU (20 mg/kg; Sigma) in 0.9% NaCl and 0.007 N NaOH, as described previously (Chang et al., 2008). The animals rapidly became adapted to this injection procedure, showing minimal signs of physical stress and no changes in body weight and the total amount of BrdU administered is known to be a saturating dose (Desouza et al., 2005; Mandyam et al., 2007). These injections were given from E12–E15, the period of peak cell birth in the hypothalamus and amygdala (Altman and Bayer, 1978; Bayer et al., 1993), or from E16–E19, the period of peak cell birth in the dentate gyrus (DG) of the hippocampus (Bayer et al., 1993). The offspring were killed at P15 and their brains were processed to reveal BrdU by immunofluorescence histochemistry, as described previously (Chang et al., 2008).
Immunofluorescence histochemistry to measure proliferation of neurons and glia
In P15 offspring of Groups 3, 4, 5, and 7, single- and double-labeling immunofluorescence was used to examine cell proliferation using BrdU, mature neurons using NeuN, astrocytes using GFAP, and microglia using Iba-1, and also to observe the coexistence of BrdU with these neuronal and glial markers in the PVN, PFLH, and CeA and with OX and MCH in the PFLH as described previously (Chang et al., 2008; Chang et al., 2012). Sections were viewed and fluorescence images were captured using a Zeiss fluorescence microscope with MetaVue software. The density of immunofluorescent cells in 8–12 images collected in each area in each animal was quantified with Image-Pro Plus software (Version 4.5; Media Cybernetics), as described below, and is reported as cell density (cells/μm2). For analysis of cells that double-labeled BrdU with the other markers or peptides, the images were captured with a 20× objective and the double-labeled cells were confirmed with a 40× objective and further validated by confocal Z-sectioning with a 40× water-immersion lens on a Zeiss LSM 510 META confocal microscope. The double-labeled cells were counted and reported as percentage of total single-labeled cells.
Digoxigenin ISH of peptides with BrdU immunofluorescence histochemistry to measure genesis of peptide neurons
In addition to using double-labeling immunofluorescence to identify BrdU cells expressing OX or MCH in the PFLH, digoxigenin-labeled ISH combined with BrdU immunofluorescence histochemistry was performed in Group 7 to determine whether the BrdU-labeled cells express ENK in the PVN and CeA as described previously (Chang et al., 2008). Briefly, the P15 brains of saline control, 0.75 mg/kg/d nicotine, or 1.5 mg/kg/d nicotine offspring of dams with BrdU injections given from E12–E15 were cut and 30 μm free-floating, alternative coronal sections were processed first for digoxigenin-labeled ISH, as described above. After the signal was visualized in NBT/BCIP, the sections were briefly washed in 0.1 m Tris-HCl containing 0.1 m NaCl and 50 mm MgCl2, pH 9.5, and PBS and then treated in 0.2 n HCl for 60 min at 37°C. The sections were then processed for BrdU immunofluorescence, with BrdU immunoreactivity revealed by Cy3-conjugated secondary antibody and viewed on a Zeiss microscope with MetaVue software. The peptide-expressing neurons with dark blue digoxigenin-labeled signal were viewed and captured first with the differential interference contrast filter and then the red rhodamine/Cy3 fluorescence filter was applied to reveal the BrdU+ signal in the same field. The images were merged and the double-labeled cells were counted and reported as a percentage of total single-labeled cells.
Antibodies for immunofluorescence histochemistry
Antibodies used were as follows: rat anti-BrdU monoclonal antibody (1:200; Novus Biologicals); mouse anti-NeuN and mouse anti-GFAP monoclonal antibody (1:50; Millipore); rabbit anti-Iba-1 polyclonal antibody (1:1000; Wako); goat anti-OX and goat anti-MCH polyclonal antibody (1:200; Santa Cruz Biotechnology); and fluorescence-conjugated secondary antibodies (1:200; Jackson ImmunoResearch Laboratories).
TUNEL to measure apoptosis
To determine whether maternal exposure to nicotine (0.75 or 1.5 mg/kg/d) compared with saline control leads to apoptosis, TUNEL was used in Group 6 to label DNA strand breaks for detection of apoptotic cells in the offspring at P15, by which time hypothalamic apoptosis is generally completed. Using an in situ cell death detection kit (fluorescein; Roche), TUNEL with positive and negative controls was performed with 30 μm free-floating coronal sections following the manufacturer's protocol.
Semiquantification of digoxigenin-labeled ISH and immunofluorescence
The density of ENK-, OX-, and MCH-expressing neurons, assayed by ISH with digoxigenin-labeled probes, and the density of BrdU-, NeuN-, GFAP-, Iba-1, OX-, and MCH-immunoreactive cells, assayed by immunofluorescence histochemistry, were measured by semiquantification with Image Pro Plus software (version 4.5; Media Cybernetics) as described previously (Chang et al., 2008; Chang et al., 2012). Briefly, for the peptide-expressing neurons in each animal, 8–12 sections at the same level were used to examine each nucleus in each animal and the entire nucleus was outlined and analyzed. The population density was used to determine the cell density in these areas. Before measurements were taken, a threshold for each nucleus was established. Using the selected sections, this threshold was set by matching the number of cells counted by the software in a defined area with the number of cells counted manually in that same area. This method, which was the same for all experiments and brain areas, yielded different threshold values (average of thresholds obtained within the same area in the 10 sections) for the different brain areas, experiments, and measurements of cells. This semiquantitative procedure allowed us to count the number of neurons in a specific area, which were then expressed as the cell density (number of cells/μm2), reflecting the density of mRNA- or protein-containing cells. In all analyses, the cells were counted only on one plane in each section and only those cells containing a nucleus in the plane (> 10 μm2) were counted, thereby excluding fractions of cells. The average cell density for the different groups was then compared and analyzed statistically.
Measurement of maternal plasma nicotine
To determine plasma nicotine in pregnant rats, tail vein blood was collected on E15. Immediately after collection, plasma was centrifuged at 2500 × g for 10 min and frozen at −20°C. Plasma nicotine analyses by HPLC was performed at the Proteomics Resource Center at Rockefeller University. Maternal plasma nicotine achieved with this administration model averaged 25 ng/ml in dams receiving 0.75 mg/kg/d and 43 ng/ml in dams receiving 1.5 mg/kg/d, which is consistent with other studies (Roy et al., 2002; Mahar et al., 2012).
Behavioral experiments
In Group 8, the consumption of different reinforcing substances, chow, and water was measured during a 4 h period at the start of the dark cycle in 4 sets of adolescent offspring (from P40 to P60) exposed prenatally to saline or nicotine (0.75 or 1.5 mg/kg/d).
Nicotine oral intake.
The first set of saline- or nicotine-exposed offspring was trained to consume a nicotine solution using a variant of the two-bottle choice procedure, with the relative positions alternated each day to prevent side preference (Dadmarz and Vogel, 2003; Biondolillo et al., 2009). In addition to water, nicotine was made available for 24 h in a second 8 oz bottle, with a nondrip sipper tube at the top of the cage (PETCO Animal Supplies). Concentration of nicotine tartrate solution was increased stepwise by 0.002% every 2 d from 0.008% to 0.02%. Total training lasted 12 d, with the animals having access to water at all times during training. Once the training was completed, animals were allowed to consume the nicotine solution and water for 4 h at dark onset on 3 consecutive test days and their intake was measured and averaged.
Ethanol oral intake.
In the second set of offspring, the effect of prenatal nicotine exposure on oral ethanol consumption was tested from P40 to P60. As described in our recent publication (Barson et al., 2013), ethanol was made available, in addition to the water, in a second 8 oz bottle with a nondrip sipper tube at the top of the cage (PETCO Animal Supplies), with the relative positions alternated each day to prevent side preference. Ethanol was provided daily (except weekends) for 8 h each day. Rats were trained to drink ethanol solution as follows: they were initially given 1% ethanol solution for 4 d and then this concentration was increased to 2% for an additional 4 d. After alcohol training was completed, rats were given 2% ethanol and water for 4 h on 3 consecutive test days and their consumption was measured and averaged. Water and chow were available ad libitum at all times. This low ethanol concentration was used because it is known to be a reliable predictor of future consumption of higher ethanol concentrations (Karatayev et al., 2010).
Fat-rich diet intake.
In the third set of adolescent offspring, the consumption of another reinforcing substance, a fat-rich diet (50% fat), was investigated as described previously (Leibowitz et al., 2004; Chang et al., 2008). Rats were trained to consume this diet (15 kcal), first by having daily access to it for 15 min over a 3 d period with chow also available; then, they were given ad libutum fat-rich diet for 4 h (with no chow present) on 3 consecutive days and consumption of this diet was measured and averaged.
Chow and water intake.
In the fourth set of offspring, the consumption of laboratory chow and water (on which the rats were maintained ad libitum) was measured during the 4 h period at dark onset on 3 consecutive days and averaged.
Statistical analysis
Comparisons between the three groups, saline, 0.75 mg/kg/d nicotine, and 1.5 mg/kg/d nicotine, were made using a univariate ANOVA, followed by Tukey's post hoc tests when appropriate. Comparisons between two groups were performed using unpaired two-tailed Student's t tests. The criterion for significance was p < 0.05 and all significance values greater than this are reported as not significant (NS). Data are reported as mean ± SEM.
Results
Effect of prenatal nicotine on peptide expression
To determine the effect of gestational nicotine exposure on the expression of orexigenic peptides in postnatal offspring, pregnant rats in Group 1 were implanted with an osmotic minipump filled with either nicotine (0.75 or 1.5 mg/kg/d) or saline and mRNA levels of the orexigenic peptides ENK, OX, and MCH were measured in specific brain areas of the offspring at P15 using ISH with radiolabeled probes. In the offspring exposed to nicotine compared with saline control (n = 7–8/group), measurements of peptide mRNA levels in the different hypothalamic areas or the CeA revealed a significant increase after both doses of nicotine (Fig. 1A), as illustrated for the higher dose in the photomicrographs (Fig. 1B). These differences were observed for ENK in the PVN (F(2,20) = 4.91, p < 0.05) and CeA (F(2,20) = 11.64, p < 0.01) and for both OX (F(2,20) = 6.51, p < 0.01) and MCH (F(2,19) = 7.29, p < 0.01) in the PFLH. Post hoc analyses showed that the offspring exposed to either dose of nicotine exhibited a significant, 15–43% increase (p < 0.05) in expression of these peptides compared with control (Fig. 1A), with a similar magnitude seen at the two doses (NS for 0.75 vs 1.5 mg/kg/d). It was confirmed in a separate set of offspring (n = 4/group) examined around puberty (P40), which in response to nicotine at 0.75 mg/kg/d exhibited a significant, 40% increase in ENK mRNA levels in the PVN compared with control (t(6) = 4.56, p < 0.01) and CeA (t(6) = 7.64, p < 0.01) and also of OX (t(6) = 5.15, p < 0.05) and MCH (F(2,19) = 7.30, p < 0.01) in the PFLH (Table 2). These data provide the first evidence that nicotine during pregnancy, at low doses, has a stimulatory effect on these orexigenic peptides and demonstrate that this effect is long lasting, being evident in pubertal offspring 6 weeks after their exposure to nicotine.
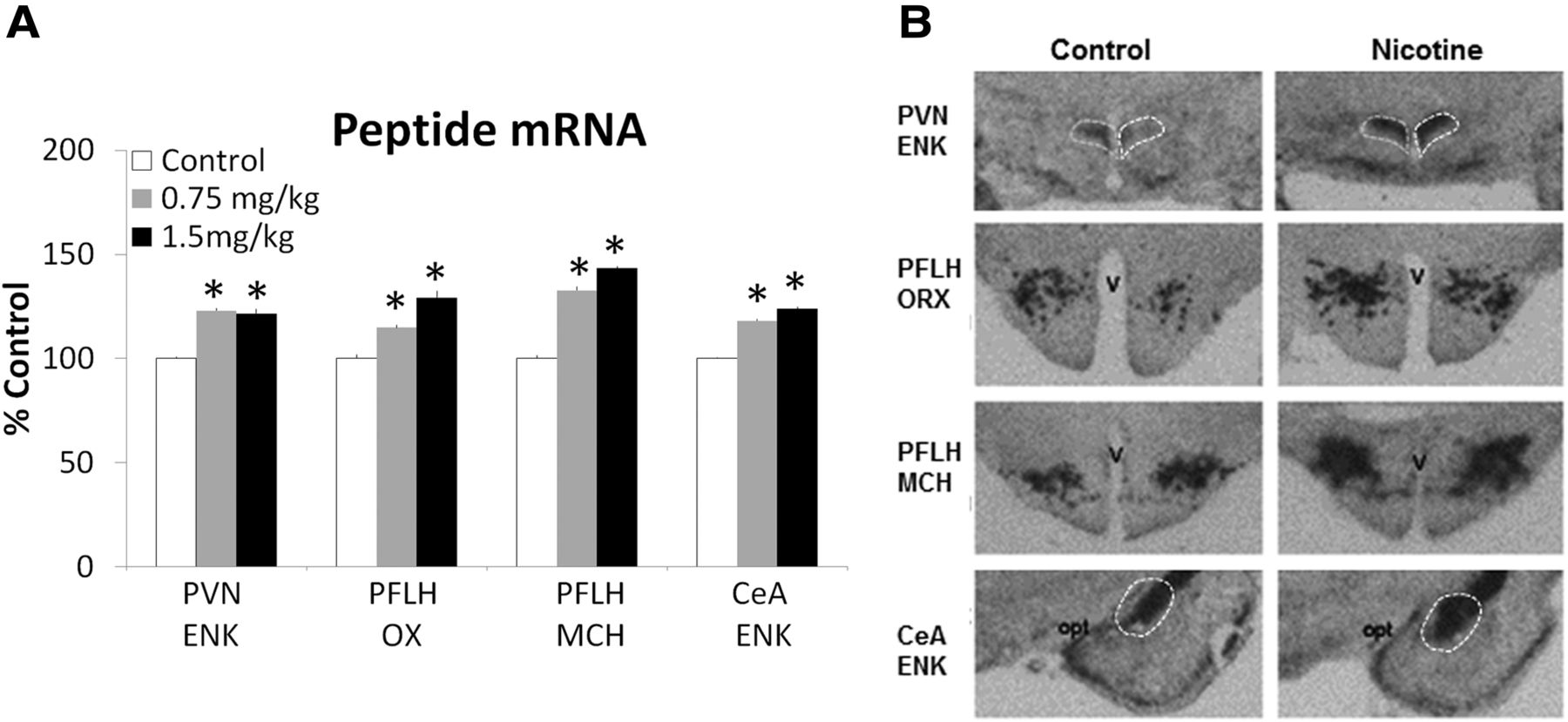
Prenatal nicotine increases peptide mRNA in P15 offspring exposed to 0.75 mg/kg/d or 1.5 mg/kg/d nicotine relative to control (n = 7–8/group), as assessed by radiolabeled ISH. A, Peptide mRNA in nicotine versus control groups expressed as percent change relative to control. Data are mean ± SEM, *p < 0.05 versus control. B, Photomicrographs illustrating the effect of 1.5 mg/kg nicotine versus control on mRNA levels of ENK in the PVN and CeA and of OX and MCH in the PFLH. opt, optic tract; V, ventricle.
Prenatal nicotine stimulates ENK expression in the PVN and CeA and OX and MCH expression in the PFLH of P15 offspring exposed to 0.75 mg/kg/d nicotine relative to control (n = 4/group), as indicated by a significant increase in mRNA levels in nicotine versus control offspring
Effect of prenatal nicotine on density of peptide-expressing neurons
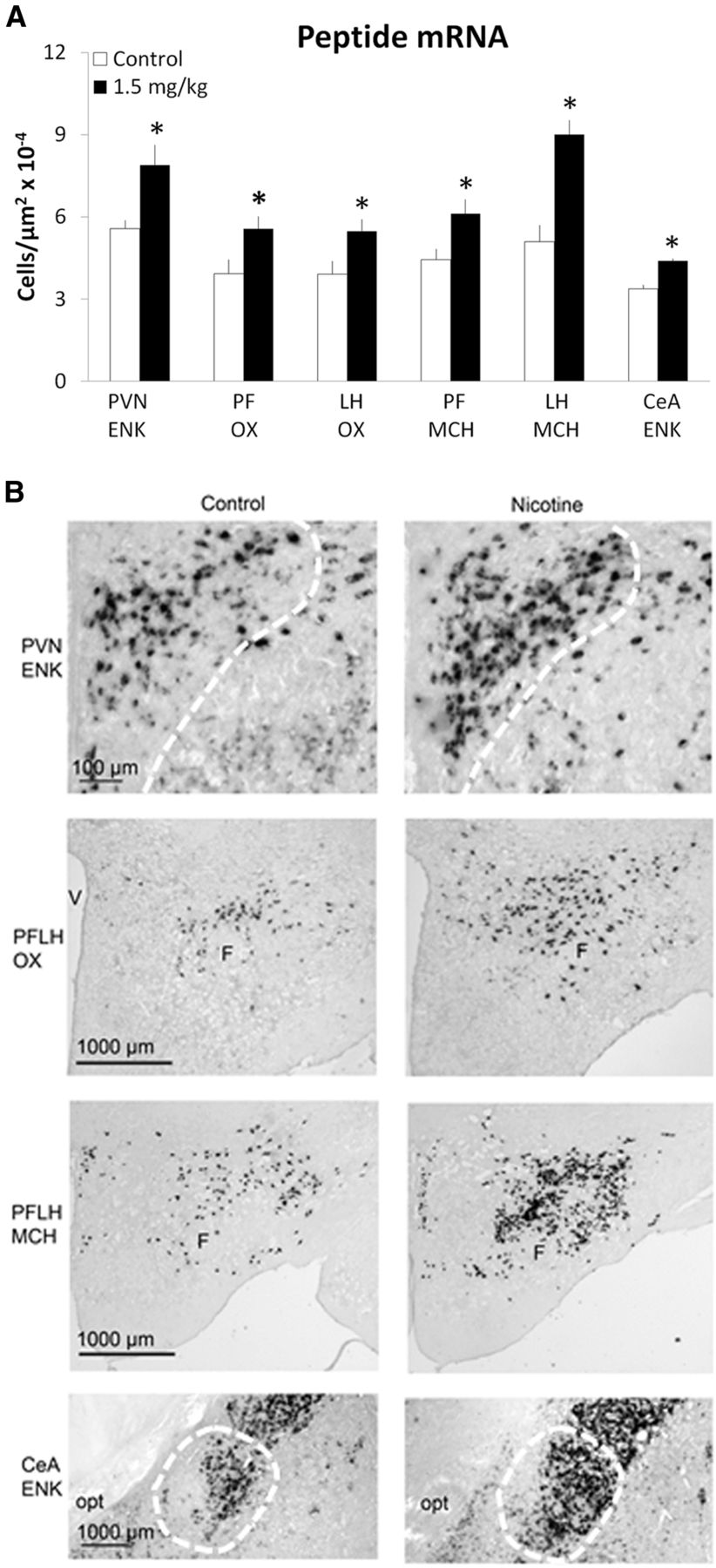
This experiment tested in Group 2 whether prenatal nicotine exposure, at a low dose that stimulates mRNA levels of the orexigenic peptides, can increase the density of peptide-expressing neurons and whether this effect can be localized more precisely to specific areas of the brain structures being studied, as measured using ISH with digoxigenin-labeled probes. In this set of offspring prenatally exposed to nicotine at 1.5 mg/kg/d or saline (n = 4–8/group) and killed at P15, measurements of ENK neurons revealed a significant difference between the groups in the PVN (t(14) = 2.88, p < 0.05) and CeA (t(8) = 6.49, p < 0.05) (Fig. 2A). The nicotine-exposed offspring exhibited a significant, 30–40% increase in the density of ENK-expressing neurons compared with control and showed this effect to be localized to the medial parvocellular region of the PVN and the lateral and capsular region of the CeA (Fig. 2B). In contrast, no change was observed in the lateral part of the PVN (t (14) = 0.06, NS) in offspring exposed to 1.5 mg/kg/d nicotine (3.61 × 10−4 ± 6.82 × 10−7 cells/μm2) compared with control (3.59 × 10−4 ± 6.96 × 10−7cells/ μm2) or in the BLA (t(14) = 1.04, NS) in offspring exposed to 1.5 mg/kg/d nicotine (6.60 × 10−6 ± 8.86 × 10−8 cells/μm2) compared with control (6.59 × 10−6 ± 1.09 × 10−7cells/μm2). There was also a significant increase in the density of neurons (+40–58%) that expressed OX in both the PF (t(14) = 2.41, p < 0.05) and LH (t(14) = 2.47, p < 0.05) and of MCH in the PF (t(14) = 2.50, p < 0.05) and LH (t(14) = 3.70, p < 0.01) (Fig. 2A,B). These data, in addition to confirming our findings in the first experiment, demonstrate the anatomical specificity of this effect and reveal the location of the peptide-expressing neurons most responsive to nicotine exposure during gestation.
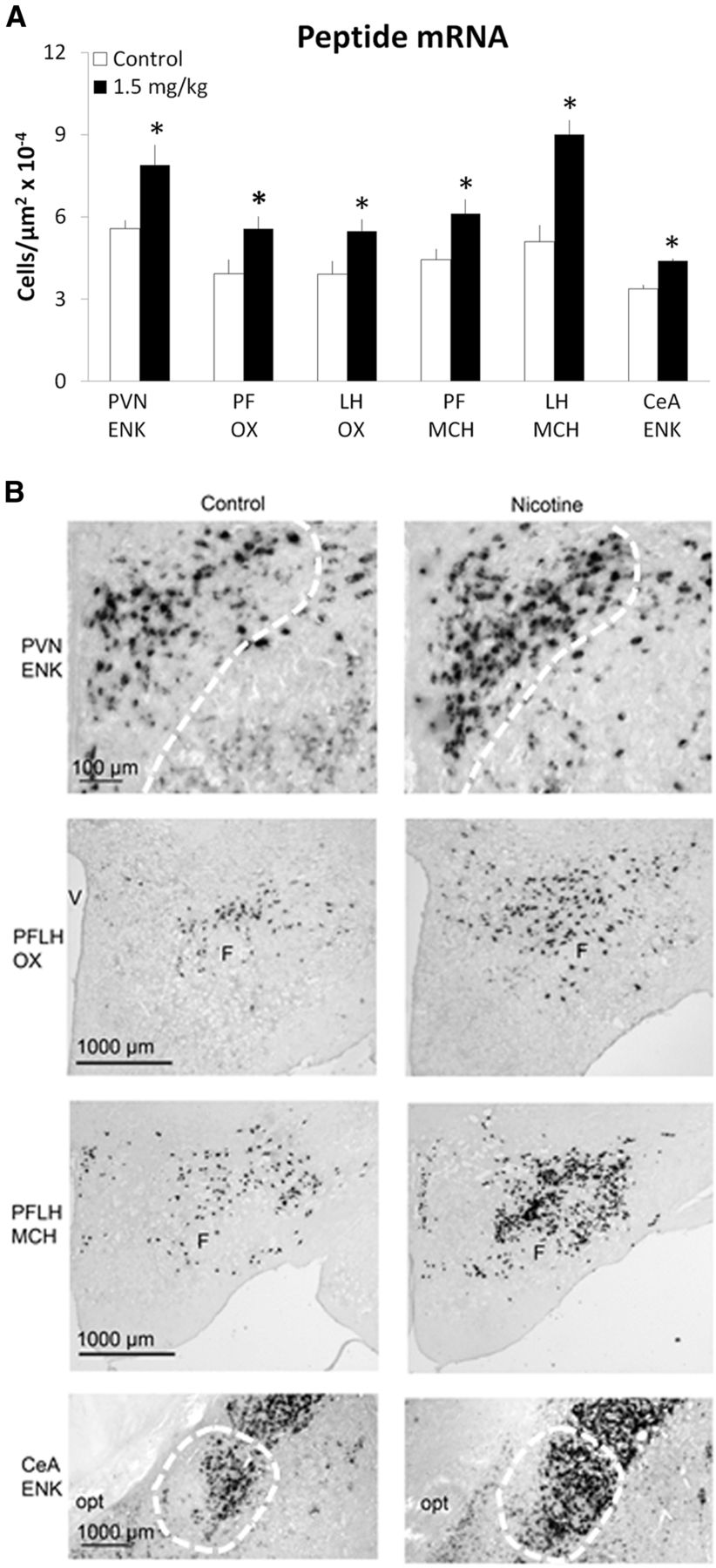
Prenatal nicotine increases the density of peptide-expressing neurons in P15 offspring exposed to 1.5 mg/kg/d nicotine relative to control (n = 4–8/group), as assessed by ISH with digoxigenin-labeled probes. A, Density of peptide-expressing neurons. Data are mean ± SEM, *p < 0.05 versus control. B, Photomicrographs illustrating this effect of 1.5 mg/kg nicotine versus control on ENK, OX, and MCH. opt, optic tract; V, ventricle; F, fornix.
Effect of prenatal nicotine on cell generation
With this nicotine-induced increase in the density of peptide-expressing neurons and mRNA levels, the next experiment investigated whether prenatal nicotine can, in fact, stimulate the generation of new cells in these brain regions. In addition to being implanted with an osmotic minipump filled with either nicotine (0.75 or 1.5 mg/kg/d) or saline control, the dams in Group 3 were injected during pregnancy with the cell proliferation marker BrdU during the period of peak cell birth in the brain areas studied and their offspring were examined at P15, when postnatal neuronal development and apoptosis in the hypothalamus and amygdala have primarily ceased (Ifft, 1972; Bayer et al., 1993). The nicotine offspring exhibited a significant increase in the density of BrdU+ cells in the different brain areas examined compared with saline control (n = 4/group). This nicotine effect was seen in the PVN (F(2,9) = 15.08, p < 0.01), PF (F(2,9) = 8.26, p < 0.01), and LH (F(2,9) = 6.40, p < 0.05) and also in the CeA (F(2,9) = 6.93, p < 0.05) (Fig. 3A), as illustrated in the photomicrographs for the higher dose of nicotine (Fig. 3B). The density of BrdU+ cells was generally increased by at least 20% (p < 0.05), with the two doses tested having a similar magnitude of effect (NS for 0.75 vs 1.5 mg/kg/d). This stimulatory effect on the generation of new cells in the PVN, PFLH, and CeA was not detected in the DG of the hippocampus (F(2,9) = 1.05, NS), consistent with a recent study showing no effect of 2 mg/kg/d nicotine in this area (Mahar et al., 2012). These results underscore the robustness of the stimulatory effect of prenatal nicotine on neuronal development and also its persistent nature, with many of the newly generated neurons still evident 2 weeks later in the absence of nicotine.

Prenatal nicotine stimulates cell proliferation in P15 offspring exposed to 0.75 mg/kg/d or 1.5 mg/kg/d nicotine relative to control (n = 4/group). A, Prenatal nicotine increases the density of BrdU+ cells. Data are mean ± SEM, *p < 0.05. B, Photomicrographs illustrating this effect of 1.5 mg/kg nicotine versus control in the PVN, PFLH, and CeA. F, fornix.
Phenotype of BrdU+ cells stimulated by nicotine exposure
This next experiment examined the phenotype of the BrdU+ cells stimulated by prenatal nicotine exposure in the hypothalamus and CeA using single- and double-labeling immunofluorescence with additional antibodies against NeuN (a marker for mature neurons) and GFAP (a marker for astrocytes) in Group 4, against Iba-1 (a marker for microglia) in Group 5, and with TUNEL staining (a marker for apoptotic cell death) in Group 6. In each of these groups, the dams were implanted with a minipump filled with nicotine (0.75 or 1.5 mg/kg/d) compared with saline control (n = 4/group) and injected during pregnancy with BrdU, with the offspring killed at P15. The nicotine-treated offspring of Group 4 compared with control exhibited a marked and highly consistent increase in mature neurons, as indicated by the greater percentage of double-labeled BrdU+/NeuN+ cells relative to total number of single-labeled cells (Fig. 4A,B). This increase in double-labeled neurons in the different brain areas, from ∼3–5 neurons in the control offspring to 30–55 neurons in the nicotine offspring, was evident specifically in the medial parvocellular region of the PVN relative to BrdU (F(2,9) = 261.75, p < 0.01) and NeuN (F(2,9) = 63.94, p < 0.05), the PF relative to BrdU (F(2,9) = 227.72, p < 0.01) and NeuN (F(2,9) = 226.79, p < 0.01), the LH relative to BrdU (F(2,9) = 289.81, p < 0.01) and NeuN (F(2,9) = 222.41, p < 0.01), and the lateral and capsular region of the CeA relative to BrdU (F(2,9) = 141.87, p < 0.01) and NeuN (F(2,9) = 295.00, p < 0.01). The two doses of nicotine were similarly effective in increasing the number of double-labeled cells (NS for 0.75 vs 1.5 mg/kd/d) in these areas. In contrast to these changes in NeuN+ mature neurons, prenatal nicotine in Group 4 produced no change in the density of GFAP-labeled astrocytes in the PVN (F(2,9) = 1.13, NS), PFLH (F(2,9) = 0.48, NS), or CeA (F(2,9) = 0.50, NS) (Table 3), and there was no evidence of BrdU+/GFAP+ double-labeled cells. In the offspring of Group 5, prenatal nicotine also failed to produce a significant increase in Iba-1 labeled microglia in the PVN (F(2,9) = 1.72, NS), PFLH (F(2,9) = 2.64, NS), or CeA (F(2,9) = 2.28, NS) (Table 4), and there was no evidence of BrdU+/Iba-1+ double-labeled cells in these areas. In the offspring of Group 6, prenatal nicotine had no effect on apoptotic cell death, with the control and nicotine-exposed offspring (0.75 and 1.5 mg/kg/d) revealing no TUNEL+ cells in the PVN, PFLH, CeA, or DG. These data demonstrate that in utero nicotine exposure has a significant, stimulatory effect on the generation and development of new neurons in the hypothalamus and CeA while having little impact on gliogenesis or apoptosis in these areas.

Prenatal nicotine stimulates neurogenesis in P15 offspring exposed to 0.75 mg/kg/d or 1.5 mg/kg/d nicotine relative to control (n = 4/group). A, Left: Prenatal nicotine increases the percentage of BrdU+/NeuN+ cells relative to total number of BrdU+ cells in the PVN, PF, LH, and CeA. A, Right: Prenatal nicotine increases the percentage of BrdU+/NeuN+ cells relative to total number of NeuN+ cells. Data are mean ± SEM, *p < 0.05. B, Photomicrographs demonstrate this effect for 1.5 mg/kg/d nicotine versus control, illustrating BrdU+ cells (red), NeuN+ cells (green), and BrdU+/NeuN+ cells (yellow). Arrowheads indicate double-labeled cells. Images on the far right are higher magnifications of images marked with a dashed square. V, ventricle; F, fornix.
Prenatal nicotine has no effect on the density of GFAP+ cells in the PVN, PFLH, and CeA in P15 offspring exposed to 0.75 mg/kg/d or 1.5 mg/kg/d nicotine relative to control (n = 4/group)
Prenatal nicotine has no effect on the density of Iba-1+ cells in the PVN, PFLH, and CeA in P15 offspring exposed to 0.75 mg/kg/d or 1.5 mg/kg/d nicotine relative to control (n = 4/group)
Effect of prenatal nicotine on genesis of peptide-expressing neurons
The next goal was to determine whether the BrdU+ neurons stimulated by prenatal nicotine exposure can express or synthesize the orexigenic peptides, as measured using ISH with digoxigenin-labeled probes in combination with BrdU immunofluorescence or double-labeling immunofluorescence. The offspring of dams in Group 7 implanted with a minipump filled with nicotine (0.75 or 1.5 mg/kg/d) compared with saline control (n = 4/group) and injected during pregnancy with BrdU were also killed at P15. Whereas the control offspring showed almost no colocalization of peptides with BrdU in the 4 areas examined (0–2 neurons), the prenatal nicotine-exposed offspring exhibited a significant increase in the genesis of peptide-expressing neurons (8–16 neurons), as indicated by a greater percentage of double-labeled peptide+/BrdU+ cells relative to single-labeled peptide+ or BrdU+ cells. There were significant group differences (Fig. 5A) in the PVN with measurements of ENK+/BrdU+ cells relative to single-labeled ENK+ (F(2,9) = 26.66, p < 0.01) and BrdU+ (F(2,9) = 15.50, p < 0.01) cells, the PF with measurements of OX+/BrdU+ cells relative to single-labeled OX+ (F(2,9) = 23.31, p < 0.01) and BrdU+ (F(2,9) = 57.00, p < 0.01) cells, and the LH with measurements of OX+/BrdU+ cells relative to single-labeled OX+ (F(2,9) = 13.09, p < 0.01) and BrdU+ (F(2,9) = 13.34, p < 0.01) cells. For MCH, this was also evident with measurements of MCH+/BrdU+ cells in the PF relative to single-labeled MCH+ (F(2,9) = 26.31, p < 0.01) and BrdU+ (F(2,9) = 19.38, p < 0.01) cells and the LH relative to single-labeled MCH+ (F(2,9) = 18.91, p < 0.01) and BrdU+ (F(2,9) = 14.03, p < 0.01) cells, as illustrated in Figure 5B. We also detected double-labeled ENK+/BrdU+ cells in the CeA of P15 offspring, where there were significant differences in the number of double-labeled cells relative to single-labeled ENK+ (F(2,9) = 8.04, p < 0.01) and BrdU+ (F(2,9) = 4.73, p < 0.05) cells. Once again, both doses of nicotine had a similar, stimulatory effect on ENK+/BrdU+ neurons in the PVN (p < 0.01 vs control; NS for 0.75 vs 1.5 mg/kg) and CeA (p < 0.05 vs control; NS for 0.75 vs 1.5 mg/kg/d), on OX+/BrdU+ neurons in the PF (p < 0.05 vs control; NS for 0.75 vs 1.5 mg/kg/d) and LH (p < 0.05 vs control; NS for 0.75 vs 1.5 mg/kg/d), and on MCH+/BrdU+ neurons in the PF (p < 0.01 vs control; NS for 0.75 vs 1.5 mg/kg/d) and LH (p < 0.01 vs control; NS for 0.75 vs 1.5 mg/kg/d). These results in postnatal offspring no longer exposed to nicotine demonstrate that in utero nicotine at relatively low doses can increase the generation of new neurons in the PVN, PFLH, and CeA that have the specific phenotype of expressing the orexigenic peptides known to be positively related to nicotine.

Prenatal nicotine stimulates of peptide-expressing systems in P15 offspring exposed to 0.75 mg/kg/d or 1.5 mg/kg/d nicotine relative to control (n = 4/group). A, Left: Prenatal nicotine increases the percentage of double-labeled, BrdU+/peptide+ neurons relative to total number of BrdU+ neurons. A, Right: Prenatal nicotine increases the percentage of double-labeled, BrdU+/peptide+ neurons relative to the total number of peptide+ neurons. Data are mean ± SEM, *p < 0.05 versus control. B, Photomicrographs demonstrate this effect for 1.5 mg/kg/d nicotine versus control. In the PVN and CeA, BrdU+ cells (red), ENK+ cells (black), and BrdU+/ENK+ cells (red nucleus in black perikaryon) are indicated by arrowheads for double-labeled cells. In the PF, BrdU+ cells (red), OX+ cells (green), and BrdU+/OX+ cells (red nucleus in green perikaryon) are indicated by arrowheads for double-labeled cells. Images on the far right are higher magnifications of images marked with a dashed square. F, fornix.
Effect of prenatal nicotine on consumption of reinforcing substances in pubertal offspring
To test the possibility that the neuronal changes induced by gestational nicotine exposure are associated with changes in ingestive behavior, three sets of offspring in Group 8 were examined during adolescence (P40–P60) for their consumption of different reinforcing substances, not only of nicotine but also of ethanol and a fat-rich diet, which are positively related to the expression of ENK and OX (Chang et al., 2004; Chang et al., 2007; Chang et al., 2010; Morganstern et al., 2010b) and known to predict and promote consumption of high concentrations of ethanol (Karatayev et al., 2010; Barson et al., 2013). In the first set of adolescent offspring tested on a nicotine solution (0.02%), the prenatal nicotine-exposed rats compared with controls (n = 6–8/group) consumed a greater amount of nicotine during the 4 h access period on 3 separate days (Fig. 6), as indicated by a significant group difference in nicotine consumption (F(2,18) = 5.58, p < 0.05), with no difference in water consumption (F(2,18) = 0.02, NS) between the 1.5 mg/kg/d (8.4 ± 0.6 g/kg) and 0.75 mg/kg/d (8.6 ± 0.9 g/kg) nicotine offspring compared with control (8.3 ± 0.9 g/kg). In the second set of adolescent rats (n = 6–8/group) tested on an ethanol solution (2%), prenatal nicotine compared with control also induced an increase in the consumption of ethanol during 4 h access (F(2,18) = 6.15, p < 0.01; Fig. 6), with no difference in water consumption (F(2,18) = 0.12, NS) between the 1.5 mg/kg/d (7.5 ± 0.4 g/kg) and 0.75 mg/kg/d (7.2 ± 0.7 g/kg) nicotine offspring compared with control (7.3 ± 0.8 g/kg). In the third set (n = 6–8/group), prenatal nicotine also increased the number of calories consumed from the fat-rich diet during the 4 h test (F(2,18) = 21.73, p < 0.01; Fig. 6). In contrast, in a fourth set of animals (n = 6–8/group), the nicotine-exposed offspring compared with control showed no change in their intake of dry laboratory chow diet (F(2,18) = 1.05, NS) or water (F(2,18) = 0.05, NS) during 4 h access (Fig. 6), indicating that the increase in consumption occurs primarily with substances having reinforcing properties. The two doses of nicotine were similarly effective in stimulating the consumption of nicotine (p < 0.05 vs control; NS for 0.75 vs 1.5 mg/kg/d), ethanol (p < 0.05 vs control; NS for 0.75 vs 1.5 mg/kg/d), and fat-rich diet (p < 0.01 vs control; NS for 0.75 vs 1.5 mg/kg/d). These results demonstrate that even low-level nicotine exposure during gestation may have profound effects in adolescent offspring on the consumption of substances prone to abuse.

The consumption of nicotine, ethanol, and fat-rich diet is increased in adolescent offspring prenatally exposed to 0.75 mg/kg/d or 1.5 mg/kg/d nicotine relative to control (n = 6–8/group), whereas the intake of chow or water is unaffected. Data are mean ± SEM, *p < 0.05 versus control.
Discussion
Recent studies suggest that exposure to drugs of abuse, such as ethanol and cocaine, and other reinforcing substances, such as dietary fat, during development have a profound impact on the programming of orexigenic peptide systems (Torres-Reveron et al., 2007; Chang et al., 2008; Chang et al., 2012). We report here for the first time that gestational exposure to nicotine at low doses significantly increases the expression of ENK in both the PVN and CeA and of OX in the PFLH of the offspring. This increase in peptide mRNA levels, previously described in adult rats after acute injection of nicotine at low doses (0.175–0.5 mg/kg/d; Houdi et al., 1998; Loughlin et al., 2006), was revealed here in P15 offspring of dams given a similar dose of nicotine (0.75 mg/kg/d) or a somewhat higher dose (1.5 mg/kg/d). Further analyses showed that prenatal nicotine stimulated another orexigenic peptide, MCH, in the PFLH, indicating that these effects of nicotine in utero are broad in scope and involve a variety of peptide systems known to promote consummatory behaviors. These results also showed that this nicotine-induced increase in peptide mRNA levels was accompanied by an increase in the density of peptide-expressing neurons that was anatomically localized, involving specific subpopulations of neurons that are unusually sensitive to the effects of nicotine on neurodevelopment. For ENK, this effect was evident specifically in the medial-parvocellular region of the PVN and the lateral and capsular region of the CeA, but not in the lateral PVN or BLA, and for both OX and MCH, it was seen in the medial and lateral regions of the PFLH. This stimulatory effect of low-dose nicotine on peptide-expressing neurons persists into puberty (P40), long after the period of nicotine exposure. This indicates that the early development of these orexigenic peptide systems is altered by nicotine exposure, possibly with long-term behavioral consequences.
Further evidence suggests that the mechanism underlying this persistent, nicotine-induced change in the peptide systems at low doses involves an increase in the proliferation of new cells and new neurons in the hypothalamus and amygdala of the offspring. This stimulation of neurogenesis was indicated by an increased density of single-labeled BrdU+ cells and of BrdU+ neurons that double-label NeuN, and it was detected in the same specific areas of the PVN, PFLH, and CeA where peptide expression levels were significantly altered. This stimulatory effect in these nuclei contrasts with results obtained in the forebrain, hippocampus, and cerebellum, where similar or slightly higher doses (0.7–2.0 mg/kg) had no impact on neurogenesis and neuronal number (Chen and Edwards, 2003; Mahar et al., 2012), suggesting that the hypothalamus and amygdala or specific subregions of these structures are unusually sensitive to nicotine's effects in utero. This effect was apparently restricted to cells of neuronal phenotype, because there was no significant change in gliogenesis as indicated by measurements of GFAP-labeled astrocytes and of microglia labeled with Iba-1. In addition, there was no effect on apoptosis with these low doses of nicotine tested. These results contrast with effects seen in the hippocampus and cerebellum with high doses of nicotine, which stimulate gliogenesis (Roy et al., 2002; Abou-Donia et al., 2006) and cause an increase in apoptosis (Anbarasi et al., 2006).
Most notable is our finding that the newly generated neurons produced by in utero nicotine exposure are able to express the orexigenic peptides, as demonstrated by an increased density ENK neurons in the PVN and CeA or OX and MCH neurons in the PFLH that colabeled BrdU, a marker of cell proliferation. The mechanisms mediating this stimulatory effect of prenatal nicotine on the development of these peptide-expressing neurons are unknown. Studies in adult animals showing ENK and OX to be stimulated by a lipid emulsion that raises circulating triglycerides and fatty acids (Chang et al., 2004), which are also elevated by nicotine during pregnancy (de Souza Mda et al., 2010), suggest that maternal lipids may have a role in the process of promoting peptide neurogenesis. This possibility is supported by studies showing that fatty acids stimulate neuronal proliferation and differentiation (Maekawa et al., 2009; Katakura et al., 2013) and increase ENK transcription in PC12 cells (Parab et al., 2007). In addition to lipids, the increased neurogenesis may result from a stimulatory effect of prenatal nicotine on neurotrophic factors in the offspring, which are elevated in certain brain areas and the blood (Son and Winzer-Serhan, 2009; Harrod et al., 2011; Wei et al., 2011), and also on nicotinic cholinergic receptors, which stimulate cell survival, perhaps via the neurotropins, while inhibiting apoptosis through actions on the JAK2 signaling pathway (Marrero and Bencherif, 2009). Although other maternal circulatory factors involved in neurogenesis may include corticosterone and leptin (Garza et al., 2008; Walker et al., 2008), low doses of nicotine similar to the ones used in the present study appear to have little impact on circulating levels of these hormones (Grove et al., 2001; Mahar et al., 2012).
This stimulatory effect of gestational nicotine on orexigenic peptide neurogenesis observed postnatally is found to be accompanied by behavioral changes during adolescence, specifically an increase in consumption of different reinforcing substances. When tested 40 d after the period of nicotine exposure, the offspring consumed significantly greater amounts of nicotine, a phenomenon previously shown in mice (Klein et al., 2003; Chistyakov et al., 2010; Schneider et al., 2012). The nicotine-exposed offspring also exhibited an increase in consumption of another drug of abuse, ethanol, at a low concentration (2%) known to predict drinking of 9% ethanol (Karatayev et al., 2010), and a palatable fat-rich diet, which is found to be closely related to ethanol intake (Carrillo et al., 2004). These findings, together with evidence that prenatal nicotine-exposed offspring exhibit increased cocaine self-administration and sensitivity to methamphetamines (Franke et al., 2008; Harrod et al., 2012), demonstrate that the effects of prenatal nicotine on consummatory behavior are broad in nature and involve a range of substances that are palatable and reinforcing. This is further supported by our finding that nicotine had no effect on the offspring's consumption of a dry laboratory chow or water.
Studies in adult animals support the possibility that the increased peptide neurogenesis and expression in the hypothalamus and amygdala, induced by low doses of gestational nicotine, is causally related to the enhanced vulnerability of adolescent offspring to excess consumption, not only of nicotine but also of other reinforcing substances such as ethanol and fat. In adult rats, administration of these peptides or their receptor antagonists significantly alters the consumption of these substances. In the hypothalamus, injection of the ENK analog [D-Ala(2),N-Me-Phe(4),Gly(5)-ol]-Enkephalin (DAMGO) in the PVN or of OX in the PFLH stimulates the drinking of ethanol and consumption of a high-fat diet (Clegg et al., 2002; Naleid et al., 2007; Barson et al., 2010; Morganstern et al., 2010a), with opposite effects produced by the receptor antagonists (Lawrence et al., 2006; Naleid et al., 2007; Moorman and Aston-Jones, 2009; Barson et al., 2010), and administration of DAMGO directly in the CeA also increases consumption of a fat-rich diet (Will et al., 2009). In addition, peripheral administration of an OX receptor 1 antagonist or of opioid antagonists is found to reduce the self-administration of nicotine (Corrigall et al., 2002; Hollander et al., 2008; Ismayilova and Shoaib, 2010; LeSage et al., 2010). Although yet to be studied with nicotine self-administration, MCH when centrally injected increases the consumption of ethanol (Duncan et al., 2005; Morganstern et al., 2011) and is endogenously elevated in the PFLH of rats prone to overconsuming fat (Morganstern et al., 2010a). The present study, revealing changes in these peptide systems in both the hypothalamus, which is involved in the consummatory and arousal aspects of food and drug intake (Barson et al., 2011), and the CeA, which mediates stress and the emotional aspects of eating and drug use (Koob, 2009), suggests that the increased neurogenesis observed after prenatal exposure to nicotine, even at low doses, has a range of behavioral consequences that increases the offspring's risk of overconsuming a variety of substances with reinforcing properties.
In conclusion, this model of prenatal exposure to low-dose nicotine presents strong evidence that the orexigenic peptide systems in brain areas involved in reward and consummatory patterns are highly sensitive to a stimulatory effect of nicotine on neurogenesis. This stimulation in the offspring, observed at an unusually low dose of 0.75 mg/kg/d and confirmed at the 1.5 mg/kg/d dose, contrasts with the suppressive effect on neurogenesis generally observed at somewhat higher doses of prenatal nicotine, >4.0 mg/kg. The stimulation of these orexigenic peptide systems is long lasting and accompanied by behavioral changes during adolescence involving increased consumption of multiple reinforcing substances. The established positive relationship between these peptides and consummatory behavior in adult animals suggests a possible causal relationship between the increased neurogenesis of peptide-expressing neurons in utero and the increased consummatory behavior in the adolescent offspring.
Footnotes
This research was supported by a Brain and Behavior Research Foundation Independent Investigator Award and by the National Institutes of Health (Grant #R01 AA12882). We thank Dr. Jessica R. Barson, Olga Lukatskaya, and Sherry Liang for their help in the preparation of this manuscript.
The authors declare no competing financial interests.
- Correspondence should be addressed to Sarah F. Leibowitz, The Rockefeller University, 1230 York Avenue, New York, NY 10065. leibow{at}rockefeller.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}