

Abstract
Idiopathic small-fiber neuropathy (I-SFN), clinically characterized by burning pain in distal extremities and autonomic dysfunction, is a disorder of small-caliber nerve fibers of unknown etiology with limited treatment options. Functional variants of voltage-gated sodium channel Nav1.7, encoded by SCN9A, have been identified in approximately one-third of I-SFN patients. These variants render dorsal root ganglion (DRG) neurons hyperexcitable. Sodium channel Nav1.8, encoded by SCN10A, is preferentially expressed in small-diameter DRG neurons, and produces most of the current underlying the upstroke of action potentials in these neurons. We previously demonstrated two functional variants of Nav1.8 that either enhance ramp current or shift activation in a hyperpolarizing direction, and render DRG neurons hyperexcitable, in I-SFN patients with no mutations of SCN9A. We have now evaluated additional I-SFN patients with no mutations in SCN9A, and report a novel I-SFN-related Nav1.8 mutation I1706V in a patient with painful I-SFN. Whole-cell voltage-clamp recordings in small DRG neurons demonstrate that the mutation hyperpolarizes activation and the response to slow ramp depolarizations. However, it decreases fractional channels resistant to fast inactivation and reduces persistent currents. Current-clamp studies reveal that mutant channels decrease current threshold and increase the firing frequency of evoked action potentials within small DRG neurons. These observations suggest that the effects of this mutation on activation and ramp current are dominant over the reduced persistent current, and show that these pro-excitatory gating changes confer hyperexcitability on peripheral sensory neurons, which may contribute to pain in this individual with I-SFN.
Introduction
Small-fiber neuropathy (SFN) is a form of neuropathy, often painful, involving small-caliber peripheral nerve fibers including myelinated Aδ and unmyelinated C fibers that mediate pain, thermal sensation, and autonomic function (Tavee and Zhou, 2009; Hoeijmakers et al., 2012a). The characteristic clinical presentation of SFN consists of autonomic dysfunction and severe pain that often occurs in a stocking-glove distribution in which the distal extremities are affected. Commonly, skin biopsy reveals a paucity of intra-epidermal nerve fiber density (IENFD), and quantitative sensory testing (QST) demonstrates abnormal thermal thresholds in SFN patients (Devigili et al., 2008; Hoeijmakers et al., 2012a; McArthur, 2012).
SFN has been recognized in various medical conditions, including diabetes mellitus, metabolic syndrome, Fabry disease, Sjögren's syndrome, celiac disease, sarcoidosis, human immunodeficiency virus, dysthyroidism, and alcohol abuse, yet a substantial proportion, varying from 24 to 93% of SFN patients in various series (Lacomis, 2002; Lauria, 2005; Devigili et al., 2008; Bednarik et al., 2009; Faber et al., 2012a; Heij et al., 2012) are diagnosed as idiopathic (I-SFN), in which no cause could be identified. Gain-of-function variants in SCN9A, which encodes the Nav1.7 sodium channel, have been identified in 28.6% of patients with biopsy-confirmed I-SFN, suggesting an etiological basis for these I-SFN cases (Faber et al., 2012a; Hoeijmakers et al., 2012b). These mutants alter biophysical properties of Nav1.7, producing increased excitability in dorsal root ganglion (DRG) neurons, which can be demonstrated by current-clamp recordings (Faber et al., 2012a; Han et al., 2012a, b; Hoeijmakers et al., 2012c). In additional I-SFN patients with no mutations in SCN9A, we searched for variants in SCN10A, which encodes Nav1.8, another sodium channel preferentially expressed in small DRG neurons (Akopian et al., 1996; Shields et al., 2012), and which has been shown to be responsible for most of the current underlying the upstroke of action potentials in these neurons (Renganathan et al., 2001). Analysis of SCN10A revealed pro-excitatory variants with diverse changes in channel gating (Faber et al., 2012b). Similar to Nav1.7 variants, these Nav1.8 mutants reduce current threshold and increase evoked firing frequencies of DRG neurons (Faber et al., 2012b). The incidence and spectrum of change in Nav1.8 in I-SFN are not fully understood.
In this study, we profiled the SCN10A sequence from an elderly male patient with painful SFN and reduced IENFD plus abnormal QST, and no underlying etiology for SFN, and identified a novel missense mutation (I1706V) that lies within the predicted binding pocket of local anesthetics in S6 of domain IV. We investigated the functional effects of this mutation by studying its biophysical properties and its impact on the excitability of DRG neurons, and we propose a mechanism whereby the mutant channels produce hyperexcitability in nociceptive neurons. We also tested the possibility of selectively blocking the mutant channel by introducing membrane-impermeant lidocaine derivative QX-314.
Materials and Methods
Patient with SCN10A variant c.5116A>G, p.Ile170 6Val.
A 61-year-old male presented with burning and tingling sensations in both legs and feet, intolerance to wearing shoes and to sheets over his feet. Cooling tended to relieve the pain. The patient also complained of discoloration and episodic swelling of his feet, hyperhidrosis, dry eyes and mouth, orthostatic hypotension, palpitations, diarrhea, and micturition problems. Family history was unremarkable. Physical examination showed no clear abnormalities. There were no signs of large nerve fiber involvement on nerve conduction studies. Peripheral nerve conduction velocities (NCV) were within the normal range (sensory nerves: median NCV 58 m/s; sural NCV 42 m/s; motor nerves: median distal motor latency (DML) 3.10 ms; NCV distal 54 m/s; proximal 59 m/s; peroneal DML 4.65 ms; distal NCV 40 m/s, proximal NCV 44 m/s; tibial DML 7.00 ms; NCV 43 m/s). On temperature threshold testing, thresholds for warmth sensation at the feet (right/left) were abnormal (thresholds according to methods of levels 46.2/40.8°C, according to methods of limits 48.8/48.5°C). Cold detection thresholds were normal (thresholds according to methods of levels 30.9/30.6°C, according to methods of limits 28.0/27.3°C (Yarnitsky and Sprecher, 1994). Skin biopsy showed an IENFD of 0.4 per mm, which is abnormal compared with normative values (fifth percentile: 2.8 per mm). Based on clinical findings and abnormal IENFD and QST, the patient was diagnosed with SFN. Extensive laboratory investigations showed no underlying cause for SFN.
SCN10A sequence analysis and exon screening.
SCN10A numbering is based on the National Center for Biotechnology Information's RefSeq database, access numbers NG 031891.1 (genomic sequence) and CCDS33736.1 (coding sequence). Mutation screening was performed for all 27 exons that constitute the SCN10A ORF. Genomic DNA was amplified by PCR with oligonucleotide primers complementary to flanking intronic sequences. Primers specific for SCN10A exons were designed using Primer3 input software (version 4.0). PCR amplification was performed in a total volume of 25 μl containing 200 ng of genomic DNA, 1× DyNAzyme Buffer (Finnzymes; Celbio), 0.2 mm each GeneAmp dNTP (Applied Biosystems), 1 U of DyNAzyme DNA I DNA Polymerase (Finnzymes), and 0.5 μm of the specific primer pair. PCRs were performed on a GeneAmp PCR System 2700 (Applied Biosystems). Thermal cycling conditions were as follows: 94°C for 5 min, followed by 35 cycles of 30 s at 94°C, 30 s at 55°C, and 1 min at 72°C, followed by 7 min at 72°C. PCR products (10 μl) were confirmed by electrophoresis on a 2% (w/v) agarose gel, containing 0.0005% ethidium bromide. Bromophenol blue was used in the loading buffer (0.04%). PCR products were purified (2.5 μl of each) using ExoSAP-IT (GE Healthcare) and sequenced using Big Dye terminator, version 3.1, Cycle Sequencing Kit (Applied Biosystems) and ABI 3100 Genetic Analyzer (Applied Biosystems). The sequences were analyzed using SeqScape, version 2.1.1, software (Applied Biosystems). DNA from 110 Caucasian control subjects (220 chromosomes) from the same geographical region as the index patient was analyzed for c.5116A>G substitution by PCR and high-resolution melting-curve analysis.
Plasmids.
The wild-type (WT) construct (pcDNA5-SCN10A) that encodes human Nav1.8 protein was purchased from Genionics. The replacement of isoleucine by valine at position 1706 within Nav1.8 protein was introduced into constructs using QuikChange II XL site-directed mutagenesis (Stratagene) and referred to as I1706V hereinafter. Identity of inserts was confirmed by sequencing at the Howard Hughes Medical Institute/Keck Biotechnology Center at Yale University, New Haven, CT.
Isolation and transfection of primary sensory neurons for patch-clamp recording.
Animal studies followed a protocol approved by the Yale University and Veterans Administration West Haven Hospital Institutional Animal Care and Use Committees. For voltage-clamp recording, DRG neurons were isolated from homozygous Nav1.8-cre mice (4–8 weeks old) that lack endogenous Nav1.8 and transfected by electroporation as previously reported (Dib-Hajj et al., 2009; Han et al., 2012b). Briefly, DRGs were harvested from homozygous Nav1.8-cre mice, incubated at 37°C for 20 min in complete saline solution (CSS) containing the following (in mm): 137 NaCl, 5.3 KCl, 1 MgCl2, 25 sorbitol, 3 CaCl2, and 10 HEPES, adjusted to pH 7.2 with NaOH containing 0.5 U/ml Liberase TM (Roche) and 0.6 mm EDTA, followed by a 15 min incubation at 37°C in CSS containing 0.5 U/ml Liberase TL (Roche), 0.6 mm EDTA, and 30 U/ml papain (Worthington Biochemical). DRGs were then centrifuged and triturated in 0.5 ml of DRG media containing 1.5 mg/ml bovine serum albumin (BSA; low endotoxin) and 1.5 mg/ml trypsin inhibitor (Sigma). After trituration, 2 ml of DRG media was added to the cell suspension, which was filtered with 70 μm nylon mesh cell strainer (Becton Dickinson). The mesh was washed twice with 2 ml of DRG media. The cells were then transfected with WT or mutant Nav1.8 constructs using a Nucleofector IIS (Lonza) and Amaxa Basic Neuron SCN Nucleofector Kit (VSPI-1003). Briefly, the cell suspension was centrifuged (100 × g for 3 min), and the cell pellet was resuspended in 20 μl of Nucleofector solution, mixed with 2 μg of hNav1.8 WT or mutant construct plus 0.2 μg of EGFP, and transfected using Nucleofector IIS and protocol SCN-BNP 6. After electroporation, 100 μl of calcium-free DMEM (37°C) was added, and cells were incubated at 37°C for 5 min to recover. The cell mixture was then diluted with DRG media containing 1.5 mg/ml BSA (low endotoxin) and 1.5 mg/ml trypsin inhibitor (Sigma), seeded onto poly-d-lysine/laminin-coated coverslips (BD), and incubated at 37°C in a 95% air/5% CO2 (v/v) incubator for 45 min for neurons to attach to the coverslips. After 45 min, 1.4 ml of DRG media was added into each well, and the DRG neurons were maintained at 37°C in a 95% air/5% CO2 (v/v) incubator for 40–55 h before voltage-clamp recording.
For current-clamp recording, DRGs from 4- to 6-week-old Sprague Dawley rats were harvested and dissociated as described previously (Dib-Hajj et al., 2009; Faber et al., 2012b; Han et al., 2012b). Briefly, DRG neurons were dissociated with a 20 min incubation in 1.5 mg/ml Collagenase A (Roche) and 0.6 mm EDTA, followed by a 18 min incubation in 1.5 mg/ml Collagenase D (Roche), 0.6 mm EDTA, and 30 U/ml papain; a suspension of DRG neurons was prepared and transfected as described above for voltage-clamp recording. After transfection, cells were recovered in calcium-free DMEM, fed with DRG media supplemented with nerve growth factor (50 ng/ml) and glial cell line-derived neurotrophic factor (50 ng/ml), and maintained at 37°C in a 95% air/5% CO2 (v/v) incubator for 40–55 h before current-clamp recording.
Whole-cell electrophysiology.
Voltage-clamp recordings were conducted 40–55 h after transfection using an EPC-10 amplifier and the PatchMaster program (v 53; HEKA Elektronik) at room temperature (22 ± 1°C). Small DRG neurons (17–30 μm) with robust green fluorescence and no apparent neurites were selected for voltage-clamp recording. Fire-polished electrodes were fabricated from 1.6 mm outer diameter borosilicate glass micropipettes (World Precision Instruments) using a Sutter Instruments P-97 puller and had a resistance of 0.7–1.5 MΩ. Pipette potential was adjusted to zero before seal formation. Liquid junction potential was not corrected. To reduce voltage errors, 80–90% series resistance compensation was applied. Cells were excluded from analysis if the predicted voltage error exceeded 5 mV. Average maximal voltage error was 2.41 ± 0.31 mV (n = 18) for the group of cells expressing the WT Nav1.8 and 2.94 ± 0.33 mV (n = 18) for I1706V mutant channels. There is no significant difference in the voltage error between two groups (p = 0.258). Linear leak currents were subtracted out using the P/N method. The average leak current did not differ between WT and mutant (WT, −106 ± 26 pA, n = 18; I1706V, −108 ± 33 pA, n = 18, p = 0.946). Sodium current recordings were initiated after a 5 min equilibration period once whole-cell configuration was achieved. Current traces were sampled at 50 kHz, and filtered with a low-pass Bessel setting of 10 kHz. The pipette solution contained the following (in mm): 140 CsCl, 10 NaCl, 0.5 EGTA, 3 Mg-ATP, and 5 HEPES, pH 7.31, with CsOH (adjusted to 313 mOsmol/L with dextrose). The extracellular bath solution contained the following (in mm): 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, 5 CsCl, 20 tetraethylammonium chloride (TEA·Cl), 0.1 CdCl2, 5 4-aminopyridine, pH 7.30, with HCl (338 mOsmol/L). Tetrodotoxin (TTX; 0.5 μm) was included in the bath solution to isolate TTX-resistant (TTX-R) currents from endogenous TTX-sensitive (TTX-S) sodium currents. DRG neurons were held at −70 mV to inactivate Nav1.9 channels (Cummins et al., 1999). Current–voltage (I–V) relationships were measured using a series of 100 ms step depolarizations (−60 to +60 mV in 5 mV increments at 5 s intervals) from holding potential. Current density was calculated by normalizing maximal peak currents with cell capacitance. Persistent currents were measured as mean amplitudes of currents between 93 and 98 ms after the onset of depolarization, and are presented as a percentage of the maximal peak current. Peak inward currents obtained from activation protocols were converted to conductance values using the equation G = I/(Vm − ENa), where G is the conductance, I is the peak inward current, Vm is the membrane potential step used to elicit the response, and ENa is the reversal potential for sodium channel, which is determined for each cell using the x-axis intercept of a linear fit of the peak inward current responses to the last six voltage steps of the activation protocol. Conductance data were normalized by the maximum conductance value and fit with a Boltzmann equation of the form G = Gmax+ (Gmin − Gmax)/(1 + exp((Vm − V1/2)/k)), where V1/2 is the midpoint of activation, and k is a slope factor. Steady-state fast inactivation was assessed with a series of 500 ms prepulses (−80 to +20 mV in 5 mV increments); the remaining noninactivated channels were activated by a 40 ms step depolarization to 0 mV. Peak inward currents obtained from steady-state fast inactivation were normalized by the maximum current amplitude and fit with a Boltzmann equation of the form I/Imax = A + (1 − A)/(1 + exp[(Vm − V1/2)/k]), where Vm represents the inactivating prepulse membrane potential, V1/2 represents the midpoint of fast inactivation, and A represents the percentage of noninactivating channels. Decaying currents were fit with a single-exponential equation of the form I = A*exp(−t/τ) + Ic, where A is the amplitude of the fit, t is time, τ is the time constant of decay, and Ic is the asymptotic minimum to which the currents decay. Recovery of hNav1.8 channels from fast inactivation, repriming, was examined using a two-pulse protocol with interpulse intervals varying from 1 to 513 ms. Recovery rates were measured by normalizing peak current elicited by the test pulse (10 ms depolarization to 0 mV) to that of the prepulse (100 ms at 0 mV) at −50 and −70 mV, respectively. Recovery time constants were calculated using monoexponential fits of the recovery fraction over recovery period. Ramp currents were elicited with slow ramp depolarization from −70 to +50 mV over a 600 ms period at a rate of 0.2 mV/ms. The amplitude of ramp current was presented as a percentage of the maximal peak current.
For studying the effects of QX-314 (Tocris Bioscience), a membrane-impermeable quaternary derivative of lidocaine, the pipette solution contained the following (in mm): 140 CsF, 10 NaCl, 1 EGTA, and 10 HEPES, pH 7.30, with CsOH (adjusted to 315 mOsmol/L with dextrose). The extracellular bath solution contained the following (in mm): 70 NaCl, 70 choline chloride, 3 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, 5 CsCl, 20 TEA·Cl, 0.1 CdCl2, and 5 4-aminopyridine, pH 7.39, with an osmolality of 338 mOsmol/L. TTX (0.5 μm) was added in the bath solution to block endogenous voltage-gated sodium currents. DRG neurons were held at −80 mV, followed by a series of 100 ms step depolarizations from −60 to +50 mV in 5 mV increments at 5 s intervals. The activation protocol was repeated for the control, vehicle treatment (10 min), and QX-314 treatment (10 min) on each neuron. Additionally, small DRG neurons were stimulated with 50 ms depolarization pulses to 0 mV from a holding potential of −80 mV at 10 s intervals for 5 min after wash-in of vehicle (i.e., saline), and subsequent QX-314 addition.
For current-clamp recordings, electrodes had a resistance of 0.7–1.5 MΩ when filled with the pipette solution, which contained the following (in mm): 140 KCl, 0.5 EGTA, 5 HEPES, and 3 Mg-ATP, pH 7.30, with KOH (adjusted to 309 mOsm with dextrose). The extracellular solution contained the following (in mm): 140 NaCl, 3 KCl, 2 MgCl2, 2 CaCl2, 10 HEPES, pH 7.30, with NaOH (adjusted to 313 mOsm with dextrose). Whole-cell configuration was obtained in voltage-clamp mode before proceeding to the current-clamp recording mode. Cells with stable (<10% variation) resting membrane potentials more negative than −35 mV and overshooting action potentials (>85 mV resting membrane potential to peak) were used for additional data collection. Input resistance was determined by the slope of a linear fit to hyperpolarizing responses to current steps from −5 to −40 pA in 5 pA increments. Threshold was determined by the first action potential elicited by a series of depolarizing current injections (200 ms) that increased in 5 pA increments. Action potential frequency was determined by quantifying the number of action potentials elicited in response to depolarizing current injections (500 ms). The expression level of Nav1.8 channels was examined by holding neurons at −50 mV (Cummins and Waxman, 1997). Cells that expressed small Nav1.8 currents (<1 nA) and unable to generate all-or-none action potentials in response to 200 ms current stimulus were excluded from analysis (Cheng et al., 2011).
Data were analyzed using Fitmaster (HEKA Elektronik) and Origin (Microcal Software) software. Unless otherwise noted, statistical significance was determined (p < 0.05) using an independent t test. Two-proportion z-test was used for comparing populations of spontaneously or repetitive firing neurons. Mann–Whitney test was used in comparison of firing frequencies between neurons expressing Nav1.8 WT and I1706V mutant in response to stimuli ranging from 25 to 500 pA. Results are presented as mean ± SEM and error bars in the figures indicate SEMs.
Results
Identification of the I1706V mutation
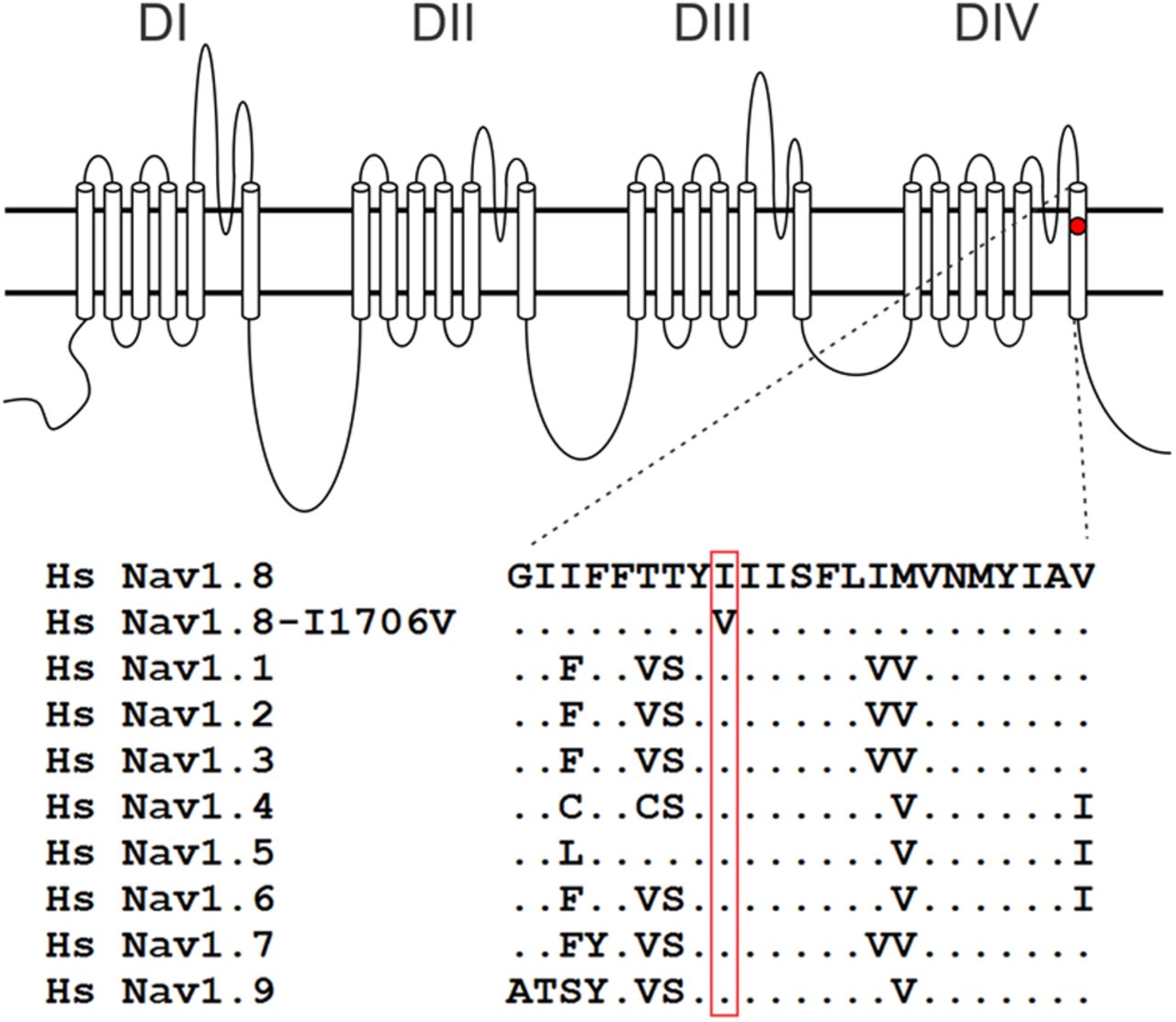
DNA analysis showed no mutation in SCN9A, encoding Nav1.7, in this patient diagnosed with I-SFN. However, a probable pathogenic variant was found in SCN10A (c.5116A>G). The c.5116A>G variant results in an amino acid substitution from isoleucine to valine at position 1706 of Nav1.8 (p.Ile1706Val). The isoleucine at this position in Nav1.8 is highly conserved in the voltage-gated sodium channel family (Fig. 1). The variant is located in the DIV/S6 domain. Prediction programs AGVGD, Polyphen-2, and Sift characterize the substitution as “c25, likely to interfere with function,” “possibly damaging,” and “nontolerated.” Splice prediction programs (SpliceSiteFinder, MaxEntScan, NNSplice, GeneSplicer, Human Splicing Finder, Fruitfly, and Softberry) predict that this variant has no effect on splicing. The variant was not found in our previous cohort of 214 SFN patients, and was not present in a control population (n = 110 individuals) from the same geographical region. The variant is not reported in the literature (dbSNP database, EVS database, and 1000 Genomes Project).

Schematic of a voltage-gated sodium channel α-subunit showing the location of the I1706V mutation and the aligned sequences for DIV/S6. I1706V is conserved in all known human voltage-gated sodium channels. Hs, homo sapiens.
Voltage-clamp characterization of DRG neurons expressing WT and I1706V mutant Nav1.8
To investigate the effect of the I1706V mutation on channel activation, Nav1.8-null mouse DRG neurons were transfected with either Nav1.8 WT or I1706V mutant construct. In the presence of 500 nm TTX in the bath, neurons were held at −70 mV to inactivate endogenous Nav1.9 currents (Cummins et al., 1999). Voltage-dependent inward currents with slow inactivation were recorded by applying 100 ms test pulses to potentials between −60 and +60 mV in 5 mV increments. Representative whole-cell currents are presented in Figure 2A and B. The average peak inward current amplitude was not significantly different between WT Nav1.8 and I1706V (WT, 4.68 ± 0.78 nA, n = 25; I1706V, 4.58 ± 0.85 nA, n = 25; p = 0.931). When normalized for capacitance, similarly, the average peak current density of I1706V (274 ± 42 pA/pF, n = 25) did not differ from WT (250 ± 32 pA/pF, n = 25; p = 0.648).

I1706V shifts activation toward hyperpolarization and decreases persistent currents. A, Inward currents recorded from a small-diameter DRG neuron expressing WT Nav1.8 sodium channels. Cells were held at −70 mV and stepped to membrane potentials from −60 to +60 mV for 100 ms in 5 mV increments. B, Inward currents recorded from a small-diameter DRG neuron expressing I1706V mutant. C, Normalized peak current–membrane voltage relationship for activation of WT (n = 18) and I1706V (n = 18) channels. D, Comparison of the voltage dependence of activation and steady-state fast inactivation for WT and I1706V channels. Activation data were obtained by normalizing the channel conductance converted from peak inward current. Steady-state fast inactivation was assessed by stepping neurons from a holding potential of −70 mV to a prepulse membrane potential varying from −80 to +20 mV in 5 mV increments for 500 ms, followed by a 40 ms test pulse to 0 mV. The peak current in response to the test pulse was normalized by the maximum response amplitude and plotted as a function of prepulse membrane potential (WT, n = 14; I1706V, n = 13). E, Voltage dependence of persistent currents in WT (n = 18) and I1706V (n = 18) channels. The amplitude of persistent current was measured as the mean value of inward currents between 93 and 98 ms after onset of depolarization, normalized to the peak inward current during the activation protocol, and plotted as a function of membrane potential. F, Voltage dependence of fast inactivation time constants for WT (n = 18) and I1706V (n = 18) channels. Time constants were obtained from single exponential fits of current decay after peak responses to activation voltage steps from −20 to +50 mV.
As shown in Figure 2C, the normalized current–voltage curve of I1706V was shifted by ∼5 mV more hyperpolarized than WT. The mean peak current was elicited at +15 mV for WT and +10 mV for I1706V. Considering the voltage potential at which the inward current is 1% of the peak as the threshold of activation, the threshold potential of WT is −35 mV with a normalized current of 1.19 ± 0.35% (n = 18) of the peak, while I1706V starts to activate at −40 mV with a normalized current of 1.37 ± 0.44% (n = 18). The reversal potential, however, is unaffected by the I1706V mutation (WT, 65.3 ± 1.1 mV, n = 18; I1706V, 65.5 ± 1.3 mV, n = 18; p = 0.878; Table 1), indicating a preservation of ion selectivity despite the position of I1706 in close proximity to the selectivity filter (Zarrabi et al., 2010).
Biophysical properties of WT and mutant Nav1.8 channels in adult mouse DRG neurons
The voltage dependence of activation and steady-state fast inactivation for WT and I1706V are illustrated in Figure 2D. The Boltzmann fits for both activation and fast inactivation were derived for each cell individually. As compared with WT, the midpoint of activation (V1/2, act) was shifted 6.4 mV more negative for I1706V (WT, −1.11 ± 1.6 mV, n = 18; I1706V, −7.47 ± 1.4 mV, n = 18; p = 0.005; Table 1), confirming that I1706V shifts activation in a hyperpolarizing direction. Additionally, I1706V steepened the activation curve with a slope factor of 7.36 ± 0.33, compared with WT (8.77 ± 0.45, p = 0.015; Table 1). The predicted window current in the subthreshold range for I1706V increased due to the hyperpolarizing shift of activation (Fig. 2D). Steady-state fast inactivation was assessed at 0 mV with a 500 ms prepulse ranging from −80 to +20 mV in 5 mV increments at a holding potential of −70 mV. In contrast to the significant difference in activation, fast inactivation exhibited no changes in either midpoint voltage (V1/2, fast) (WT, −29.9 ± 1.2 mV, n = 14; I1706V, −29.9 ± 1.9 mV, n = 13; p = 0.999) or slope factor (WT, 6.33 ± 0.30; I1706V, 6.09 ± 0.40; p = 0.628; Table 1).
We have previously shown that a fraction of Nav1.8 channels is resistant to fast inactivation in the suprathreshold range (i.e., more depolarized than −10 mV; Faber et al., 2012b). Interestingly, the noninactivating component decreases by 60% in I1706V (WT, 6.58 ± 0.43%, n = 14; I1706V, 2.61 ± 0.40, n = 13; p < 0.001; Table 1). The presence of a subpopulation of noninactivating channels can result in persistent sodium currents. Consistently, the average peak persistent currents, described as the percentage of the peak inward current, was reduced by 30% in I1706V (7.68 ± 0.67%, n = 18) compared with WT (11.0 ± 0.77%, n = 18; p = 0.003; Fig. 2E, Table 1). At +20 mV, for example, persistent currents were 7.51 ± 0.63% (n = 18) for WT and 2.48 ± 0.75% (n = 18) for I1706V channels (Fig. 2E), both of which reflect percentages of noninactivating channels at the same voltage (Fig. 2D). Similar to the hyperpolarizing shift of activation in the mutant, the voltage of peak persistent current was shifted from 0 mV for WT to −10 mV for I1706V. Although the decline in peak persistent current is statistically significant, the persistent current from −60 to −20 mV in I1706V does not differ from WT. For example, the fractional persistent current at −20 mV is 5.29 ± 0.59% for WT and 5.17 ± 0.79% for I1706V (p > 0.05). Since WT and I1706V channels exhibit persistent currents of similar magnitude in membrane potentials more negative than −20 mV or the subthreshold range, the anti-excitatory effect for neuron activity resulting from reduction in persistent current is minimal.
For currents evoked at potentials between −20 and +50 mV where the persistent currents were significantly smaller in I1706V than WT, we measured the kinetics of fast inactivation. Despite the significant reduction of persistent currents, monoexponential fits of the current decay phase reveal that fast inactivation takes place at the same rate for WT and I1706V at all potentials tested (Fig. 2F).
We further examined the response of WT and I1706V channels to slow depolarizations, using ramp stimuli at a rate of 0.2 mV/ms from the holding potential of −70 to +50 mV over 600 ms. The ramp current elicited by slow depolarization was normalized to the peak inward current recorded during the activation for each cell. Representative ramp current traces for WT and I1706V are shown in Figure 3A, in which the ramp current of WT (black line) peaks at −13.7 mV and reaches its amplitude 26.7% of the peak inward current, while the ramp current of I1706V (red line) peaks at −17.9 mV and is 23.8% of its peak current. The average ramp currents were 23.7 ± 1.5% (n = 14) for WT and 18.7 ± 1.9% (n = 14) for I1706V (p = 0.052) (Fig. 3B; Table 1). There was a trend toward reduction of peak ramp current for mutant channel, but it did not reach statistical significance. In addition, the peak ramp current occurred at a more hyperpolarized potential for I1706V (−13.8 ± 1.4 mV, n = 14) as compared with WT (−8.48 ± 1.8 mV, n = 14, p = 0.030) (Fig. 3C; Table 1).

I1706V leads to hyperpolarizing shift of the voltage dependence of slow ramp currents. A, To elicit ramp currents, small-diameter DRG neurons held at −70 mV were stimulated with a depolarizing voltage ramp that increased to +50 mV over 600 ms at a rate 0.2 mV/ms. The current recorded during the ramp was expressed as a percentage of the peak inward current obtained during the initial activation protocol and plotted as a function of membrane potential. Data from DRG neurons expressing WT and I1706V channels are represented by a black line and a red line, respectively. B, Fractional ramp current at maximum for I1706V (n = 14) was smaller than WT (n = 14) channels without reaching statistical significance (p = 0.052). C, Voltage at which the peak response of ramp occurs for I1706V (n = 14) was 5.3 mV more hyperpolarized than WT (n = 14) channels (p = 0.030). D, Recovery from inactivation for WT (n = 11) and I1706V (n = 10) channels at −50 mV. The average time constant is not significantly different between WT and mutant (inset, p = 0.849). E, Recovery from inactivation for WT (n = 7) and I1706V (n = 8) channels at −70 mV. The average time constant has a slight trend of decrease in I1706V but does not reach statistical significance (inset, p = 0.093). *p < 0.05.
Finally, we investigated the recovery of sodium channels from fast inactivation using a two-pulse protocol at 0 mV involving a varied interpulse interval at −50 and −70 mV, respectively. The amplitude of inward current elicited during the second pulse was normalized to that in the first pulse. The ratio of current amplitude was plotted against the interpulse interval and fit with single exponentials to measure the rate of recovery from inactivation. The average repriming kinetics at −50 and −70 mV are illustrated in Figure 3, D and E, respectively. I1706V did not have a significant impact on the recovery rate at either −50 mV (WT, 11.3 ± 1.9 ms, n = 11; I1706V, 10.6 ± 2.6 ms, n = 10; p = 0.849) or −70 mV (WT, 3.97 ± 0.91 ms, n = 7; I1706V, 2.34 ± 0.28 ms, n = 8; p = 0.093).
Current-clamp characterization of DRG neurons expressing WT and I1706V channels
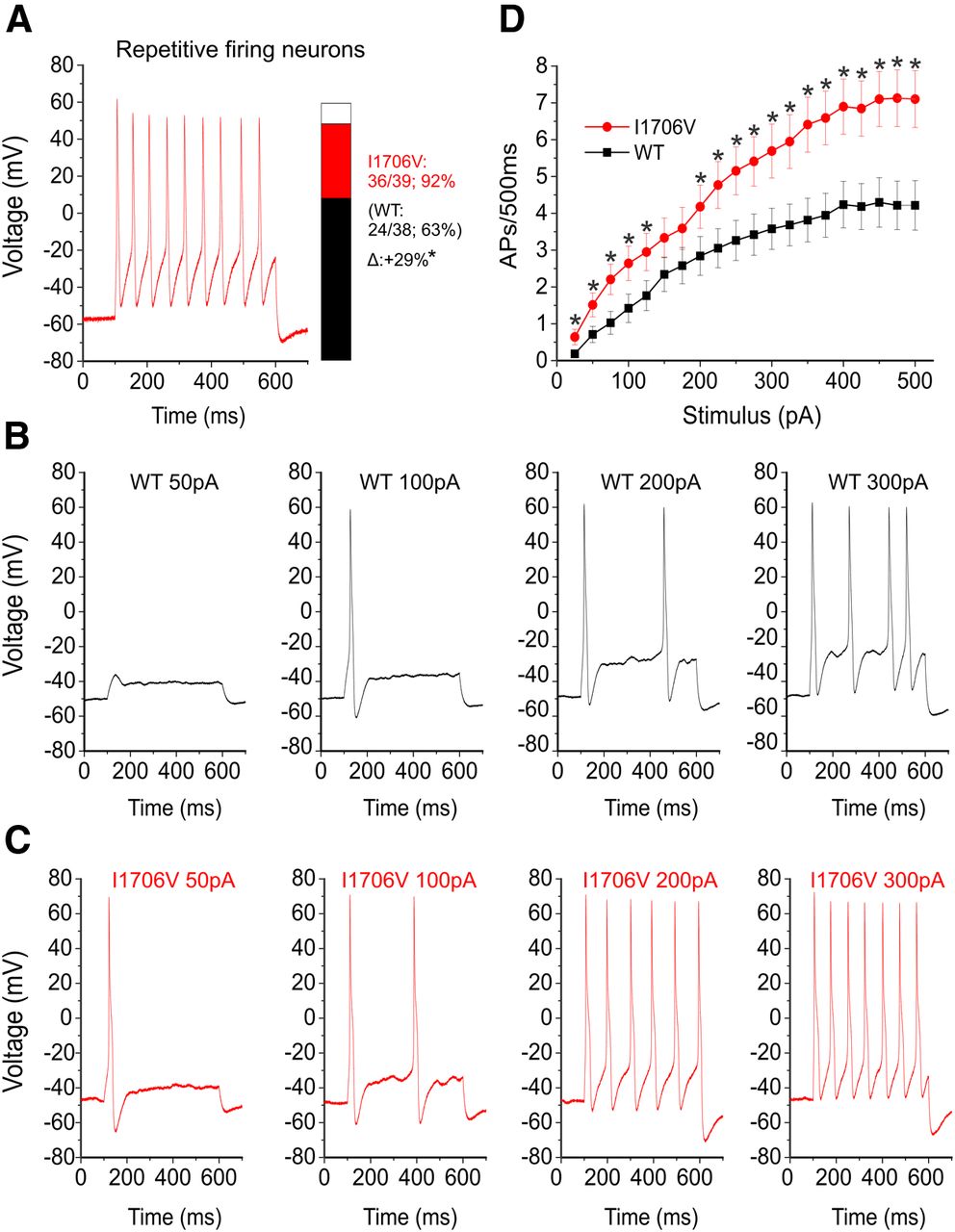
To assess the effects of I1706V on DRG neuronal excitability, we performed current-clamp studies in two groups of small DRG neurons electroporated with WT or I1706V channels. The population of spontaneously firing neurons for I1706V (26%) is not significantly different from WT (21%, p = 0.555) (Fig. 4A, Table 2). Because it is difficult to measure the responses to injected currents in neurons producing spontaneous action potentials, we excluded these neurons from the analysis of resting membrane potential, input resistance, action potential amplitude, half-width, and voltage threshold as well as current threshold for both WT (10 of 48 neurons) and I1706V (14 of 53 neurons). There were no significant differences in input resistance (p = 0.597), action potential amplitude (p = 0.277), or half-width (p = 0.399) between WT (n = 38) and I1706V (n = 39) (Table 2). The resting membrane potential of neurons expressing I1706V, however, was depolarized by 5.3 mV (WT, −51.1 ± 1.4 mV, n = 38; I1706V, −45.8 ± 1.3 mV, n = 39; p = 0.006; Table 2, Fig. 4B). The average voltage at which takeoff occurs for an all-or-none action potential (i.e., voltage threshold) was hyperpolarized from −25.5 ± 1.2 mV (n = 38) for WT to −31.0 ± 0.91 mV (n = 39) for I1706V (p < 0.001) (Table 2, Fig. 4C). Current threshold at which an all-or-none action potential is elicited was assessed by injecting a series of currents in increments of 5 pA over 200 ms to neurons expressing either WT or I1706V. There is a marked reduction by 53% of current threshold for single action potentials in I1706V-transfected neurons (WT, 139 ± 16 pA, n = 38; I1706V, 66.0 ± 9.9 pA, n = 39; p < 0.001) (Table 2, Fig. 4D). Representative responses of WT and I1706V are illustrated in Figure 4, E and F, respectively. While an injection of 150 pA was still considered subthreshold for WT (Fig. 4E), an input as low as 35 pA was able to produce an overshooting action potential for this cell expressing I1706V (Fig. 4F).

I1706V mutation reduces current threshold, negatively shifts voltage threshold of action potential, and results in depolarization of resting membrane potential. A, Representative recording of spontaneous firing in a DRG neuron expressing I1706V mutant channels. Trace was recorded for 30 s without current injection. Inset, Bar graph showing the proportion of spontaneous firing cells for DRG neurons expressing I1706V (red) and WT (black); numbers to the right of the bar graph show values for WT (lower value in parentheses) and I1706V (upper value). B, Resting membrane potential (RMP) of neurons expressing WT (n = 38) was depolarized by 5.3 mV as compared with that of I1706V (n = 39) channels (p = 0.006). C, Voltage threshold at which the all-or-none action potential takes off was depolarized by 5.5 mV in I1706V as compared with WT channels (p < 0.001). D, Current threshold was significantly reduced after expressing I1706V channels (p < 0.001). E, Responses of a current-clamped DRG neuron transfected with Nav1.8-WT DNA to a series of subthreshold and suprathreshold depolarizing current steps. Starting at a subthreshold stimulus intensity, the current amplitude was increased in 5 pA increments to an intensity well beyond threshold. The RMP for this cell was −55 mV, and the current threshold was 155 pA. F, The same threshold protocol was applied to a DRG neuron transfected with the I1706V mutant DNA. The RMP for this cell was −47 mV, and the current threshold was 35 pA. Arrows with numbers indicate the current step amplitude used to elicit the labeled response. *p < 0.05.
Action potential characterization for WT and mutant Nav1.8 channels in adult rat DRG neurons
We further examined the numbers of action potentials evoked by graded (25–500 pA) prolonged current injections (500 ms) in both groups of neurons. There was a significant difference with regard to repetitive firing between groups. Compared with 63% of neurons expressing Nav1.8-WT that can fire repetitively, 92% of neurons expressing I1706V were capable of generating multiple action potentials in response to 500 ms depolarizing stimuli (p = 0.002; Table 2, Fig. 5A). Representative action potentials triggered by increasing current injections of 50, 100, 200, and 300 pA in neurons transfected with either WT or I1706V channels were illustrated in Figure 5B and C. Cells expressing I1706V (Fig. 5C) tended to fire with a higher frequency than WT (Fig. 5B) in response to similar stimuli. The average numbers of action potentials in WT and I1706V elicited by graded current injections in 25 pA steps are plotted in Figure 5D. Compared with WT, neurons expressing I1706V produced significantly higher firing frequencies at almost every stimulus across the broad range studied.

I1706V expression increases excitability in DRG neurons. A, Representative recordings of repetitively firing action potentials evoked by 500 pA of injected current for 500 ms in a DRG expressing I1706V channels. Inset, Bar graph showing the proportion of neurons expressing WT (black) and I1706V (red) channels that fire multiple action potentials in response to the maximally injected current (500 pA). Numbers to the right of the bar graph show values for WT (lower value in parentheses) and I1706V (upper value). *p < 0.05. B, Responses of a neuron expressing WT channels to 500 ms depolarizing current steps that are 50, 100, 200, and 300 pA. C, The same stimulus protocol for a neuron expressing I1706V mutant channels. D, Summary of firing frequency data. The total number of action potentials elicited by the indicated depolarizing current steps from 25 to 500 pA in 25 pA increments was compared between two groups of neurons expressing WT (n = 38) or I1706V (n = 39) channels. *p < 0.05.
Effects of externally applied QX-314 for WT and I1706V channels
Previous studies demonstrated that the isoleucine residue homologous to position 1706 in Nav1.8 channel has its side chain oriented toward the pore to guard the extracellular access of local anesthetics (Ragsdale et al., 1994). QX-314, a permanently charged derivative of lidocaine that is membrane-impermeant, was found to block variants of voltage-gated sodium channels with the analogous isoleucine replaced by smaller amino acid residues like alanine (Ragsdale et al., 1994; Wang et al., 1998; Sunami et al., 2001). In I1706V mutant channel, isoleucine, which has a spatial volume of 167Å3, is substituted by a smaller residue valine (140Å3) (Zamyatnin, 1972; Pommié et al., 2004). We therefore tested the effect of QX-314 on I1706V by voltage-clamp whole-cell recordings. Each of the small-diameter neurons (<30 μm; WT, n = 6; I1706V, n = 9) was recorded in three conditions in order: control and wash-in of vehicle for 10 min, followed by adding external QX-314 (1 mm) for an additional 10 min. Voltage-gated Nav1.8 TTX-R inward currents were elicited by a series of 100 ms test pulses from −60 to +50 mV from a holding potential of −80 mV in 5 mV increments. After whole-cell currents reached steady state, vehicle (i.e., saline) was applied from the bath for 10 min, followed by an identical series of activation voltage commands. Subsequently, 1 mm QX-314 dissolved in vehicle (saline) was added to the bath for 10 min and sodium currents were then recorded. Representative current traces from neurons expressing WT channels in the presence of vehicle or QX-314 are shown in Figure 6A. External QX-314 lead to a 2% reduction of peak sodium currents in this cell (Vehicle, 6.68 nA; QX-314, 6.54 nA). In regard to WT, the average peak currents are 4.54 ± 1.0 nA for the control, 4.68 ± 1.1 nA post vehicle application, and 4.85 ± 1.0 nA post-QX-314 treatment (n = 6, Fig. 6C). The vehicle-treated peak currents are not significantly different from the control (p = 0.377, paired t test) or QX-314-treated currents (p = 0.130, paired t test; Fig. 6C). In Figure 6B, the peak current of a neuron expressing I1706V mutant is decreased from 9.88 to 8.18 nA after extracellular QX-314 was applied for 10 min, resulting in a 17% blockade. The average peak currents are 4.51 ± 0.86 nA for I1706V control, 4.90 ± 0.94 nA for vehicle treated, and 3.55 ± 0.90 nA for QX-314 treated (n = 9; Fig. 6D). There is no significant difference in peak current amplitude between the control and vehicle groups for I1706V (p = 0.193, paired t test). In contrast, QX-314 significantly decreased the peak current by ∼30% (p = 0.001, paired t test; Fig. 6D). The normalized current–voltage curves for WT and I1706V illustrated in Figure 6,E and F, demonstrated that external QX-314 blocked I1706V currents without any effect on WT currents. On average, externally applied quaternary blocker QX-314 barely altered the normalized peak current of WT (vehicle, 1.05 ± 0.042; QX-314, 1.06 ± 0.028; n = 6, p = 0.871), while it significantly inhibited I1706V by 29% (vehicle, 1.05 ± 0.029; QX-314, 0.711 ± 0.065; n = 9, p < 0.001) (Fig. 6G).

Extracellular QX-314 specifically blocks I1706V without affecting WT channel. A, Representative inward currents recorded from a small-diameter DRG neuron expressing WT channels after 10 min wash-in of vehicle (saline), followed by another 10 min treatment of 1 mm QX-314 in the bath. Cells were held at −80 mV and stepped to membrane potentials from −60 to +50 mV for 100 ms in 5 mV increments. B, The same activation protocol was applied to a DRG neuron expressing I1706V channels in the presence of vehicle and QX-314. C, The average peak inward current of neurons expressing WT channels (n = 6) was not altered by external QX-314 (p = 0.130, paired t test). Each line connects the data points of pretreament and post-treatment QX-314 at the same neuron. D, Extracellular QX-314 significantly reduced peak current amplitude of neurons expressing I1706V channels (p = 0.001, paired t test). E, Normalized peak current–membrane voltage (I–V) relationship for activation of WT channels (n = 6) before treatment, after 10 min wash-in of vehicle, and after 10 min wash-in of 1 mm QX-314. F, Normalized I–V curved of I1706V channels (n = 9) before treatment, after 10 min wash-in of vehicle, and after 10 min wash-in of 1 mm QX-314. External QX-314 blocked I1706V currents without significant effect on WT currents. G, Normalized peak inward currents of neurons expressing WT (n = 6) or I1706V (n = 9) channels in the presence of vehicle or 1 mm QX-314. Externally applied quaternary blocker QX-314 barely altered the normalized peak current of WT (p = 0.871), but it significantly decreased the normalized current of I1706V by ∼30% (p < 0.001). There was no significant difference (p = 0.991) in the normalized peak current treated by vehicle between WT and I1706V channels. H, Action of externally applied QX-314 on WT (vehicle, n = 8; QX-314, n = 6) and I1706V (vehicle, n = 11; QX-314, n = 14) channels, respectively, for the time indicated. Currents were evoked by depolarizing pulses to 0 mV for 50 ms at 10 s intervals for 5 min. The data in each experiment were normalized with respect to the control currents. I, Representative current traces of a DRG neuron expressing WT channels treated with vehicle, followed by 1 mm QX-314 application in the bath. WT current slightly run up over time. J, Representative current traces of a DRG neuron expressing I1706V channels washed-in with vehicle for 5 min, followed by external QX-314 treatment for an additional 5 min. I1706V current (green line) was markedly reduced compared with vehicle treatment (red) or control (black).
We further tested effect of QX-314 by applying stimuli to small DRG neurons via 50 ms depolarization pulses to 0 mV from a holding potential of −80 mV at 10 s intervals for 5 min. Consistently, QX-314 barely affected DRG neurons expressing WT but rapidly blocked DRG neurons expressing I1706V. At 5 min, the normalized current of I1706V at 0 mV in the presence of QX-314 was 0.762 ± 0.044 (n = 14), indicating a 24% blockade (Fig. 6H). Representative current traces predrug and postdrug treatment are presented for WT (Fig. 6I) and I1706V neurons (Fig. 6J), respectively. WT current shows slight run-up from 7.01 to 7.20 nA after QX-314 was added in the bath for 5 min (Fig. 6I). In contrast, I1706V current was markedly reduced by 21% from 7.45 to 5.92 nA (Fig. 6J).
Discussion
In this study we report the functional profile of I-SFN-related mutation in Nav1.8, I1706V, identified in a patient suffering from painful neuropathy with burning pain in the legs and feet. Abnormal IENFD and QST confirmed the diagnosis of SFN, and normal physical examination of nerve conduction studies excluded large fiber involvement. We have previously described gain-of-function variants of Nav1.7 in 30% of patients with painful I-SFN (Faber et al., 2012a). In voltage-clamp experiments, many of these mutants impair fast or slow inactivation of Nav1.7 sodium channel (Estacion et al., 2011; Faber et al., 2012a; Han et al., 2012b; Hoeijmakers et al., 2012c), and some increase resurgent current (Faber et al., 2012a; Han et al., 2012a). DRG neuron hyperexcitability produced by these mutations appears to account for pain in these patients (Faber et al., 2012a); there is also evidence suggesting that sustained sodium influx through the mutant channels may trigger reverse Na/Ca exchange that leads to axonal injury (Persson et al., 2013). Among I-SFN patients with no mutations in Nav1.7, we have searched for variants in Nav1.8, which is preferentially expressed in small-diameter DRG neurons. We previously reported the presence of the L554P variant of Nav1.8, which accelerates the recovery from fast inactivation near resting membrane potential and enhances the response to slow ramp depolarization, and the A1304T variant of Nav1.8, which hyperpolarizes the voltage dependence of activation (Faber et al., 2012b). These pro-excitatory gating changes confer hyperexcitability on DRG neurons, suggesting a basis for the pain reported by these patients. It is interesting, in this regard, that Bierhaus et al. (2012) have presented evidence suggesting a role of Nav1.8 sensitization by methylglyoxal in nociceptor firing and pain in diabetic neuropathy.
In contrast to accelerated recovery from inactivation as seen with the L554P Nav1.8 mutation, but similar to the A1304T Nav1.8 variant, biophysical analysis of I1706V revealed an enhancement of activation, which is shifted so that the activation midpoint is more hyperpolarized for I1706Vmutant channels; in contrast to A1304T, the I1706V mutation also hyperpolarizes the voltage threshold for channel opening. In an early study of the adult rat skeletal muscle isoform Nav1.4, a mutation at the corresponding position (I1575E) shifted activation midpoint in a hyperpolarization direction by 6.8 mV (Sunami et al., 2001). It is worth mentioning that valine and glutamic acid have almost identical spatial volume (V, 140.0 Å3; E, 138.4 Å3) despite obvious different polarities (Zamyatnin, 1972; Kyte and Doolittle, 1982). It is not clear whether the unique mutation site of I1706V accounts for the large hyperpolarizing shift (6.4 mV) of activation. S6 helices form the pore of sodium channels (Catterall, 2000) and mutations in these segments would be predicted to alter activation. It is notable that ΔL955, a Nav1.7 mutation within S6 of domain II, displays a large shift in activation (−40 mV;Cheng et al., 2011; Yang et al., 2013). Consistent with the shift of activation, the response to slow ramp depolarizations in I1706V showed a similar hyperpolarizing shift (5.3 mV). These biophysical changes are paralleled by a 5.5 mV hyperpolarizing shift in the voltage for action potential takeoff in DRG neurons expressing I1706V.
In current-clamp recordings, the I1706V mutation produces hyperexcitability in small DRG neurons, manifested by >50% reduction in the current threshold for single action potential, a 29% increase in the population of repetitive firing neurons, and a marked increase in firing frequency in response to graded depolarizing stimuli. These pro-excitatory changes presumably are attributable to hyperpolarizing shifts of activation and peak ramp response. However, not all the biophysical changes caused by I1706V mutation are predicted to contribute to hyperexcitability. The fraction of I1706V channels resistant to fast inactivation in suprathreshold Nav1.8 activation range (from −10 to +20 mV) was decreased, which is correlated with a reduction in normalized persistent current in the comparable voltage range (−20 to +50 mV). The peak response to slow ramp depolarization that occurred within this range also shows a reduction trend. An early study on the equivalent mutant in Nav1.2 channel (I1760A) expressed in tsA-201 cells showed a similar trend of reduction in persistent current compared with WT channels (Linford et al., 1998). However, in the more physiological range from −60 to −20 mV, the normalized persistent current in I1706V does not differ from WT. Thus changes in activation and ramp current appear to be dominant over reduced persistent current in terms of contributing to DRG neuron hyperexcitability.
We did not find a difference in average peak current or current density between WT and I1706V channels. Nav1.8 contributes 60–80% of the inward membrane current flowing during the rising phase of the action potential in DRG neurons (Renganathan et al., 2001; Blair and Bean, 2002). Thus it is not surprising that the amplitude of the action potentials in I1706V did not differ from that of WT. In contrast, previous studies on ND7 cells found that another mutation (I1706A) at the same site within human Nav1.8 channels showed a 50% reduction in current density, without an effect on activation (Browne et al., 2009). It is not clear whether the difference between I1706V and I1706A mutant channels is related to cell background; for example, the current density of Nav1.8 WT within mouse DRG neurons in the present study (250 ± 32 pA/pF) was three times as large as that in ND7 cells (79 ± 7 pA/pF) (Browne et al., 2009). Additionally, the nature of the substituted residue (valine compared with alanine) may have contributed to the reduction in current density in I1706A.
It has long been established that the isoleucine residue homologous to position 1706 in Nav1.8 channel plays an important role in the interaction with local anesthetics (Catterall, 2000). This particular isoleucine is highly conserved in voltage-gated sodium channels across species and situated within the upper third region of DIV-S6. It has been reported that I1706 is located in a narrow region of the pore with its side chain oriented toward the pore, guarding the extracellular access of local anesthetics (Ragsdale et al., 1994). This suggestion is supported by the observation that QX-314, a charged quaternary amine derivative of lidocaine that is impermeable to cell membrane, is able to block Nav1.2 and Nav1.4 channels from the extracellular side when the corresponding isoleucine is substituted by smaller amino acid residues, such as alanine, cysteine, or glutamic acid (Ragsdale et al., 1994; Wang et al., 1998; Sunami et al., 2001). Increased accessibility of QX-314 in these mutant channels has been attributed to relief of steric hindrance or an alteration of microhydrophobicity environment (Wang et al., 1998). Additionally, the DIV-S6 segment is believed to be less flexible as compared with S6 segments in other domains due to its lack of a glycine hinge (Zhao et al., 2004). Binshtok et al. (2007) recently reported that externally administrated QX-314 can pass through TRPV1 channel when coapplied with capsaicin. We observed that external QX-314 significantly decreased by 30% the peak current of I1706V channels, with little effect on WT Nav1.8 channels. Consistent with this finding, previous recordings of Nav1.2 and Nav1.4 show 20–65% reduction in currents of mutants harboring an alanine replacement of the corresponding isoleucine when treated with extracellular QX-314 (Ragsdale et al., 1994; Wang et al., 1998; Sunami et al., 2001).
Nav1.8 is expressed within nociceptive and non-nociceptive DRG neurons (Shields et al., 2012), and hyperexcitability of the former appear to explain the pain reported by our patients, however, Nav1.8 expression has not been detected within autonomic ganglion neurons (Akopian et al., 1996; Rush et al., 2006). The complaints of the patient described here, in addition to pain, included dry eyes and mouth, hyperhidrosis, diarrhea, and constipation, and occasional episodes of orthostatic dizziness and palpitations. These complaints cannot be directly attributed to Nav1.8 mutation-mediated dysfunction of autonomic neurons, which do not express Nav1.8. However, Nav1.8 is present within trigeminal neurons (Thun et al., 2009), and it has been proposed that hyperactivity of corneal nociceptors can trigger a sensation of ocular dryness (Belmonte and Gallar, 2011; Rosenthal and Borsook, 2012). Nav1.8 is also present within nodose ganglion neurons (Schild and Kunze, 1997; Matsumoto et al., 2007) and in nerve fibers within the myocardium (Facer et al., 2011) and intracardiac neurons (Agarwal et al., 2004; Verkerk et al., 2012). Sympathetic fibers run with nociceptors in the same Remak bundle, and it is possible that increased activity in nociceptive axons may have affected nearby sympathetic axons by ephaptic transmission (Katz, 1942; Rasminsky, 1980) or some other form of cross talk involving, e.g., increased extracellular potassium concentrations (Delio et al., 1987; Lisney and Devor, 1987). Further studies will be needed to determine whether complaints of autonomic symptoms in patients with Nav1.8 mutations and SFN are due to direct effects of expression of mutant channels in tissues such as myocardium, to indirect, reflex effects of mutant channels within sensory neurons such as those of the nodose ganglion, or to cross talk.
Footnotes
This work was supported in part by grants from the Rehabilitation Research Service and Medical Research Service, Department of Veterans Affairs, and the Erythermalgia Association. The Center for Neuroscience and Regeneration Research is a Collaboration of the Paralyzed Veterans of America with Yale University. We thank Lynda Tyrrell and Palak Shah for technical assistance and Dr. Mark Estacion, Dr. Xiaoyang Cheng, Dr. Chongyang Han, Dr. Dmytro Vasylyev, and Dr. Hyesook Ahn for valuable comments.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Stephen G. Waxman, Neuroscience Research Center, Building 34, VA Connecticut Healthcare System (127A), 950 Campbell Avenue, West Haven, CT 06516. Stephen.Waxman{at}yale.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}