

Article Figures & Data
Figures
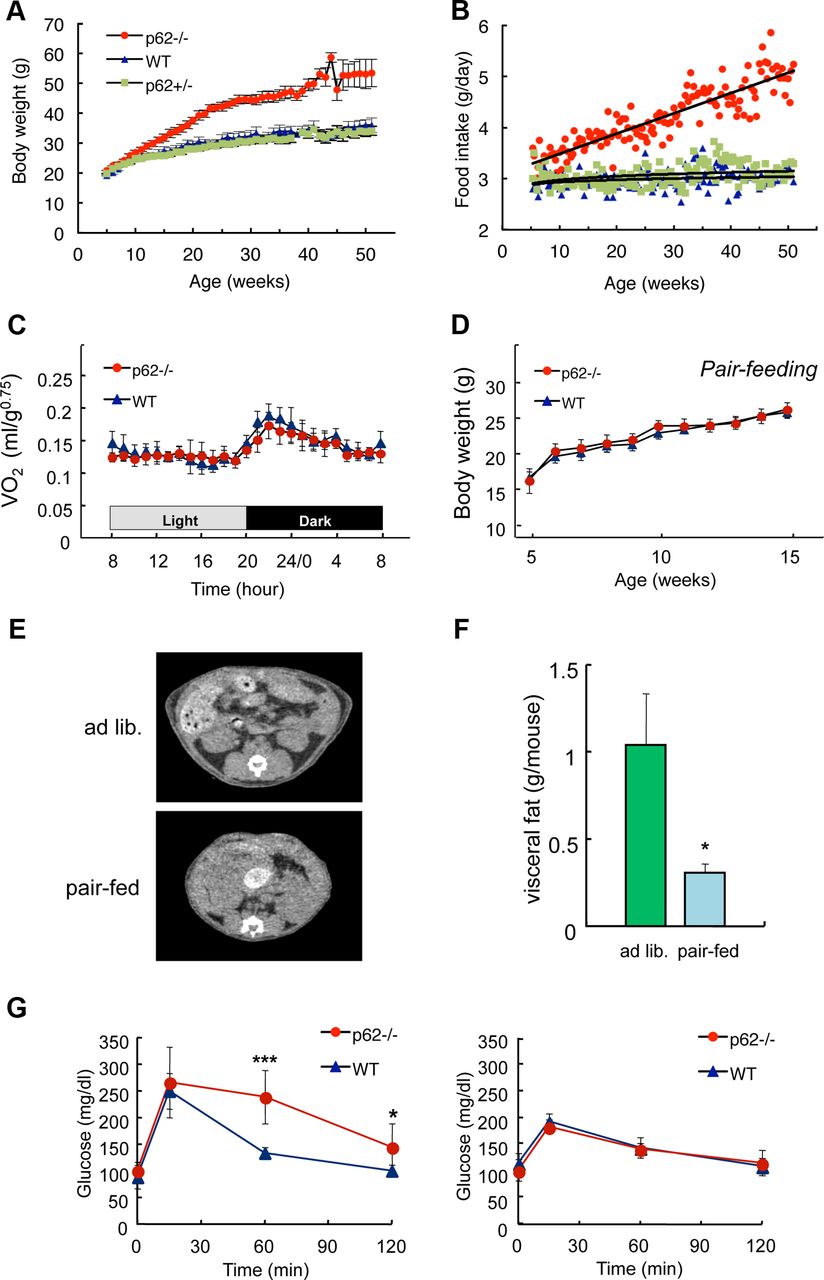
- Figure 1.
Hyperphagia causes mature-onset obesity in p62−/− mice. A, Weight change (mean ± SD; n = 5) and (B) food intake (mean ± SD; n = 5) of p62−/− (red), p62+/− (green), and control p62+/+ (wild-type, blue) male mice fed a standard diet ad libitum. C, Daily oxygen consumption rates of body weight-matched wild-type (blue, 30.3 ± 2.8 g) and p62−/− (red, 29.8 ± 1.6 g) mice (13–16-weeks-old; mean ± SD; n = 6). D, Pair-feeding experiment. Pellets were provided at 3.0 ± 0.1 g/mouse/d. A 3.0 g portion of food was given to each mouse in a separate cage at 18:00, and the body weight (mean ± SD; n = 5) was measured every week. E, F, Computed tomography (CT) analysis of the visceral fat content. Restricted daily food intake (3.0 g/d/mouse) for 10 weeks from the age of 5–15 weeks (Fig. 1D) suppressed visceral fat accumulation compared with that under ad libitum-feeding conditions in the p62−/− mice. The amount of visceral fat (F) was calculated from CT data. G, Glucose tolerance tests in ad libitum fed (left, 25–28-weeks-old) or diet restricted for 10 weeks (right, 20-weeks-old) wild-type and p62−/− mice. Blood glucose levels after intraperitoneal injection of glucose (1 g/kg body weight) are shown. All data in F and G represent the mean (n = 5–6) ± SD. Asterisks represent statistical significance; ***p < 0.001, *p < 0.05.
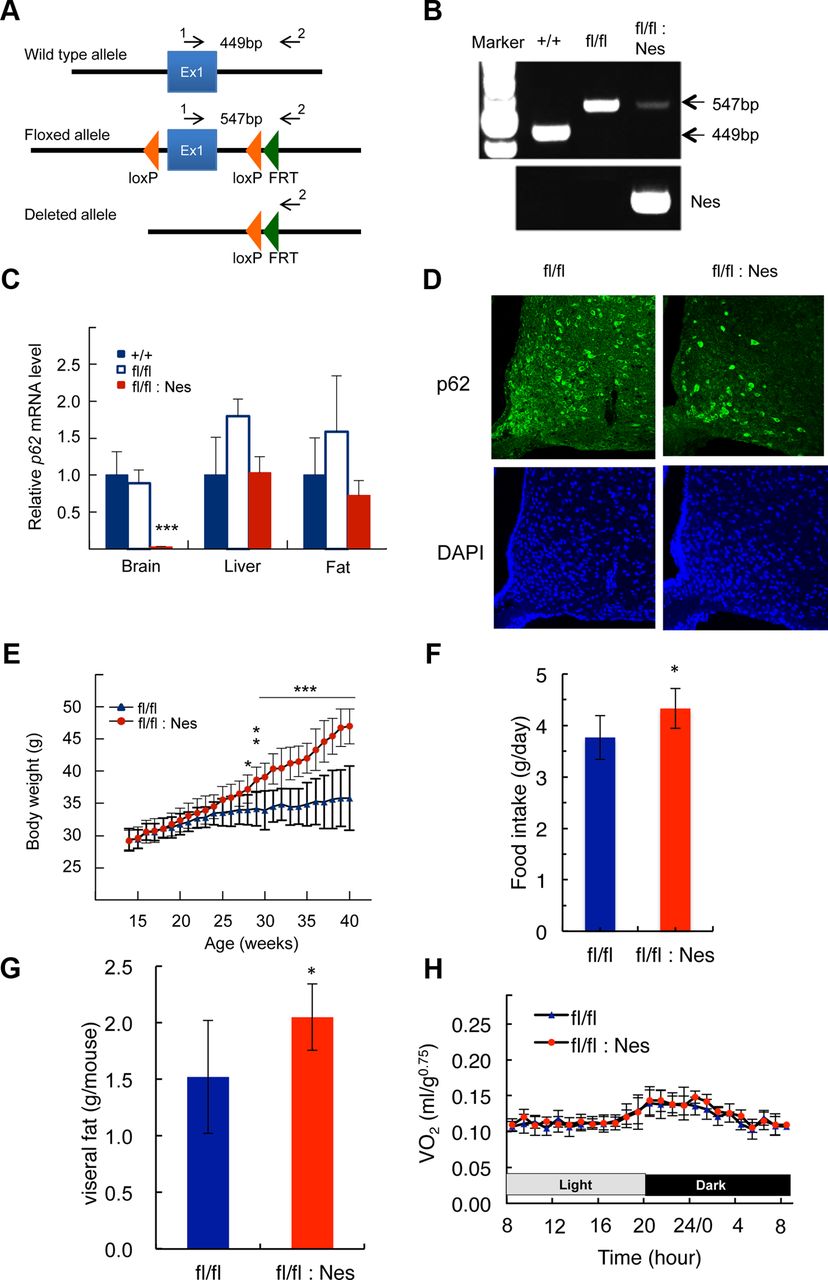
- Figure 2.
Construction of brain-specific p62−/− mice and their characterization. A, Schematic representation of the wild-type, floxed, and deleted allele of p62. Arrows represent the primers used to determine genotype. B, PCR analysis for genotyping using hypothalamic genomic DNA. C, p62 mRNA expression levels in the whole brain, liver, and epididymal fat tissue measured by quantitative real-time PCR (n = 5). D, Immunofluorescence staining of p62 in the arcuate nucleus of the hypothalamus. E, Weight gain curve of male p62flox/flox (n = 10) and p62flox/flox;Nes-Cre (n = 9) mice. F, G, The amount food intake and visceral fat in >30 weeks old mice (n = 9). H, Daily oxygen consumption rates of body weight-matched p62flox/flox (blue, 26.7 ± 1.6 g) and p62flox/flox;Nes-Cre (red, 27.7 ± 2.0 g) mice (12–18-weeks-old; n = 5). All data represent the mean ± SD. Asterisks represent statistical significance; ***p < 0.001, **p < 0.01, *p < 0.05.
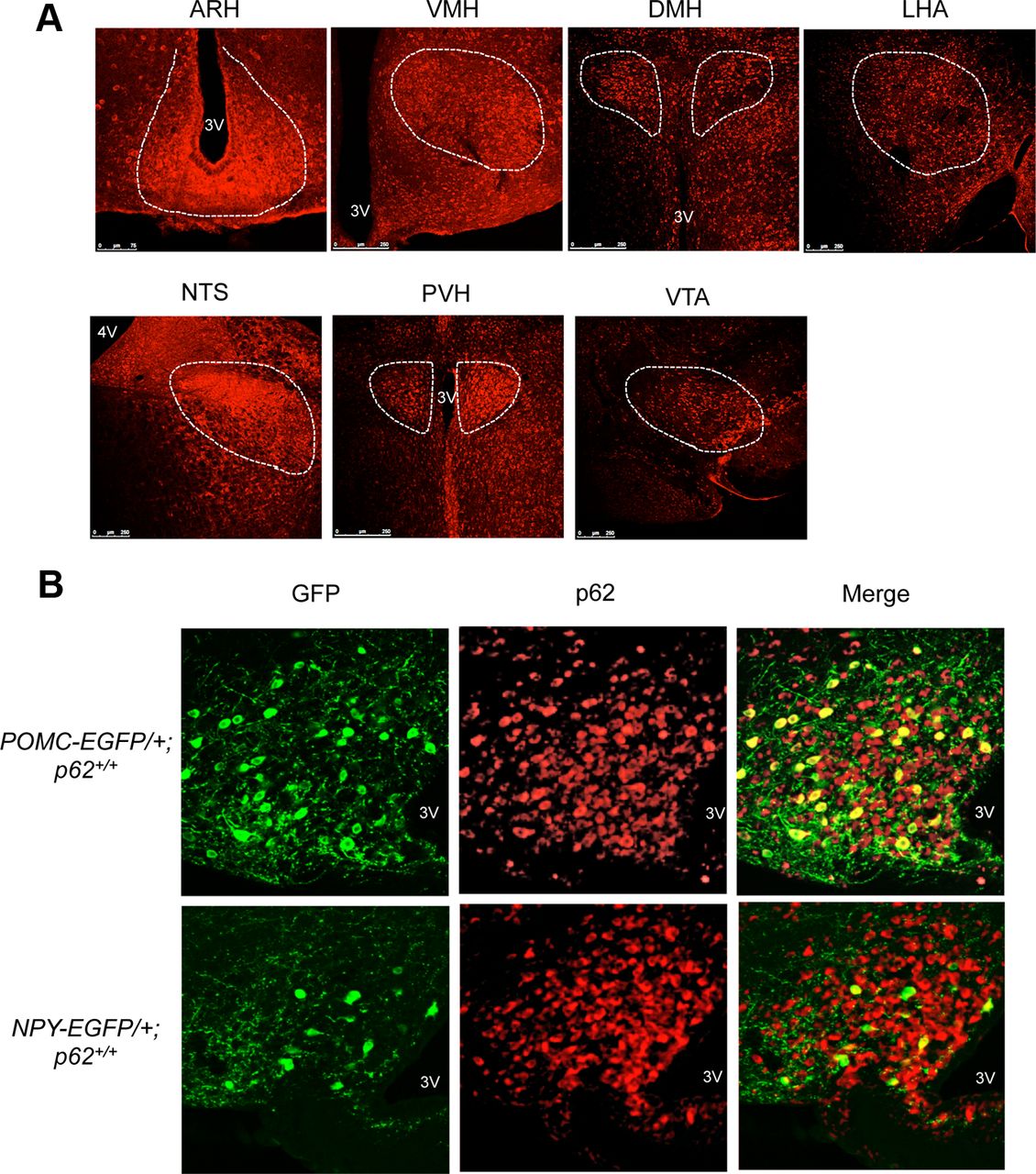
- Figure 3.
p62 expression in the hypothalamus. A, Immunofluorescence analysis showing that p62 is expressed in the brain regions implicated in the feeding behavior. p62 staining was abundantly expressed in the arcuate nucleus (ARH), ventromedial hypothalamus (VMH), dorsomedial hypothalamic nucleus (DMH), lateral hypothalamic area (LHA), nucleus tractus solitarius (NTS), paraventricular nucleus (PVH), or ventral tegmental area (VTA). 3V, Third ventricle; 4V, fourth ventricle. B, Double-staining with anti-p62 and anti-GFP antibodies in POMC (top) and NPY (bottom) neurons in POMC-EGFP and NPY-EGFP mice, respectively.
- Figure 4.
Serum leptin levels and effects of ICV injection of leptin, MTII, and NPY on eating behavior. A, Serum leptin levels in 3-week-old p62−/− (n = 11) and wild-type (n = 12); 10-week-old p62−/− (n = 10) and wild-type (n = 10); and 30-week-old p62−/− (n = 5) and wild-type (n = 5) mice. Means ± SDs are shown. Asterisks represent statistical significance; ***p < 0.001 and *p < 0.05 between the two groups. B, Serum leptin levels versus body weight of 3- and 10-week-old wild-type and p62−/− mice. C, Either leptin (0.3 μg/mouse) or physiological saline (vehicle) was ICV injected into body weight-matched wild-type (25.2 ± 1.7 g) and p62−/− (25.1 ± 4.6 g) mice after 16 h fasting. Food intake was measured for 24 h after ICV injection (10–15-weeks-old; mean ± SEM; n = 9–11). D, Effect of injecting MTII (5 nmol) on food intake 4 h after a 16 h fasting period (15–17-weeks-old; mean ± SEM; n = 5–6). E, Rapid effect of NPY (0.3 nmol/mouse) injection on food intake under ad libitum-feeding conditions was measured at 1 h after injection (10–17-weeks-old; mean ± SEM; n = 6–7). Asterisks represent statistical significance compared with vehicle injection; ***p < 0.001, **p < 0.01, *p < 0.05.
- Figure 6.
Leptin-mediated electrophysiological activation of POMC neurons. A, The spontaneous firing of POMC neurons was increased by leptin (50 nm) exposure in both wild-type and p62−/− neurons. Brain slices containing the hypothalamus from POMC-EGFP mice (3–4-weeks-old) were used for whole-cell patch-clamp recording. The recordings show the duration before and after leptin application. Representative traces are shown. B, Changes in membrane voltage (left) and frequency (right) determined from the recordings were not significantly different between wild-type and p62−/− mice. Values represent the mean ± SEM (wild-type, n = 3; KO, n = 8).
- Figure 7.
Defect in intracellular distribution of the transcription factor Stat3 in ARH neurons of p62−/− mice and MEFs. A, Immunoblotting of Stat3 and phosphorylated Stat3 in the hypothalamus (top). Animals (8–12-weeks-old; body weight-matched: wild-type, 26.0 ± 1.2 g; p62−/−, 25.5 ± 0.5 g) were fasted for 16 h and killed 0.5 and 1 h after intraperitoneal injection of leptin (1 mg/kg body weight). The levels of phosphorylated Stat3 were quantified relative to the levels of Stat3 in three independent animals (bottom). Results were shown mean ± SEM; *p < 0.05. B, Merged images of anti-Stat3 (red) and DAPI (blue, nucleus) staining in ARH neurons. Body weight-matched wild-type and p62−/− mice (8–12-weeks-old) were fasted for 16 h and killed 1 h after intraperitoneal injection of leptin (1 mg/kg body weight) or an equal amount of vehicle (control). Stat3 signals were concentrated into the nucleus after leptin stimulation in wild-type mice but were less in p62−/− mice. Similar results were observed in at least three independent animals. Scale bar, 10 μm. C, Distribution of Stat3 1 h after leptin or vehicle injection in ARH, POMC, or NPY neurons was scored from three to four independent animals. In addition to wild-type and p62−/− mice, POMC-EGFP and NPY-EGFP transgenic mice with (+/+) or without (−/−) p62 were used (8–12-weeks-old). Triple immunostaining was performed using anti-Stat3 (red), anti-GFP (green), and DAPI (blue, nucleus). Antibody stained POMC neurons are shown in Figure 3. Black columns represent the percentage of neurons in which anti-Stat3 staining of the nucleus was greater than or equal to that of the cytoplasm (N ≥ C). Total numbers of neurons counted in each fraction from 3 to 4 different animals are shown in parentheses. N, Nucleus; C, cytosol. “a” represents significantly different from wild-type control; p < 0.05. ***p < 0.001, *p < 0.05 compared with their controls (n = 3–4). D, Merged image of anti-Stat3 (red), POMC-EGFP (green), and nuclear (blue) staining of ARH neurons in p62−/− mice (8–12-weeks-old), 1 h after intraperitoneal injection of a high-dose leptin (5 mg/kg body weight). Stat3 signals were concentrated in the nucleus after leptin stimulation both in POMC (arrowhead) and other ARH neurons (arrow). Similar results were observed in three independent animals. Scale bar, 10 μm. E, MEFs from both types of mice were cultured and serum starved for 24 h, then Stat3 was visualized by immunofluorescence.



Tables
- Table 1.
Number of POMC-EGFP and NPY-EGFP neurons in the hypothalamus ARH in wild-type and p62−/− mice
Neuron type Age (weeks) No. of neurons in ARH (wild-type) (p62−/−) POMC 6–8 3952 ± 584 (n = 4) 4441 ± 720 (n = 3) 13–17 3792 ± 192 (n = 3) 3412 ± 352 (n = 3) 21–25 3912 ± 744 (n = 3) 4888 ± 448 (n = 3) NPY 6–8 1040 ± 176 (n = 3) 1559 ± 368 (n = 3) 13–17 1007 ± 48 (n = 3) 944 ± 128 (n = 3)* 21–25 1000 ± 320 (n = 4) 768 ± 56 (n = 4)* ↵*p < 0.01 compared with 6- to 8-week-old p62−/− mice.
Values represent mean ± SD.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}