

Abstract
Mitochondria carry out many of the processes implicated in maintaining axon health or causing axon degeneration, including ATP and reactive oxygen species (ROS) generation, as well as calcium buffering and protease activation. Defects in mitochondrial function and transport are common in axon degeneration, but how changes in specific mitochondrial properties relate to degeneration is not well understood. Using cutaneous sensory neurons of living larval zebrafish as a model, we examined the role of mitochondria in axon degeneration by monitoring mitochondrial morphology, transport, and redox state before and after laser axotomy. Mitochondrial transport terminated locally after injury in wild-type axons, an effect that was moderately attenuated by expressing the axon-protective fusion protein Wallerian degeneration slow (WldS). However, mitochondrial transport arrest eventually occurred in WldS-protected axons, indicating that later in the lag phase, mitochondrial transport is not required for axon protection. By contrast, the redox-sensitive biosensor roGFP2 was rapidly oxidized in the mitochondrial matrix after injury, and WldS expression prevented this effect, suggesting that stabilization of ROS production may mediate axon protection. Overexpression of PGC-1α, a transcriptional coactivator with roles in both mitochondrial biogenesis and ROS detoxification, dramatically increased mitochondrial density, attenuated roGFP2 oxidation, and delayed Wallerian degeneration. Collectively, these results indicate that mitochondrial oxidation state is a more reliable indicator of axon vulnerability to degeneration than mitochondrial motility.
Introduction
Axons are vulnerable to changes in energy production, organelle transport, and oxidative stress, all of which accompany axon degeneration (Coleman, 2005). The relative importance of each of these processes in degeneration has been difficult to parse, given their interdependence and the challenge of monitoring them in vivo. Mitochondria perform many of the processes implicated in degeneration, including ATP and reactive oxygen species (ROS) generation, as well as calcium buffering and protease activation. Therefore, it is not surprising that changes in mitochondrial function and transport are frequently observed during axon degeneration (Court and Coleman, 2012); however, the specific role that mitochondria play in these processes remains poorly understood.
Mitochondria have been linked to Wallerian degeneration (WD), a stereotyped program by which injured axons degenerate that shares features with axon pathology in neurodegenerative diseases (Coleman, 2005). Recent studies suggest that mitochondria may directly initiate WD, potentially through activation of the mitochondrial permeability transition pore (mPTP; Barrientos et al., 2011). WD can be dramatically delayed by the Wallerian degeneration slow protein (WldS), a mutant mouse protein created by a spontaneous genomic rearrangement (Perry et al., 1991; Coleman et al., 1998; Mack et al., 2001). Recent evidence suggests that the protective effects of WldS are mediated by mitochondria (Avery et al., 2012; Fang et al., 2012). For example, mitochondria isolated from WldS-expressing axons exhibit increased ATP production (Yahata et al., 2009) and calcium buffering (Avery et al., 2012), and a recent study found a correlation between mitochondrial transport and axon protection by WldS (Avery et al., 2012). However, whether mitochondrial transport is necessary or sufficient for axon protection after injury is not clear. Moreover, conflicting data indicate that mitochondria can either promote or inhibit axon degeneration in different contexts (Fang et al., 2012; Kitay et al., 2013).
To examine the role of mitochondria in axon degeneration we have imaged them in zebrafish peripheral sensory neurons undergoing WD. This model can be used to monitor the entire process of WD in vivo (O'Brien et al., 2009a; Martin et al., 2010) and to characterize dynamic subcellular events (Plucińska et al., 2012). By imaging mitochondria after axotomy in these neurons, we have characterized mitochondrial density, morphology, transport, and redox state during WD. Axonal injury led to mitochondrial transport arrest and oxidation of the redox-sensitive biosensor roGFP2 (Hanson et al., 2004) in the mitochondrial matrix, suggesting that mitochondrial ROS production may be relevant to axon degeneration. Increased mitochondrial transport was neither necessary nor sufficient for axon protection. By contrast, expression of WldS or the transcriptional coactivator PGC-1α prevented roGFP2 oxidation and delayed axon degeneration. Collectively, our results suggest that stabilization of mitochondrial redox state may regulate axon protection after injury.
Materials and Methods
Animal care
Fish were raised on a 14/10 h light/dark cycle at 28.5°C. Embryos were maintained in a 28.5°C incubator. Experiments were performed at larval stages, when male and female zebrafish cannot be distinguished. All experiments were approved by the Chancellor's Animal Research Care Committee at the University of California, Los Angeles.
Injections
For transient, mosaic gene expression in sensory neurons, ∼10 pg of each transgene was injected at the one-cell stage into wild-type AB or nacre (Lister et al., 1999) embryos or embryos from the transgenic lines described below. Embryos were screened for fluorescent transgene expression in Rohon-Beard (RB) neurons at 24–48 h postfertilization (hpf), using a Zeiss Discovery.V12 SteREO fluorescence dissecting microscope.
Transgenes
The stable transgenic lines used to visualize sensory neurons have been described previously (Sagasti et al., 2005; Palanca et al., 2013). Briefly, the isl1(ss) enhancer from the islet-1 gene (Higashijima et al., 2000) was used to drive expression of Gal4VP16, which activated expression of EGFP or DsRed under control of the Gal4 upstream activation sequence (14 copies, 14XUAS; Köster and Fraser, 2001). For studies of mitochondrial transport after injury, fish from a previously described stable transgenic line expressing GFP under the control of the Fru.trkA enhancer (Palanca et al., 2013) were also used, because their reporter expression was sparse, allowing the identification of embryos expressing GFP in a single RB neuron. For developmental experiments in wild-type embryos, sensory neurons were visualized by transient injection of the isl1(ss):Gal4VP16–14XUAS, reporter plasmids used to generate the stable lines described above. The Tol2/Gateway zebrafish kit (Kwan et al., 2007) was used for construction of all remaining plasmids. In all cases transgene expression was driven by the CREST3 enhancer (gift from Hitoshi Okomoto, RIKEN Brain Science Institute, Tokyo, Japan; Uemura et al., 2005). Like isl1(ss), the CREST3 enhancer is derived from the islet-1 gene and drives expression in peripheral sensory neurons.
WldS.
For coexpressing GFP and WldS in sensory neurons, we used a previously described transgene (CREST3:Gal4VP16–14xUAS–GFP–14xUAS–WldS; Martin et al., 2010). To achieve equimolar expression of DsRed and WldS, we used the viral T2A sequence, which causes ribosomal skipping (Donnelly et al., 2001; Tang et al., 2009). This sequence was inserted into the Gateway system (Invitrogen) behind a multiple cloning site. WldS was amplified using primers to add XhoI and HindIII restriction sites to the 5′ and the 3′ ends of the sequence, respectively, and cloned into the system to generate CREST3:Gal4VP16–14XUAS–WldS-2A-DsRed.
PGC-1α.
A plasmid encoding mouse PGC-1α, which was previously expressed in zebrafish (Hanai et al., 2007), was a gift from Shintaro Imamura (National Research Institute of Fisheries Science, Yokohama, Japan). The PGC-1α coding sequence was amplified using primers to add XhoI and BamHI to the 5′ and 3′ ends of the sequence, respectively. The sequence and the p3E–MCS-2A-DsRed and p3E–MCS-2A-GFP Gateway donor vectors were cloned with XhoI and BamHI sites to generate p3E–PGC-1α-2A-DsRed and p3E–PGC-1α-2A-GFP. These donor vectors were then recombined to generate CREST3:Gal4VP16–14XUAS–PGC-1α-2A-GFP and CREST3:Gal4VP16–14XUAS–PGC-1α-2A-DsRed.
MitoDsRed.
To visualize mitochondria, a cox8 mitochondrial targeting sequence was added to DsRed (mitoDsRed; gift from Meghan Johnson and Carla Koehler, UCLA, Los Angeles, CA). The reporter UAS–mitoDsRed–polyA was used to generate a MitoDsRed transgenic line using the Tol2 system (Kawakami, 2004). The plasmid, which contained Tol2 sites flanking UAS–mitoDsRed–polyA, was injected with transposase mRNA into zebrafish eggs at the one cell stage. These fish were raised and crossed to isl1(ss):Gal4VP16:14XUAS-GFP fish. Progeny from this cross were then screened for the expression of GFP and mitoDsRed in sensory neurons. These double-transgenic fish were then crossed to wild-type AB fish to obtain a line of single-transgenic UAS-mitoDsRed fish.
Mito-roGFP2.
To monitor changes in the mitochondrial redox potential, roGFP2 (Hanson et al., 2004) was localized to the mitochondrial matrix with a cox8 targeting sequence (mito-roGFP2; gift from Meghan Johnson and Carla Koehler). Mito-roGFP2 was cloned into the Gateway system to generate UAS–mito-roGFP2–polyA. This transgene was coinjected with one of the CREST3:DsRed reporters described above, for expression of DsRed (± WldS or PGC-1α) in the axon and mito-roGFP2 in the mitochondrial matrix.
Synaptophysin-GFP.
The transgene encoding the zebrafish synaptophysin-EGFP fusion protein (Syp-GFP) has been described (Meyer and Smith, 2006), and was a gift from Martin Meyer (King's College, London, UK).
Morpholinos
Morpholino oligonucleotides targeting the zebrafish pink1 gene (NM_001008628; Anichtchik et al., 2008) were acquired from GeneTools. Sequences were as previously described (ATG targeting, 5′-GCT GAG AAC ATG CTT TAC TGA CAT T-3′; 5′-untranslated region targeting, 5′-ATA TTG ACT ATG AGA GGA AAT CTG A-3′) and 7 ng of each morpholino were coinjected into one-cell stage embryos (Anichtchik et al., 2008), in combination with the plasmids described above. Control embryos were injected with 14 ng of the standard GeneTools control morpholino (5′-CCT CTT ACC TCA GTT ACA ATT TAT A-3′).
Imaging axons and mitochondria
Embryos were dechorionated, anesthetized in 0.01–0.02% tricaine, mounted in 1.2% low-melt agarose (Promega, V2111) in sealed chambers (O'Brien et al., 2009b), and imaged on a heated stage kept at 28.5°C. For time-lapse analysis of mitochondrial movement and WD, GFP-, and mitoDsRed-expressing embryos were imaged at various intervals for up to 3 h on a confocal microscope (Zeiss LSM 510). Images were compiled into projections and movies with QuickTime software.
Mitochondrial imaging.
To monitor mitochondrial morphology and density, GFP reporter transgenes were injected into UAS–mitoDsRed-expressing embryos. Embryos were imaged at 2–4 d post-fertilization (dpf) using a confocal microscope with a 40× oil objective and 3× digital zoom. Mitochondrial and vesicular transport imaging was limited to a single optical section. Before each time-lapse movie, an image was taken using both 488 and 543 nm lasers, so that the axon could be visualized for later analysis. Mitochondria or vesicles were imaged every second for 6 min. In the case of axotomy, a time-lapse movie was made before axon transection for comparison. Only axons in which at least one mitochondrion moved before injury were then cut and imaged for postinjury analysis. For evaluation of relative redox potential, mito-roGFP2-expressing embryos were imaged with a 20× air objective and 4× digital zoom. Images were taken before and at various intervals after injury, scanning at 543 nm to visualize red axons, and at 405 and 488 nm to monitor roGFP2 oxidation.
Axon transection
Axons were cut using a Zeiss microscope (LSM 710) equipped with a multiphoton laser (O'Brien et al., 2009b). Embryos were imaged with a 20× water objective and 488/543 nm laser scanning to identify the axonal region of interest, then 1–5 scans of the multiphoton laser (tuned to 910 nm for green axons, 850 nm for red axons) were used to transect an axonal region of interest at 100× digital zoom.
Data analysis
Mitochondrial morphology.
Morphology was quantified from 2D projections of confocal stacks generated from images taken at 40×, 3× digital zoom. In the green channel, all axons within an image were traced using ImageJ software. Line length was calibrated to convert pixels to distance, and the Measure plugin for ImageJ was used to determine the length and diameter of each mitochondrion in an image. All mitochondria within an image were quantified, and the ratio of length/width was taken as a measure of mitochondrial morphology. Mean morphology was compared with unpaired t tests. To further analyze morphology, GraphPad Prism software was used to divide mitochondria into bins according to their length/width ratio (bin width: 1.0; i.e., all mitochondria with a length/width ratio between 1 and 2 were in Bin 1), and the relative frequency of each bin was determined as a percentage of the total, and represented in a histogram. Note that all discussions of mitochondrial “length” refer to this ratio, to control for any differences in expression levels that might increase or decrease mitoDsRed fluorescence intensity and thereby affect measurement.
Mitochondrial transport.
The percentage of total mitochondria that moved in a 50 μm axon segment during a 6 min time-lapse movie was quantified using the kymograph macro for ImageJ. Kymograph color was inverted so that mitochondria appeared black. A mitochondrion was considered to be moving only if it traveled at least 2 μm, at a speed of at least 0.1 μm/s, to be consistent with published work (Misgeld et al., 2007). Speed was calculated as the slope of distance (x) over time (y) on the kymograph. Vesicular transport was quantified using the same parameters. For measures of transport after axotomy, data from all axons were binned according to time after injury, and statistical analysis was performed.
Mitochondrial density.
Mitochondrial density in axons of wild-type and WldS-expressing embryos, and in control and PTEN-induced putative kinase 1 (PINK1) morphants, was determined by counting all mitochondria (stationary and motile) in a kymograph. In PGC-1α-expressing neurons, mitochondrial distribution was irregular. For this reason, mitochondrial density was determined from still images in multiple regions of the axon, and values therefore primarily represented the density of stationary mitochondria. Mitochondrial density in still images from wild-type axons was evaluated as a control.
Relative redox potential.
The excitation maxima of roGFP2 are ∼400 and ∼475–490 nm; under reducing conditions, the 475–490 nm excitation peak dominates, whereas oxidation results in relatively higher excitation at 400 nm (Hanson et al., 2004). Using confocal images taken before and at various time points after injury, projections were generated to calculate the 405/488 fluorescence ratio of mitochondria in the Z stack. Measures were taken from all mitochondria within 100 μm of the eventual injury site, and values were pooled to generate a mean fluorescence ratio for each axon before injury. All postinjury values for a given axon were calculated as a percentage of the initial. Data from all axons were then binned according to time after injury, and statistical analysis was performed.
Statistical analysis.
GraphPad Prism software was used for statistical analysis. Planned, unpaired Student's t tests were used to compare independent groups. A paired t test was used to compare mito-roGFP2 oxidation in individual axons early and late in the lag period before degeneration. One-way ANOVA was used to compare three or more groups, with Dunnett's post-tests used for comparison to the control. Two-way ANOVA was used to evaluate the effect of time and genotype/location on changes in transport and roGFP2 oxidation after injury, followed by Bonferroni post-tests to evaluate significance at each time point.
Results
Monitoring mitochondria in living zebrafish sensory neurons
To study morphology and motility of mitochondria in a live, vertebrate system, we created reporters to label them in zebrafish somatosensory neurons (Fig. 1A). DsRed fused to the cox8 mitochondrial matrix targeting sequence (mitoDsRed) was placed under the control of UAS elements and used to generate a stable transgenic line (UAS:mitoDsRed; Fig. 1B). The CREST3 enhancer element, which drives expression in peripheral sensory neurons (Uemura et al., 2005; Palanca et al., 2013), was used to drive Gal4VP16, and this transgene was injected into mitoDsRed embryos to achieve mosaic expression in small numbers of sensory neurons (Fig. 1C). Mitochondrial density and transport were evaluated at 2 and 4 dpf in 50 μm segments of individual peripheral axons. Cells were monitored for 6 min at one frame per second (Fig. 1D), and transport was quantified using kymograph analysis (Fig. 1E). Mitochondrial density was slightly but significantly lower at 4 dpf (2 dpf: 280 ± 0.24 mitos/mm; 4 dpf: 208 ± 0.19 mitos/mm; n ≥ 16 cells, p < 0.05) but mitochondrial motility, defined as the percentage of mitochondria that moved at any point within a 6 min time-lapse movie, did not differ significantly between 2 and 4 dpf (2 dpf: 23.2 ± 4.0%; 4 dpf: 20.1 ± 5.1%; n ≥ 16 cells, p = 0.59). Among motile mitochondria, the speed of uninterrupted runs in the retrograde direction did not change between 2 and 4 dpf (2 dpf: 0.69 ± 0.06 μm/s; 4 dpf: 0.58 ± 0.06 μm/s; n ≥ 16 mitos in n ≥ 11 cells, p = 0.383); however, anterograde velocity was higher at 4 dpf (2 dpf: 0.50 ± 0.03 μm/s; 4 dpf: 0.65 ± 0.04 μm/s; n ≥ 51 mitos in n ≥ 11 cells, p = 0.003). The mitoDsRed system can thus be used to detect quantitative changes in axonal mitochondria during development, similar to the recently described MitoFish system (Plucińska et al., 2012).
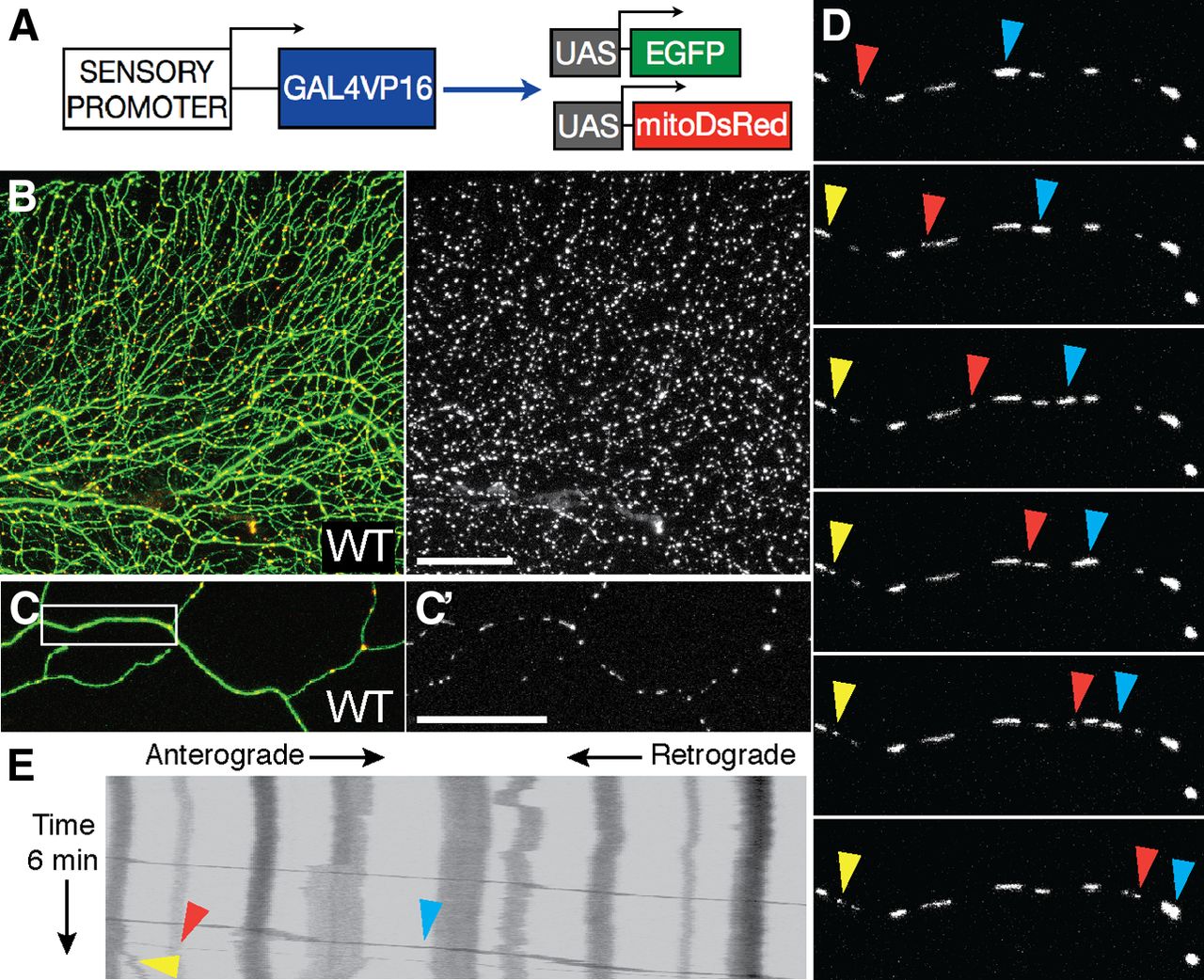
In vivo imaging of mitochondrial transport in zebrafish somatosensory neurons. A, Transgenes to visualize GFP in neurons and DsRed in the mitochondrial matrix. B, When a stable UAS–mitoDsRed transgenic fish line was crossed to a previously described line in which the isl1(ss) sensory promoter drives Gal4 and GFP expression in somatosensory neurons, red fluorescent mitochondria (B′) were visible in green fluorescent axons. C, C′, To permit quantification of mitochondrial transport in a single cell, the CREST3:Gal4-UAS–GFP reporter transgene was injected into UAS–mitoDsRed embryos at the one-cell stage for transient mosaic gene expression. D, A single optical section was repeatedly imaged at a frequency of 1 Hz to quantify mitochondrial motility (the percentage of mitochondria that moved within a 6 min movie), and to characterize transport speed and direction. Images in D represent still frames every 1 min. E, Kymograph representing transport in a representative axon. Arrows point to moving mitochondria in D. Scale bars, 50 μm. White box in C represents axonal segment in D.
Mitochondrial transport arrest after injury is local and specific
Recent studies in invertebrate and in vitro systems have suggested that mitochondrial number and transport decrease upon injury (Avery et al., 2012; Fang et al., 2012). To determine whether mitochondrial transport was also affected in a vertebrate system in vivo, we transected peripheral axons of sensory neurons with a laser at 3 dpf. At this stage WD in peripheral sensory neurons is highly stereotyped, and fragmentation occurs ∼90 min after axotomy (Martin et al., 2010). MitoDsRed-expressing axons were imaged before (Fig. 2A) and after (Fig. 2B) laser transection. Mitochondrial transport was monitored along 50 μm axon segments either immediately adjacent (0–50 μm) or 100–200 μm distal to the site of injury. Within 2 min postaxotomy (mpa), mitochondrial transport completely terminated in the axonal segment immediately distal to the site of injury and never recovered (Fig. 2C,D,I). By contrast, transport in axonal segments further from the cut site was not immediately affected (Fig. 2E,F,I). However, motile mitochondria in this region spent significantly less time moving in the anterograde direction (away from the cut site), and a greater amount of time paused between runs (Fig. 2J), and the velocity of uninterrupted runs in the retrograde direction was decreased (Fig. 2K). Thus, although degeneration is synchronous throughout the distal fragment (Fig. 2G,H; Martin et al., 2010), acute transport arrest was limited to the region nearest the site of injury. However, all mitochondrial transport ultimately stopped in the distal axon before fragmentation.

Axonal injury causes mitochondrial transport arrest in a separated axon fragment. A, B, Mitochondrial transport was imaged in a 50 μm axon segment before (A) and after (B) GFP- and mitoDsRed-expressing axons were severed with a laser (yellow arrow points to injury site). Transport was monitored either immediately distal to the injury (white arrowheads) or 100–200 μm distal (blue arrowheads). C–F, Representative kymographs from 50 μm axonal segments before and after axotomy at the two locations. Red arrowhead in F points to motile mitochondrion in more distal region 20 min post-axotomy (mpa). G, H, The distal axon ultimately fragmented synchronously: the entire detached axon was intact 65 mpa (G), and had fragmented by 75 mpa (H). I, Quantification of mitochondrial motility before and at various time points after axotomy. Adjacent to the injury, motility stopped abruptly (percentage motile before: 27.00 ± 4.35%; 2–10 mpa: 4.50 ± 2.64%; n = 13 axons, p = 0.0001) and did not recover (10–90 mpa: 2.70 ± 0.86%; p < 0.0001). Further from the injury (100–200 μm), mitochondrial transport did not immediately terminate (percentage motile before: 26.51 ± 5.25% motile; 2–10 mpa: 21.68 ± 4.57, n ≥ 8 axons; p = 0.5386); however, the characteristics of transport were acutely affected (J, K). J, At the 100–200 μm distal site, motile mitochondria spent a higher percentage of time paused immediately after axotomy (before: 43.28 ± 7.91%; 2–10 mpa: 74.48 ± 7.73%), and proportionally less time moving in the anterograde direction, away from the cut site (before: 39.45 ± 7.96%; 2–10 mpa: 8.55 ± 3.71%, n ≥ 21 mitochondria from ≥9 axons, p < 0.001). By 10–30 mpa, however, this difference was no longer significant (Bonferroni post-test, p > 0.05). K, Quantification of the speed of uninterrupted runs (not counting time paused) revealed that retrograde transport was reduced acutely in mitochondria after injury (retrograde speed before: 0.67 ± 0.09 μm/s; 2–10 mpa: 0.36 ± 0.06 μm/s; n ≥ 8 mitochondria from ≥5 axons, p = 0.0286), but anterograde speed was not affected (before: 0.46 ± 0.05 μm/s; 2–10 mpa: 0.40 ± 0.04 μm/s; n ≥ 8 mitochondria from ≥5 axons). Later in the lag phase, retrograde transport velocity recovered (10–30 mpa: 0.51 ± 0.08 μm/s; p = 0.238). Despite this transient recovery, transport eventually stopped throughout the distal axon (I) before fragmentation. Scale bars, 50 μm.
The eventual loss of all distal mitochondrial movement after injury could be caused by a global impairment of axonal transport, or the effect could be specific to mitochondria. To distinguish between these possibilities, we imaged vesicular transport by expressing a synaptophysin-GFP fusion protein (Meyer and Smith, 2006) in DsRed-labeled axons (Fig. 3A–C). In contrast to the acute mitochondrial transport arrest, vesicular transport was unaffected immediately after axotomy, and persisted at baseline rates until shortly before axon fragmentation (Fig. 3D–F). Mitochondrial transport arrest after axotomy is therefore not likely caused by generalized microtubule breakdown.

Vesicular transport is not affected by axonal injury. A–C, DsRed and a synaptophysin-GFP fusion protein were expressed in axons to visualize vesicle transport before and after axotomy in 2 dpf embryos. Image in (C, C′) is a single optical section taken from boxed region in B. D, E, Representative kymographs illustrating vesicle motility before and 10 min postaxotomy. F, In contrast to the acute mitochondrial transport arrest, vesicular transport was not acutely affected by injury (percentage motile before: 50.22 ± 4.14%; 2–10 mpa: 50.17 ± 5.70%; n = 9 axons, p = 0.99), and persisted at baseline rates until shortly before axon fragmentation (90–120 mpa: 24.54 ± 6.81%, p = 0.0014). Scale bar, 50 μm.
Mitochondrial transport in the proximal axon persists after axotomy
To examine whether mitochondrial transport arrest correlates with axon degeneration, we monitored mitochondrial motility in the proximal portion of the transected axon (0–50 μm proximal to the injury, Fig. 4A), which remains connected to the cell body and does not degenerate. Overall mitochondrial motility in the proximal axon was not altered at any point after axotomy (Fig. 4B–E). Motile mitochondria did, however, spend a higher percentage of time paused for the first hour after axotomy (Fig. 4F), and the duration and speed of retrograde movement were also reduced (Fig. 4G). However, the net direction of transport was not significantly different (Fig. 4H). Together, these changes in the proximal axon stump favored mitochondrial retention near the injury site. One hour later the differences in mitochondrial transport were no longer statistically significant (Fig. 4G). Despite the mild and transient changes in the characteristics of transport, overall mitochondrial motility in the proximal axon was thus preserved after axotomy, supporting the possibility that mitochondrial transport arrest in the distal axon might underlie degeneration.

Mitochondrial transport persists in the proximal axon after injury. A, Mitochondrial motility was monitored in the 50 μm axonal segment immediately proximal to the site of injury (white arrowheads) at various time points after axotomy (yellow arrow points to injury site). B–D, Representative kymographs before and after axotomy. E, Overall mitochondrial motility in the proximal axon was not significantly different from baseline at any point after axotomy (p = 0.2158). F, Motile mitochondria spent a higher percentage of time paused for the first 30 min after axotomy (before: 33.10 ± 6.55%; 2–30 mpa: 63.38 ± 5.86%; n ≥ 29 mitos from ≥6 axons, p = 0.0014). G, The net direction of transport in the proximal axon was unchanged before and after axotomy (p = 0.7959). H, The speed of uninterrupted runs in the retrograde direction was reduced during the first 30 min after axotomy (before: 0.60 ± 0.07 μm/s; 2–30 mpa: 0.41 ± 0.06 μm/s; n ≥ 22 mitos from ≥6 axons, p = 0.0118), but the anterograde velocity was unchanged (p = 0.2138). NS, Not significant. Scale bar, 50 μm.
WldS mildly affects baseline mitochondrial morphology and transport
Expressing WldS in zebrafish somatosensory axons (Fig. 5) dramatically delays degeneration after transection (Fig. 6A,B; Martin et al., 2010). To determine whether WldS alters mitochondrial motility in zebrafish sensory axons we coexpressed WldS and GFP in mitoDsRed-expressing embryos (Fig. 5A–C). Overall mitochondrial density in uninjured WldS-expressing axons did not differ from wild-type axons (WT: 2.20 ± 0.16 mitos/μm; WldS: 2.33 ± 0.25 mitos/μm; n ≥ 18 cells, p = 0.64), but the average mitochondrial morphology (ratio of length/width) was slightly elongated (Fig. 5B–D). Overall mitochondrial motility in uninjured WldS-expressing axons was not significantly different from wild-type (Fig. 5E). The amount of time motile mitochondria spent either paused or moving in either direction was also not different from wild-type (Fig. 5F). The speed of anterograde runs was higher in WldS mitochondria than wild-type, but retrograde speed was unchanged (Fig. 5G). Thus, WldS expression in zebrafish somatosensory neurons appears to affect mitochondria only mildly.

WldS mildly affects mitochondrial morphology and transport in uninjured axons. A, Transgenes used to visualize mitochondria in WldS-expressing axons. A sensory promoter drove expression of either GFP (WT, B, B′) or WldS and GFP (C, C′) in UAS-mitoDsRed embryos. B–D, Mitochondria in WldS-expressing axons were, on average, elongated compared with wild-type mitochondria (WT average length/width ratio: 1.70 ± 0.07; WldS: 2.12 ± 0.09, p = 0.0002). This difference was primarily attributable to the presence of fewer small, spherical mitochondria (length/width ratio of 1; WT example indicated by arrowheads in B′), as indicated by the histogram in D. E, Overall mitochondrial motility in uninjured WldS-expressing axons was not significantly different from wild-type (WT % motile: 20.71 ± 4.96%; WldS 13.32 ± 2.66%; n ≥ 18 axons, p = 0.18). F, The amount of time motile mitochondria spent paused or moving in either direction was not different from wild-type (p > 0.05). G, Mitochondrial transport in the anterograde direction was significantly faster in WldS-expressing axons (WT anterograde speed: 0.37 ± 0.03 μm/s; WldS: 0.55 ± 0.06 μm/s; n ≥ 18 mitochondria from ≥11 axons), but retrograde speed was not significantly different (WT: 0.41 ± 0.07 μm/s; WldS: 0.35 ± 0.06 μm/s; n ≥ 18 mitochondria from ≥11 axons; p = 0.5282). Scale bar, 20 μm.

WldS moderately and temporarily mitigates mitochondrial transport arrest after injury. A, B, WldS-expressing axons were transected and imaged at various time points after injury. B, WldS-expressing axons remained intact 24 hpa, despite persistent detachment from the cell body. C, Representative kymographs from movies of mitochondrial transport at various time points before and after axotomy. All mitochondrial transport was evaluated in 50 μm axonal segments immediately distal to the injury. D, As in wild-type axons, acute mitochondrial transport stopped in the distal axon immediately after transection in WldS-expressing axons (percentage motile before: 23.28 ± 3.40%; 2–10 mpa: 3.85 ± 1.73%, n = 13 cells, p < 0.0001), but shortly thereafter transport temporarily recovered to baseline values (10–30 mpa: 17.43 ± 3.67%, n = 13 cells). Eventually, mitochondrial transport terminated altogether in WldS-expressing axons (percentage motile 50–90 mpa: 6.53 ± 2.97%; p = 0.0010). Asterisks represent statistical significance (p < 0.01) compared with wild-type motility (replotted from Fig. 2; two-way ANOVA, Bonferroni post-test). E, After recovering from acute transport arrest, motile mitochondria spent a lower percentage of time moving in the anterograde direction than before injury (before: 37.72 ± 8.32%; 10–30 mpa: 9.58 ± 5.48%; n ≥ 18 mitos from 12 cells; p = 0.0143). F, The net direction of mitochondrial movement also changed, with mitochondria traveling a higher percentage of their total distance in the retrograde direction toward the cut site (retrograde before: 32.85 ± 8.33%; 10–30 mpa: 65.97 ± 9.5%; n ≥ 18 mitos from ≥ 9 cells, p = 0.013). G, Run velocity also did not change in either direction (anterograde before: 0.45 ± 0.07 μm/s; 10–30 mpa: 0.33 ± 0.04 μm/s; p = 0.2633; retrograde before: 0.31 ± 0.06 μm/s; 10–30 mpa: 0.32 ± 0.05 μm/s; p = 0.9170). Scale bar, (in A, B) 50 μm.
WldS modestly and temporarily mitigates mitochondrial transport arrest after axotomy
Although there was no significant difference in baseline mitochondrial motility in WldS-expressing axons, it is possible that preservation of motility after injury underlies its protective effect (Avery et al., 2012). To test this hypothesis, we evaluated mitochondrial transport before and after axotomy. As in wild-type axons, mitochondria in the portion of the axon immediately distal to the injury stopped moving acutely after transection in WldS-expressing axons, but shortly thereafter mitochondrial transport recovered to baseline values (Fig. 6C,D). At that time, motile mitochondria moved more in the retrograde direction than before injury, as measured both by time (Fig. 6E), and by distance (Fig. 6F). The velocity of uninterrupted runs in either direction was not different (Fig. 6G). Mitochondrial transport during this phase thus favors retrograde movement toward the cut site, similar to the effect seen in the wild-type proximal axon.
The early transport recovery observed in transected WldS-expressing axons was temporary. At all later time points, mitochondrial motility was reduced relative to baseline (Fig. 6D). This observation suggests that long-term protection is not mediated by long-term transport preservation. To further examine this idea, we monitored axonal mitochondria in WldS-expressing neurons 24 h postaxotomy (hpa) and compared mitochondrial motility to uncut axons from age-matched controls. Although distal axons from these WldS-expressing cells were still intact (Fig. 6B), all mitochondria were essentially stationary (4 dpf uncut: 13.71 ± 3.16% motile mitochondria; 4 dpf, 24 hpa: 0.45 ± 0.45% motile; 14/15 axons had no motile mitos, p = 0.0004). This finding demonstrates that mitochondrial motility is not required for the maintenance of axonal integrity. It also suggests that WldS may not protect axons by regulating mitochondrial transport.
Some studies suggest that uneven mitochondrial distribution induces local hypoxia that initiates axon degeneration (Misko et al., 2012). It is therefore possible that the early attenuation of mitochondrial transport arrest by WldS allows mitochondria to redistribute to a more uniform pattern. To examine this possibility, we first measured mitochondrial distribution in wild-type and WldS-expressing axons before axotomy, and found no difference in average intermitochondrial distance or the variation from the mean (WT before: 4.81 ± 0.33 μm; WldS before: 4.36 ± 0.35 μm, n ≥ 73 mitos in ≥6 axons, p = 0.370). We then measured mitochondrial spacing between 50 and 90 mpa, after the period of transport recovery in WldS had occurred. There was no difference in distribution between wild-type and WldS-expressing cells at that time (WT: 4.53 ± 0.35 μm; WldS: 4.73 ± 0.36 μm, n ≥ 68 mitos in ≥6 axons, p = 0.717). These results suggest that WldS does not protect axons by altering mitochondrial distribution after injury.
PINK1 knock-down increases mitochondrial transport after axotomy without delaying degeneration
Observing mitochondria in WldS-expressing axons demonstrated that ongoing motility is not required for protection. To test whether early motility might be sufficient to protect axons from degeneration we sought to prevent their arrest after axotomy with an independent manipulation. Damaged mitochondria accumulate PINK1 on their surface (Matsuda et al., 2010), which ultimately leads to their dissociation from microtubules (Liu et al., 2012). We hypothesized that if transport arrest after axotomy is mediated by PINK1 stabilization on depolarized mitochondria, PINK1 knock-down might promote recovery of transport after transection. We used a previously characterized morpholino (Anichtchik et al., 2008) to knock down PINK1 expression and conducted experiments at 2 dpf to ensure a robust morphant phenotype. We observed the same gross morphological changes previously reported in these morphants, such as cerebral edema (Fig. 7A,B). Axonal mitochondria were elongated in PINK1 morphants (Fig. 7C,D), consistent with the fact that PINK1 promotes mitochondrial fission (Deng et al., 2008; Poole et al., 2008). There was no difference in baseline mitochondrial motility between PINK1 and control morphant embryos (Fig. 7E). Between 2 and 10 min after axotomy, differences in motility between PINK1 and control morphants were not significant (p = 0.12). Mitochondrial motility later recovered modestly in PINK1 morphants (Fig. 7F), but this increased motility was not accompanied by axon protection. On the contrary, axon degeneration was slightly accelerated, with a shorter lag phase before fragmentation in PINK1 morphants compared with controls (Fig. 7G). PINK1 knock-down therefore phenocopies the postinjury mitochondrial transport recovery observed in WldS-expressing axons, but does not provide axon protection. Together our results demonstrate that axon motility is neither required nor sufficient for axon protection.

PINK1 knock-down increases mitochondrial motility after injury and accelerates degeneration. A, B, UAS-mitoDsRed embryos were coinjected with control or PINK1 morpholinos and the sensory:Gal4:UAS-GFP transgene used in Figures 1, 2. Phenotypes such as cerebral edema (B, arrowhead) have previously been reported with this PINK1 morpholino. C–D, Control (C, C′) and PINK1 morphants (D, D′) were imaged at 2 dpf. Mitochondria were on average longer in PINK1 morphants than controls (D, arrowheads). E, There was no difference in baseline mitochondrial motility between PINK1 and control morphant embryos (Control MO: 21.93 ± %; PINK1 MO: 26.48 ± 2.70%; n ≥ 5 axons, p = 0.5285). F, Immediately after axotomy, mitochondrial transport was reduced in both groups (WT 2–10 mpa: 0 ± 0% motile; PINK1: 8.32 ± 4.90%; n ≥ 9 axons). Between 10 and 50 mpa, motility was significantly higher in PINK1 morphants than controls (10–30 mpa: p = 0.028; 30–50 mpa: p = 0.038). G, Axon degeneration after injury was accelerated in PINK1 morphants (quantified as the duration of the lag period between injury and axon fragmentation; Control MO: 135.0 ± 6.3 min; PINK1 MO: 87.5 ± 2.5 min; n = 8 axons per group; p < 0.0001). Scale bar, 10 μm.
Axonal injury alters the redox state of mitochondria
Because mitochondrial motility per se does not appear to be instructive in WD, we investigated another feature of mitochondrial function. In addition to their roles in ATP synthesis and calcium buffering, mitochondria are the primary site of ROS production in the cell. Because oxidative stress is associated with aging and neurodegeneration (Balaban et al., 2005; Lin and Beal, 2006) and has been proposed to play a role in axon degeneration (Press and Milbrandt, 2008; Fischer and Glass, 2010; Calixto et al., 2012), we determined whether mitochondrial redox state was affected by axotomy, using the ratiometric, redox-sensitive roGFP2 (Hanson et al., 2004) localized to mitochondria (Fig. 8A). The emission intensity of roGFP2 at two excitation wavelengths (∼400 nm and ∼475–490 nm) is a function of the redox state of the subcellular compartment to which it is localized: an increase in the 405/488 fluorescence ratio of roGFP2 reflects oxidation (Hanson et al., 2004). We evaluated mito-roGFP2 fluorescence before and after transecting wild-type axons with a laser. Mito-roGFP2 was rapidly and significantly oxidized in the distal axon after injury (2–10 mpa: 161.9 ± 13.1% initial) and remained elevated but stable until shortly before fragmentation, when it was further oxidized (Fig. 8B). In the proximal axon, by contrast, the early increase in oxidation was mild (2–10 mpa: 107.9 ± 10.5% initial). Later, mito-roGFP2 oxidation in the proximal axon fell below baseline, and was significantly lower than was observed in the distal axon at all later time points (Fig. 8C).

Mito-roGFP2 oxidation after injury correlates with axon degeneration. A, Transgenes were generated to express DsRed +/− WldS in sensory neurons and the redox-sensitive biosensor roGFP2 in the mitochondrial matrix. B, Mito-roGFP2 was rapidly oxidized in the distal axon after injury (2–10 mpa: 167.21 ± 13.91%, n = 16 axons), and remained elevated but stable for most of the lag period before fragmentation. Shortly before fragmentation of the distal axon, however, mito-roGFP2 was further oxidized (90–120 mpa: 289.62 ± 57.79%; n = 5 axons). In the distal axon of WldS-expressing cells, mito-roGFP2 was slightly oxidized acutely after axotomy (2–10 mpa: 121.8 ± 14.62%; n = 22 axons); however, this effect was still significantly lower than mito-roGFP2 oxidation in wild-type axons (p < 0.05, two-way ANOVA with Bonferroni post-tests), and WldS entirely suppressed the long-term oxidation (p < 0.001). C, Mito-roGFP2 was not significantly oxidized in the proximal axon any point after axotomy (2–10 mpa: 107.9 ± 10.49%; 10–30 mpa: 102.6 ± 11.13%; 50–90 mpa: 91.93 ± 12.21%). Later in the lag phase, mito-roGFP2 oxidation returned to baseline levels in WldS-expressing axons, and was significantly lower than was observed in the distal axon at all time points (p < 0.001).
To determine whether WldS expression might regulate mitochondrial redox state, we examined mito-roGFP2 fluorescence in severed WldS-expressing axons. Unlike the change in wild-type axons, mito-roGFP2 was only mildly and temporarily oxidized after injury in WldS-expressing axons (2–10 mpa: 121.0 ± 14.6% initial), and this effect was significantly lower than that seen in wild-type (Fig. 8B; p < 0.05). At later time points, mito-roGFP2 oxidation returned to baseline in WldS-expressing axons, and was persistently lower than in wild-type axons (Fig. 8B,C; WT 10–50 mpa: 162.4 ± 10.7% initial; WldS: 96.0 ± 8.3%; p < 0.0001). We also monitored roGFP2 oxidation in control and PINK1 morphants at 2 dpf. PINK1 knock-down did not suppress the redox changes after injury (CON MO 10–50 mpa: 145.5 ± 14.0%; PINK1 MO: 127.5 ± 14.5%; p = 0.380).
Together, these results demonstrate that injury changes the oxidation state of axonal mitochondria, and the ability to prevent that change correlates with axon protection.
Previous studies have indicated that mitochondria are more depolarized in PINK1 morphants than controls (Anichtchik et al., 2008). To determine whether PINK1 knockdown affected WldS-mediated protection, we expressed WldS in PINK1 and control morphants, and evaluated axonal survival after axotomy. In WldS-expressing control morphants, all axons (n = 10) remained intact 24 h after axotomy. In PINK1 morphants, 1/10 axons degenerated within 12 h after axotomy. By 24 hpa, however, an additional 3/10 had undergone fragmentation. PINK1 knock-down therefore reduces the protective effect of WldS in transected axons. These results support the hypothesis that mitochondrial function may be relevant to axon protection by WldS.
PGC-1α delays axon degeneration and roGFP2 oxidation after injury
The transcriptional coactivator PGC-1α regulates mitochondrial biogenesis (Wu et al., 1999; Lehman et al., 2000) and ROS detoxification (St-Pierre et al., 2006). Because mitochondrial redox state correlated with axon degeneration, we tested whether PGC-1α overexpression could affect WD. We generated reporters to express mouse PGC-1α and either mitoDsRed or mito-roGFP (Fig. 9A). PGC-1α expression increased mitochondrial content (Fig. 9B–F): PGC-1α-expressing axons had a greater density of mitochondria (Fig. 9F), and mitochondria had a dramatically elongated morphology (Fig. 9C,E,G), likely reflecting an increase in mitochondrial biogenesis and/or fusion. PGC-1α had no effect on baseline mitochondrial transport (WT % motile: 19.62 ± 4.82%; PGC-1α: 22.157 ± 3.82%; n ≥ 19 axons; p = 0.315).

PGC-1α alters axonal mitochondrial properties and delays degeneration. A, Transgenes used to image mitochondria in PGC-1α-expressing neurons. B–G, Mouse PGC-1α expression in zebrafish sensory neurons caused a significant increase in mitochondrial content, quantified as a higher density of stationary mitochondria (F; WT: 110 ± 9.0 mitos/mm; PGC-1α: 161 ± 10.3 mitos/mm; n ≥ 9 axons) and an elongated mitochondrial phenotype (C, E, G; arrowheads in C′ and E′ point to elongated mitochondria). H, PGC-1α-expressing axons exhibited significantly less roGFP2 oxidation compared with wild-type immediately after axotomy (WT 2–10 mpa: 161.9 ± 13.14% initial; PGC-1: 79.98 ± 7.27% n ≥ 11 axons; p < 0.001) and later in the lag period (WT 10–50 mpa: 162.4 ± 10.65% initial; PGC-1α: 94.74 ± 14.99% initial; n ≥ 19 axons; p = 0.005). I, Quantification of mitochondrial transport after axotomy of PGC-1α-expressing axons. Mitochondrial transport was acutely and persistently reduced after axotomy in PGC1α-expressing cells (motility before: 30.72 ± 5.80%; 2–10 mpa: 16.83 ± 3.19%; 10–30 mpa: 10.52 ± 2.70%; 30–50 mpa: 12.30 ± 2.43%; 50–90 mpa: 9.77 ± 3.06%; 90–120 mpa: 2.32 ± 1.60%; n ≥ 9 axons; p < 0.001). However, motility after injury was significantly higher than in wild-type axons at 2–10 mpa (p = 0.0069). This trend persisted throughout most of the lag period, though it was not always statistically significant (10–30 mpa, p = 0.1485; 30–50 mpa, p = 0.0020; 50–90 mpa, p = 0.1751). Asterisks represent significance compared with wild-type. J, Changes in mitochondrial transport and function correlated with a delay in axon degeneration, with a longer lag period before fragmentation in injured PGC-1α-expressing axons (WT: 94.6 ± 4.7 min; PGC1α: 176.8 ± 16.6 min; n ≥ 15 axons; p < 0.0001). K, Mitochondrial roGFP oxidation data from wild-type and PGC-1α-expressing axons at time points between 10 and 30 min after injury were pooled. Linear regression analysis revealed a negative correlation between roGFP2 oxidation and the lag period before degeneration (R2 = 0.2855; p = 23 axons; p = 0.009). Scale bars: B, C, 50 μm; D, E, 20 μm.
To determine whether PGC-1α improved axon survival after injury, we axotomized PGC-1α-expressing sensory neurons, which exhibited markedly decreased mito-roGFP2 oxidation after injury compared with wild-type (Fig. 9H). This result suggests that PGC-1α might help regulate mitochondrial redox homeostasis. PGC-1α did not prevent acute reduction in mitochondrial transport (percentage motile before: 30.72 ± 5.80%; 2–10 mpa 16.83 ± 3.19%, p = 0.040, n = 10 axons), and motility remained significantly reduced after injury (Fig. 9I). Despite this reduction, however, mitochondrial motility after injury was higher in PGC-1α-expressing axons, compared with wild-type axons (Fig. 9I). Like WldS, PGC-1α therefore mitigated the effects of axonal injury on mitochondrial transport and function. To determine whether these effects correlated with axon protection, we monitored axonal integrity after injury. PGC-1α delayed the onset of fragmentation relative to wild-type (Fig. 9J). To quantify this correlation, we combined roGFP2 oxidation data from wild-type and PGC-1α-expressing mitochondria at 30 mpa, and compared these values to the lag period for each axon (Fig. 9K). Linear regression analysis confirmed that the correlation was significant (R2 = 0.2855; p = 0.009). These results suggest that effects on mitochondrial oxidation state are functionally relevant to axon degeneration and protection.
Discussion
In vivo imaging of mitochondrial transport and oxidation state after injury
Mitochondrial defects are associated with multiple neurodegenerative disease processes (Sheng and Cai, 2012) but the extent to which different mitochondrial functions contribute to degeneration is unclear. We have described a model that allows mitochondrial transport and physiology to be visualized noninvasively in zebrafish larvae. Like the recently described MitoFish (Plucińska et al., 2012), in which zebrafish mitochondria were labeled with CFP, mitoDsRed expression in zebrafish peripheral sensory neurons permits quantitative analysis of mitochondrial morphology and transport during development and neurodegeneration. We also used mito-roGFP2 (Hanson et al., 2004) to monitor dynamic changes in mitochondrial redox state in a living, vertebrate system. The development and characterization of these and other genetically encoded reporters in zebrafish neurons (Drerup and Nechiporuk, 2013) may provide insight into many biological processes underlying axon degeneration and protection.
Mitochondrial motility can be dissociated from axonal integrity after injury
Mitochondrial transport defects are associated with many forms of neurodegeneration (Coleman and Perry, 2002), including WD of injured Drosophila axons (Avery et al., 2012). The extent to which such defects are instructive in degeneration, however, is unclear. Mitochondrial, but not vesicular, transport terminated in zebrafish peripheral sensory neurons after injury. Transport in the proximal axon persisted, though transport behaviors shifted to favor retention of mitochondria at the injury site. Because the proximal axon does not degenerate, this correlation raised the possibility that mitochondrial arrest might underlie axon degeneration. Further experiments, however, dissociated mitochondrial transport from axon degeneration. In contrast to recent findings in flies (Avery et al., 2012), we detected no difference in overall mitochondrial motility in uninjured cells expressing the axon-protective protein WldS. Moreover, transport arrest was observed both acutely and several hours after injury in WldS-expressing axons. This result confirmed that, as was recently observed in a neurodegenerative disease model system (Marinkovic et al., 2012), long-term mitochondrial transport is not required for axon survival, and revealed that long-term mitochondrial transport preservation does not mediate the protective effect of WldS.
Phases of mitochondrial transport arrest
A more detailed analysis of mitochondrial transport in wild-type and WldS-expressing axons allowed us to distinguish different phases within the lag period, and revealed a transient effect of WldS on mitochondrial transport. The first phase occurred immediately after injury and was not suppressed by WldS. This acute arrest was limited to the region adjacent to the injury site, and may be mediated by rapid calcium influx (Yawo and Kuno, 1985; Ziv and Spira, 1995; Adalbert et al., 2012; Avery et al., 2012). Calcium-induced mitochondrial transport arrest is an adaptive response promoting local calcium buffering and ATP synthesis at sites of high demand (Saotome et al., 2008; Macaskill et al., 2009; Wang and Schwarz, 2009) and may minimize acute damage. PGC-1α-expressing cells also exhibited reduced transport after injury, though to a lesser degree than in wild-type axons. Because PGC-1α increases mitochondrial density, it is possible that sufficient calcium buffering is provided by the same number, but a smaller percentage, of mitochondria.
Observation of mitochondria later in the lag phase revealed a temporary recovery of transport in WldS-expressing axons. This recovery did not occur in wild-type axons, suggesting that it might be relevant to axon protection. Mitochondrial distribution, which is relevant in another disease model (Misko et al., 2012), was not affected by WldS expression at baseline or after injury. It is therefore unlikely that early transport recovery provided long-term protection by regulating mitochondrial spacing. The early effects of WldS on transport may confer protection by promoting a more robust network of mitochondria. Indeed, transport behaviors in this recovery phase favored mitochondrial retention at the cut site, mirroring mitochondrial motility in the proximal axon after injury. These transport behaviors may provide a selective benefit, providing local calcium buffering and energy production that support the entire mitochondrial network. By contrast, PINK1-deficient mitochondria, which are more depolarized (Anichtchik et al., 2008) and more vulnerable to oxidative stress (Wang et al., 2006; Mai et al., 2010) than controls, also exhibit a moderate recovery of transport after injury. However, axons in PINK1 morphants degenerate more rapidly than in control morphants. The mitochondrial transport recovery in PINK1 morphants is therefore not equivalent to that observed in WldS-expressing axons. This may reflect a functional difference between mitochondria in PINK1-deficient and WldS-expressing axons, either at baseline or after injury.
Dynamic changes in mitochondrial redox state after injury
Mitochondrial transport eventually terminated throughout the distal axon in WldS-expressing cells, and did not recover. Mitochondria were immobile 24 h after injury despite persistence of the distal axon. This finding does not preclude the possibility that the early and transient recovery of mitochondrial transport provides long-term effects, but suggests that other aspects of mitochondrial function may be more critical at later stages. To determine whether ROS production might be relevant to axon degeneration and protection, we evaluated mitochondrial redox state before and after axotomy using the redox-sensitive biosensor mito-roGFP2 (Hanson et al., 2004). After axotomy, mito-roGFP2 was rapidly oxidized in the disconnected axon branch, consistent with increased ROS production in the mitochondrial matrix. This effect was also observed in the distal axons of control and PINK1 morphants, but not in wild-type proximal axons (still attached to the cell body) or in WldS-expressing axons. Because mitochondrial ROS production can be induced by calcium (Yan et al., 2006), increased calcium buffering by WldS-expressing mitochondria (Avery et al., 2012) could contribute to this difference. It is also possible that WldS directly affects mitochondrial ROS production. Consistent with this possibility, overexpression of Nmnat (the critical catalytic component of the WldS fusion protein) prevents degeneration of cultured neurons exposed to hydrogen peroxide (Press and Milbrandt, 2008). Moreover, antioxidant treatment protects against Wallerian-like degeneration in Caenorhabditis elegans and mice (Calixto et al., 2012).
Oxidation of roGFP2 remained elevated but stable in wild-type distal axons for most of the lag phase. Shortly before axon fragmentation, however, mito-roGFP2 oxidation rose further. This increase, which was not observed in WldS-expressing axons, may reflect formation of the mPTP, which occurs under conditions of calcium overload (Haworth and Hunter, 1979; Hunter and Haworth, 1979a,b), particularly in the presence of ROS (Kowaltowski et al., 1996; Vercesi et al., 1997). The permeability transition results in further ROS production (Hansson et al., 2008), loss of membrane potential, and calcium efflux into the cytoplasm, where it activates calcium-dependent cysteine proteases that degrade the cytoskeleton (George et al., 1995).
PGC-1α expression delays mito-roGFP2 oxidation and axon degeneration
The transcriptional coactivator PGC-1α has many targets that regulate mitochondrial biogenesis, bioenergetics, and ROS detoxification (Wu et al., 1999; Lehman et al., 2000; St-Pierre et al., 2006; Cantó and Auwerx, 2009). Impairments in PGC-1α-mediated transcription have been associated with Huntington disease and Parkinson disease, and PGC-1α has been proposed as a therapeutic target (Cui et al., 2006; Weydt et al., 2006; Zheng et al., 2010; Shin et al., 2011; Tsunemi and La Spada, 2012). PGC-1α expression delayed axon degeneration of zebrafish sensory neurons after transection. Because the distal axon is not connected to the cell body after axotomy, injury-induced transcriptional activation by PGC-1α cannot contribute to this protection. However, PGC-1α increased mitochondrial density, and a larger number of mitochondria may be able to buffer more calcium. Additionally, PGC-1α may upregulate ROS detoxification programs at baseline, making mitochondria resistant to ROS buildup after injury. Indeed, PGC-1α overexpression protects cultured neurons from ROS-mediated degeneration, an effect associated with increased mRNA expression of multiple enzymes involved in ROS detoxification (St-Pierre et al., 2006). Nevertheless, detachment from the cell body prevents the distal axon from enjoying further effects of PGC-1α activation after injury. This limitation could explain why the delay of WD in PGC-1α-expressing axons, although significant, was not comparable to that provided by WldS expression. In many neurodegenerative disease models, axon degeneration proceeds via a WD-like mechanism without physical separation from the cell body. PGC-1α overexpression may provide improved protection in these models, as axons could still benefit from upregulation of protective gene expression after the initial insult.
Footnotes
K.C.O. was supported by a training grant from the UCLA Training Program in Neural Repair (NINDS T32 NS07449:13). M.E.V. was supported by the Jules Stein EyeSTAR Training Program. This work was supported by grants to A.S. from the National Institutes of Dental and Craniofacial Research (RO1 DE018496), and the American Parkinson Disease Association Pilot Fund (20082501). We thank Meghan Johnson and Carla Koehler for the mitoDsRed and mito-roGFP constructs and Jeffrey Rasmussen for comments on the paper.
- Correspondence should be addressed to Dr Alvaro Sagasti, University of California Los Angeles, 621 Charles East Young Drive South, Los Angeles, CA 90095. sagasti{at}mcdb.ucla.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}