

Abstract
The principle by which unmyelinated primary sensory neurons transducing thermal, itch and pain perception are specified in early development is unknown. These classes of sensory neurons diversify from a common population of late-born neurons, which initiate expression of Runt homology domain transcription factor RUNX1 and the nerve growth factor receptor TrkA. Here, we report that signals emanating from within the mouse dorsal root ganglion mediated partly by early-born neurons destined to a myelinated phenotype participate in fating late-born RUNX1+/TrkA+ neurons. Inductive factors include FGFs via activation of FGF receptor 1 (FGFR1). Consistently, FGF2 is sufficient to induce expression of RUNX1, and Fgfr1 conditional mutant mice display deficits in fating of the common population of late-born RUNX1+/TrkA+ neurons that develop into unmyelinated neurons. Thus, the distinct lineages of sensory neurons are acquired in response to increasing FGF levels provided by a rising number of born neurons.
Introduction
The existence of distinct types of sensory neurons with different response profiles to stimuli underlies the ability for discrimination between different modalities of sensation and allows for the aware perception of the environment by the somatic sensory system. Depending on the modality of sensation, dedicated neuronal types arise from different branches in a hierarchical process of diversification from a common progenitor during development. All DRG sensory neurons are formed from neural crest cells (NCCs) undergoing neurogenesis under the control of either the bHLH transcription factor Neurogenin 2 (NGN2) or NGN1, with the former appearing slightly earlier in development than the latter (Ma et al., 1999; Perez et al., 1999; Bertrand et al., 2002; Lo et al., 2002; Lallemend and Ernfors, 2012).
A vital function of the somatosensory system is providing information about the occurrence or threat of injury. Hence, the sensation of pain by its aversive nature contributes to the capacity to detect and protect from potentially damaging stimuli. Unmyelinated DRG neurons arise from neurogenesis induced by NGN1 during mid-gestation in mouse, and the resulting precursors differentiate progressively to subdivide into functionally distinct populations of neurons responsible for transducing pain (nociceptors), itch (pruriceptors), and temperature (thermoceptors) (Lallemend and Ernfors, 2012). Although the NGN2-mediated wave of neurogenesis results almost exclusively in myelinated neurons, both myelinated and unmyelinated neurons within the DRG develop from the NGN1-induced neurogenesis. Thus, the Neurogenins do not appear to specify sensory neurons to distinct subtypes (Zirlinger et al., 2002; Marmigère et al., 2006). Instead, the fate of unmyelinated sensory precursors is defined by the tropomyosin-receptor kinase A (TrkA) expression, which is induced in close association to cell cycle exit of NCCs and neurogenesis. Shortly thereafter starts expression of the runt-related transcription factor 1 (RUNX1) (Kramer et al., 2006). It is the lineage of TrkA+/RUNX1+ neurons that, in a developmental process extending well into postnatal life, segregates into distinct types of unmyelinated neurons. Consistently, TrkA signaling and RUNX1 activities play critical roles for development of defining functional characteristics of unmyelinated neurons (Patel et al., 2000; Chen et al., 2006; Luo et al., 2007; Abdel Samad et al., 2010; Lopes et al., 2012).
Although much is known about the central role of RUNX1 and TrkA for the progressive segregation of immature precursor neurons into distinct types, the mechanisms that instruct and define the lineage of unmyelinated neurons have not been addressed previously. The strict temporal appearance of presumptive myelinated and unmyelinated precursor neurons during development argues that this might not be a stochastic process (Frank and Sanes, 1991; Ma et al., 1999; Maro et al., 2004). Here, we tested the hypothesis that fate decision and phenotypic differentiation of unmyelinated sensory neurons are governed by instructive cues. Such cues would be predicted to result from spatial and temporal changes in environmental signals surrounding or from within the coalescing DRG. For this purpose, we developed a candidate approach where soluble signals known to be expressed within, or in tissues associated with, the coalescing DRG were examined for their ability to induce RUNX1. Our results show that FGFs expressed within the DRG, via MAPK/ERK and PI3K pathways, induce RUNX1 expression in early sensory neurons. This inductive activity of FGFs correlates with the dynamics of expression of Fgf2 and Fgfr1 in vivo during DRG development. Consistently, conditional mutant mice for Fgfr1 display deficits during development of RUNX1 and TrkA-expressing neurons.
Materials and Methods
Animals.
Chick embryos were staged according to Hamburger and Hamilton stages. In our experiments, embryonic day (E) 4.5, 5, 5.5, 6, and 6.5 correspond to Hamburger and Hamilton stages 25, 27, 28, 29, and 30, respectively. Wild-type (WT) C57BL/6 mice were used unless specified otherwise. Wnt1Cre (Danielian et al., 1998), Rosa26Tomato strain (Gt(ROSA)26tm9(CAG-tdTomato), Rosa26YFP) were obtained from The Jackson Laboratory, and TrkCCre, Isl2DTA, and Fgfr1lox/lox have been described previously (Yang et al., 2001; Trokovic et al., 2003; Fünfschilling et al., 2004). Animals of either sex were included in this study. All experiments were approved by the regional research ethics committee.
DRG culture.
Chick or mouse DRGs were microdissected and cultured as explants on Growth Factor Reduced Matrigel (BD Biosciences, 5% in PBS) coated coverslips in a medium consisting of DMEM/F-12 GlutaMax supplemented with 10% FBS, 0.2% penicillin/streptomycin, and 20 μg/ml gentamicin (all from Invitrogen). For immunofluorescence, DRGs were fixed 20 min on ice (4% PFA in PBS), washed with PBS, and processed for immunostaining. For AraC experiments, lumbar DRGs, which develop with a 12–24 h delay compared with brachial regions, were used to ensure that precursor cells in the DRGs were eliminated long before RUNX1 is induced. NGF, neurotrophin-3 (NT3), GDNF, hepatocyte growth factor (HGF), Sonic hedgehog (Shh), IGF1, and FGFs were obtained from R&D Systems and used at the following concentrations if not stated otherwise: 50 ng/ml for NGF, NT3, GDNF, and FGFs; 20 ng/ml for IGF1 and HGF; and 1 μg/ml for Shh. Inhibitors from Calbiochem were used at the following concentrations: AraC, 10 μm; PD98059 and LY294002, 10 μm (similar results obtained with 20 μm); staurosporine, 10 nm; PD166866 and picropodophyllin (PPP), 1 μm. Phorbol 12,13-dibutyrate (Sigma) was used at 0.1 μm. Each in vitro experiment included the analysis of 4 or 5 DRGs and was repeated at least twice.
qPCR.
Total RNA from cultured or freshly dissected embryonic mouse or chick DRGs (20–80 DRGs per sample) was extracted using the RNeasy Mini kit (QIAGEN) following the manufacturer's instructions, including DNase digestion on the column. Reverse transcription was performed using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems) according to manufacturer's instructions from 1 μg of total RNA. Negative controls were prepared by excluding the reverse polymerase. Real-time quantitative PCR (qPCR) was conducted in a 10 μl reaction containing 1 μl of 1:100 dilution of reverse transcription reaction, 5 μl of SYBR Green PCR Master Mix (Applied Biosystems), and 0.5 μl of each primer (5 μm; MWG-Biotech AG). UDG was incubated for 10 min at 50°C, and cDNA was denatured for 10 min at 95°C and amplified in 40 cycles in a two-step program as follows: 15 s at 95°C; 60 s at 60°C. Primers used were the following: ATPsy (Gallus gallus), forward, 5′GGTCTGTTAGATCATCTGCTGGC3′, reverse, 5′ACGCAAAGGCTCCATCACTTC3′; RUNX1 (Gallus gallus), forward, 5′AACCAAGTCGCGAGGTTCAA3′, reverse, 5′TTGATGGCTCTGTGGTACGTG3′; ER81 (Gallus gallus), forward, 5′CAATGTCAGTGCCTATGATCAGAA3′, reverse, 5′GGTGACACAGGAGTGCTGCAT3′; Tbp (Mus musculus), forward, 5′GGGGAGCTGTGATGTGAAT3′, reverse, 5′CCAGGAAATAATTCTGGCTCA3′; Runx1 (Mus musculus), forward, 5′CAGGTAGCGAGATTCAAC3′, reverse, 5′TTTGATGGCTCTATGGTAGGT3′; Fgf1 (Mus musculus), forward, 5′AGTTCTTCAGTGCTGAGCCTA3′, reverse, 5′GGTTGAACCTCTCGGTCAGG3′; Fgf2 (Mus musculus), forward, 5′GAGGAGTTGTGTCTATCAAGG3′, reverse, 5′ATCCGAGTTTATACTGCCCA3′; Fgf3 (Mus musculus), forward, 5′CTACCAAGTACCACCTCCAG3′, reverse, 5′GTAGTGATCCGAAGCATACAG3′; Fgf4 (Mus musculus), forward, 5′GACAGTCTTCTGGAGCTCTC3′, reverse, 5′CACAGTCTAGGAAGGAAGTGG3′; Fgf5 (Mus musculus), forward, 5′AATATTTGCTGTGTCTCAGGG3′, reverse, 5′CTGAACTTACAGTCATCCGT3′; Fgf6 (Mus musculus), forward, 5′TTGCCATGAACAGTAAAGGA3′, reverse, 5′TTACCCGTCCATATTTGCTC3′; Fgf7 (Mus musculus), forward, 5′ACCCAGGAGATGAAGAACAG3′, reverse, 5′TTTGCATAGAGTTTCCCTTCC3′; Fgf8 (Mus musculus), forward, 5′GCTCATTGTGGAGACCGA3′, reverse, 5′CGCCGTGTAGTTGTTCTC3′; Fgf9 (Mus musculus), forward, 5′GGAATTTATCAGTATAGCAGTGGG3′, reverse, 5′GTTTATAGAGGTTGGAAGAGTAGG3′; Fgf10 (Mus musculus), forward, 5′ACCAACTGCTCTTCTTCCTC3′, reverse, 5′CAACAACTCCGATTTCCACTG3′; Fgf11 (Mus musculus), forward, 5′CATCGTCACCAAACTGTTCTG3′, reverse, 5′GTAATTCTCAAAGACGCACTCC3′; Fgf12 (Mus musculus), forward, 5′ATGTGATCTATTCCTCAACCCT3′, reverse, 5′CGTGTAGTGATGGTTCTCTG3′; Fgf13 (Mus musculus), forward, 5′TTACCAAACTATACAGCCGA3′, reverse, 5′TTGAACTCCTTGAATAGCCA3′; Fgf14 (Mus musculus), forward, 5′GAAGGGCAAGTTATGAAAGG3′, reverse, 5′ATCATGCAAGGATGGTTCTC3′; Fgf15 (Mus musculus), forward, 5′ATGTCTCCAACTGCTTCCTC3′, reverse, 5′AAACAGTCCATTTCCTCCCT3′; Fgf16 (Mus musculus), forward, 5′TGGATCGAAGAAACTCACAC3′, reverse, 5′TTTAGTCCTGTATCCCTCCC3′; Fgf17 (Mus musculus), forward, 5′GGCAAATCCGTGAATACCAG3′, reverse, 5′CCGAATGTATCTGTCTCCAC3′; and Fgf18 (Mus musculus), forward, 5′TCTACCTGTGTATGAACCGA3′, reverse, 5′TGACTGTGGTGTATTTGAAGG3′; Fgf20 (Mus musculus), forward, 5′CACAGTCTCTTCGGTATCCT3′, reverse, 5′CCAGTTCTCTTCAAATTGTTCC3′; Fgf21 (Mus musculus), forward, 5′CTCTACACAGATGACGACCA3′, reverse, 5′GACACCCAGGATTTGAATGAC3′; Fgf22 (Mus musculus), forward, 5′ATAGTGGAGATCCGTTCTGTC3′, reverse, 5′TCAAGACGAGACCAAGACTG3′; Fgf23 (Mus musculus), forward, 5′GGACCAGCTATCACCTACAG3′, reverse, 5′GATCCATACAAAGGAACCTTCG3′; Igf1 (Mus musculus), forward, 5′GTCTTCACACCTCTTCTACC3′, reverse, 5′TACATCTCCAGTCTCCTCAG3′; and Igf2 (Mus musculus), forward, 5′GGTGCTTCTCATCTCTTTGG3′, reverse, 5′ ACTCTTCCACGATGCCAC 3′.
Experiments were performed in triplicates. Quantification was done using the ΔΔCt method (Pfaffl, 2001), relative to the reference genes (Tbp for mouse or ATPsy for chicken).
Immunostaining.
Animals were collected, decapitated, and fixed 1–6 h on ice (4% PFA in PBS) depending on the stage, washed in PBS, cryopreserved in 30% sucrose in PBS, embedded in OCT (Tissue-Tek), and sectioned at 14 μm. Sections or DRGs (after culture) were incubated 48 h at 4°C with primary antibodies diluted in blocking solution (2% donkey serum, 1% Triton X-100 in PBS). Primary antibodies used were as follows: rabbit anti-RUNX1 (Chen et al., 2006), goat anti-TrkA (R&D Systems; AF1056, 1:500), rabbit anti-TrkA (Millipore, 06–574, 1:500), mouse anti-ISL1 (Developmental Studies Hybridoma Bank; 39.4D5; 1:200), goat anti-RET (R&D Systems; AF482), goat anti-TrkC (R&D Systems; AF1404), and mouse anti-NF200 (Sigma, N0142; 1:500). After washing with PBS, Alexa Fluor secondary antibodies (Invitrogen; 1:500 in blocking solution) were applied overnight (at 4°C). Samples were then washed in PBS and mounted in Dako fluorescent mounting medium. Staining was documented by confocal microscopy (Carl Zeiss LSM 5 EXCITER) using identical settings between control and experimental images. Optical sections were 2 μm in 20× overview pictures. Whole-mount immunofluorescent staining was performed as previously described (Huber et al., 2005), and z-stacks were collected and 2D projections were created using the Zeiss LSM image processing software.
In situ hybridization.
Tissue was prepared as for immunofluorescence. Before hybridization, slides were air dried for 2–3 h at room temperature. Plasmids containing 750 bp specific regions of Fgf2, Fgf11, Fgf13, Fgf18, Fgfr1, Fgfr2, Fgfr3, Fgfr5, Igf1, and Igf2 were synthesized by Epoch Life Science. A 750 bp region of Fgf1 was cloned from cDNA of adult DRG and TOPO-cloned into pCRII (Invitrogen). T7 polymerase (Promega) was used to synthesize digoxigenin-labeled antisense riboprobes according to the manufacturer's instructions (Roche) and purified by LiCl precipitation. Sections were processed as previously described (Marmigère et al., 2006). Slides were washed in PBS before immunostaining.
EdU injections.
Intraperitoneal injection of pregnant females with EdU (100 mg/kg, Invitrogen) was performed at E10 and E12 d of gestation. Injected females were killed 2 h after injection for analysis. EdU incorporation was subsequently resolved using Alexa Fluor 488 azide according to the manufacturer's instructions (Invitrogen).
Image analysis.
Image analysis was done using the ImageJ software. For quantification of RUNX1-positive cells in culture, a macro was developed to calculate the average intensity of RUNX1 signal over pixels positive for ISL1. For sections, single-cell analysis was performed for every positive cell, and average intensity per cell (in arbitrary units) was calculated for each section from different DRG of particular axial levels of different animals.
Results
A time-dependent signaling from early-born neurons participates in induction of RUNX1 expression in later-born sensory neurons
To search for soluble signals with RUNX1-inductive properties, we first established the precise temporal developmental sequence of initiation of RUNX1 expression in vivo in the chick and mouse. In embryonic chicken DRG, transcripts for RUNX1 were first detected at very low levels at E5, consistent with previous data (Lallemend et al., 2012) and gradually increased to reach a maximum at E6.5 (Fig. 1A). In mouse DRG, Runx1 expression was virtually absent at E11.5, increased to moderate levels at E12.5, and reached very high levels at E13.5 (Fig. 1B). The levels of Runx1 mRNA correlated with actual presence of RUNX1 protein, which was absent at E11.5 and clearly observed in large numbers of TrkA+ cells at E12.5, with variable intensity of staining (Fig. 1C), as previously described (Chen et al., 2006). The present study was focused on these RUNX1+/TrkA+ neurons, which generate the large majority, if not all, of unmyelinated sensory neurons. A small population of TrkA+ neurons that develop into thinly myelinated neurons is born along with the early-born low-threshold mechanoreceptors. These develop independent of RUNX1 and are observed as TrkA+ at E11.5 (Fig. 1C) (Bachy et al., 2011).
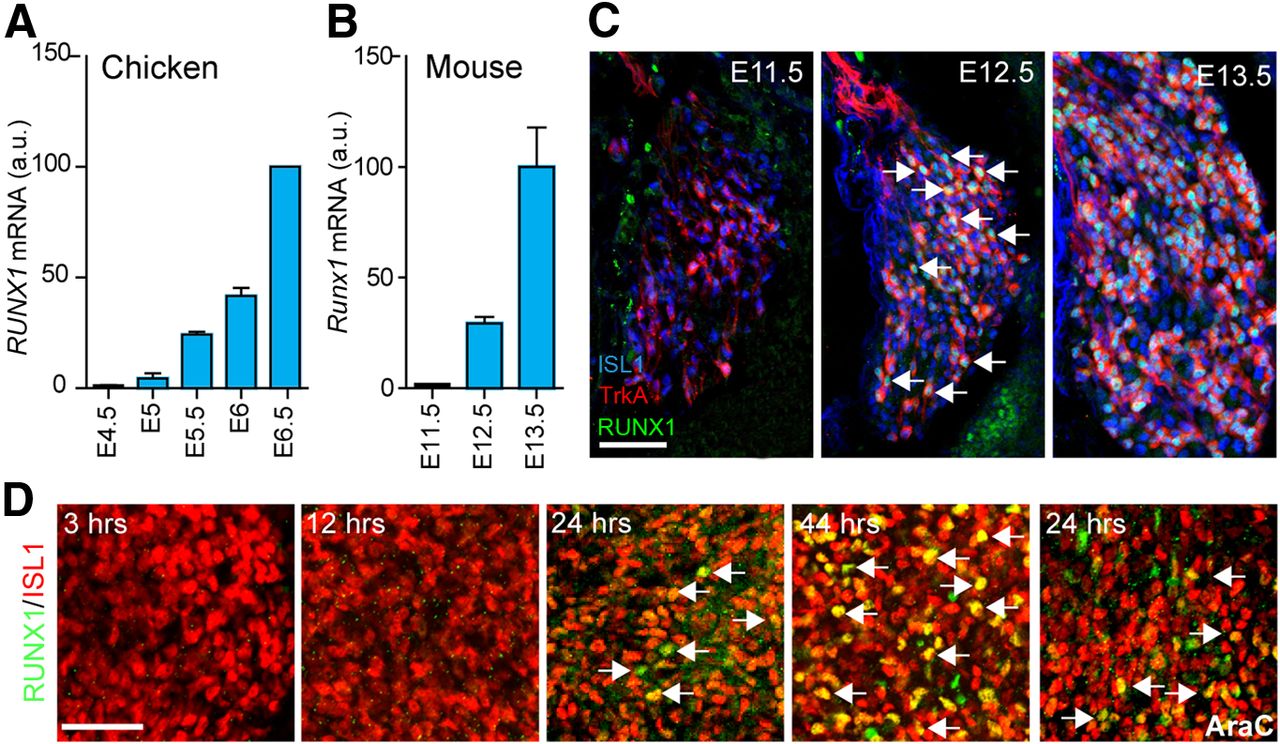
RUNX1 induction in chicken and mouse DRG. A, B, qPCR analysis of Runx1 mRNA relative expression in embryonic chicken (A) and mouse (B) brachial DRG shows the onset of expression at E5 in chicken and between E11.5 and E12.5 in mouse. Data are presented as mean ± SEM. C, Triple immunostaining for ISL1, TrkA, and RUNX1 on brachial DRG sections from mouse embryo showing RUNX1 protein first detected at E12.5 in cells that already express TrkA and at E13.5 in a large majority of TrkA-positive cells. D, DRG cultures from E11.5 mouse recapitulate the dynamics of RUNX1 induction seen in vivo. Note expression of RUNX1 in neurons (arrows) after 24 h, which is not affected by AraC. a.u., Arbitrary units. Scale bars: C, D, 50 μm.
We examined whether the precise temporal profile of RUNX1 expression and hence, commitment for development into unmyelinated DRG neurons, is acquired in the DRG independent of putative signals from adjacent tissues. For this purpose, we cultured brachial DRG isolated from E11.5 mouse embryos for 3, 12, 20, and 44 h with the pan-caspase inhibitor Q-VD-OPh alone (i.e., without the addition of any soluble signals). A small number of cells with induction of RUNX1 at low levels were observed at 12 h with increasing numbers and levels at 24 and 44 h in vitro, with ∼70% of the ISL1+ neurons being RUNX1+ throughout the ganglia at the 44 h time point (Fig. 1D). To distinguish between a possible intrinsic mechanism and exogenous factors inducing RUNX1, we analyzed the expression of RUNX1 in lumbar DRG neurons from E11.5 embryos cultured for 24 h in the presence of AraC, which swiftly removes dividing progenitor cells that may affect non–cell-autonomously the induction of RUNX1 in DRG neurons. AraC did not affect induction of RUNX1 (Fig. 1D).
We next examined whether early-born DRG neurons that develop into myelinated neurons and originate from the first wave of neurogenesis instruct acquisition of RUNX1 in later-born neurons diversifying into unmyelinated neurons. We therefore made use of a TrkCCre mouse line (Fünfschilling et al., 2004). Analysis of P0 TrkCCre;Rosa26YFP offspring generated by breeding TrkCCre mice to the Rosa26YFP reporter strain (Srinivas et al., 2001) revealed all cells that at any stage expressed Cre from the TrkC locus (Fig. 2A,B). All TrkC+ neurons were YFP+; in addition, many YFP+ cells were found, which were TrkC− (Srinivas et al., 2001; Fünfschilling et al., 2004). Consistent with an expression of Cre primarily in presumptive myelinated neurons, TrkA+/NF200− cells, including RUNX1+ cells, were largely negative for YFP, whereas nearly all NF200+ neurons (including TrkC+/RET−, RET+/NF200+, TrkB+/RET−, TrkA+/NF200+ populations) were YFP+, although rarely some NF200+ neurons, mostly TrkA+, were YFP− (Fig. 2A,B). To establish a role of early-born TrkC-expressing neurons for development of later-born unmyelinated neurons, we generated TrkCCre;Isl2DTA embryos, in which the expression of the Cre recombinase in the majority of the neurons generated between E10 and E11 allowed for the expression of the diphtheria toxin in most or all of these early-born sensory neurons, resulting in their death before induction of Runx1 in the later-born TrkA+ neurons. Consistent with the YFP reporter results, E12.5 TrkCCre;Isl2DTA embryos displayed a selective loss of virtually all presumptive myelinated neurons, including TrkC+, RET+, and TrkB+ neurons, which represent partly different classes of sensory neurons (i.e., proprioceptive and various low threshold mechanoreceptive neurons) (Fig. 3A) (Lallemend and Ernfors, 2012). In contrast, TrkA+ and RUNX1+ neurons were present, although in reduced numbers (Fig. 3A,B). Quantification revealed a loss of 42% of ISL1+, 82% of RUNX1+, and 50% of TrkA+ neurons at E12.5 (Fig. 3B). Because nearly all myelinated neurons selectively are ablated in these mice, a proportional increase of TrkA+ and RUNX1+ neurons compared with all ISL1+ neurons is expected. However, the percentage of RUNX1+ and TrkA+ neurons among all ISL1+ neurons was 32.8% and 54.9% in WT mice and 10.5% and 47.4% in TrkCCre;Isl2DTA mice, respectively. Furthermore, the few remaining RUNX1+ neurons expressed significantly reduced levels of RUNX1 (Fig. 3A,B). Thus, RUNX1 and TrkA fail to be induced in some of the immature neurons, which normally should express RUNX1 and TrkA. The deficit in RUNX1 levels persisted until birth (Fig. 3A,C). Moreover, DRG neurons were virtually all TrkA+ in the mutant at birth, most of them expressing RUNX1, and their number was reduced by 64% compared with the control animals (Fig. 3A,C). Together, that Cre recombination is expected to occur only within ∼17% of this population when analyzing P0 TrkCCre;Rosa26YFP mice (Fig. 2B) and that ISL1+ neurons in mutant mice represent 58% at E12.5 and only 32% at P0 of control mice (Fig. 3B,C), an increased naturally occurring cell death of the late TrkA+/RUNX1+ neurons between E12.5 and P0 is suggested in the absence of the early-born neurons. As neuronal cell death in the developing nervous system depends on target-derived neurotrophins, we analyzed whether removal of early-born DRG neurons could impact the growth of peripheral axons. Such deficit could contribute to the excessive neuronal loss by limiting the access to growth factors in later-born neurons. At E14.5, TrkA+ axons project massively in WT forelimbs, innervating the tips of the embryonic digits and arborizing from the main trunks (Fig. 3D). In contrast, in TrkCCre;Isl2DTA embryos, the stereotypical projection pattern observed in control animals was dramatically reduced. Invariably, in the mutant, TrkA+ axons failed to extend to the distal forelimb and to arborize; in addition, the second, third, and fourth digits nearly completely failed to be innervated (Fig. 3D). Interestingly, in the mutant, the large-diameter myelinated axons (mostly motor axons) visualized by NF165 staining showed similar growth deficits (Fig. 3D), whereas ISL1+ motor neuron position and number within the ventral spinal cord were not affected in TrkCCre;Isl2DTA embryos at E12.5 (Fig. 3E). In accordance with these results, motor neurons in the ventral spinal cord were negative for TrkC expression from E9.5 until birth in WT mice (data not shown). Combined, these analyses reveal that early-born neurons contribute to specification of later-born TrkA+ neurons, including their normal levels of RUNX1 expression, and are necessary for the proper limb innervation.

Cre activity in DRG from TrkCCre;R26YFP mice. A, Triple immunostaining for YFP/TrkC/ISL1, YFP/RET/NF200, YFP/TrkA/NF200, and YFP/RET/TrkB on P0 brachial DRG sections from TrkCCre;R26YFP mice. Reporter expression in 100% of TrkC-expressing cells and in the majority of myelinated (NF200+) neurons. B, Pie chart summarizing reporter expression in the major types of DRG neurons. YFP expression in ∼88% and 17% of the myelinated and unmyelinated neurons, respectively. In the quantitative experiments, the different types of myelinated neurons have been classified according to their expression of TrkA, TrkB, TrkC, RET, and NF200. There is a small number of YFP+/TrkA+/NF200− cells in B. Quantification was performed in two different animals (five DRG sections/animal). Scale bar: A, 100 μm.

Early-born sensory neurons contribute to the development of later-born TrkA/RUNX1 neurons. A, Immunostaining for ISL1 and TrkC, RET, TrkB, TrkA, or RUNX1 (E12.5) and for TrkA/RUNX1 (P0) on brachial TrkCCre;Isl2DTA DRG sections. At E12.5, a complete absence of TrkC+ cells, very few if any RET+ and TrkB+ cells, and a decrease of TrkA+ and RUNX1+ cells. There is low expression of RUNX1 in the mutant animals (insets). B, C, Quantification of A at E12.5 (B) and P0 (C). Number of cells are in the left and intensity of RUNX1 fluorescence in the two right-most bars of the graphs. D, Whole-mount TrkA and NF165 (insets) immunostaining of E14.5 TrkCCre;Isl2DTA and WT embryos reveals severe impairments of forelimb innervation in the absence of early-born DRG neurons. Fiber extension and arborization throughout the limb are limited in the mutant embryos, with the lateral aspect of both the anterior (A) and posterior (P) regions of the limb and the second, third, and fourth digits lacking innervation. E, Immunostaining of brachial ventral neural tube shows similar pattern of ISL1+ motor neurons (MNs) in E12.5 TrkCCre;Isl2DTA and WT embryos. Data are presented as mean ± SEM. n = 2–4 animals per genotype. *p < 0.05 (two-tailed, unpaired t test, statistical comparison with WT). **p < 0.01 (two-tailed, unpaired t test, statistical comparison with WT). ***p < 0.001 (two-tailed, unpaired t test, statistical comparison with WT). a.u., Arbitrary units. Scale bars: A, D, E, 100 μm.
Induction of RUNX1 by FGFs and IGFs produced in the DRG
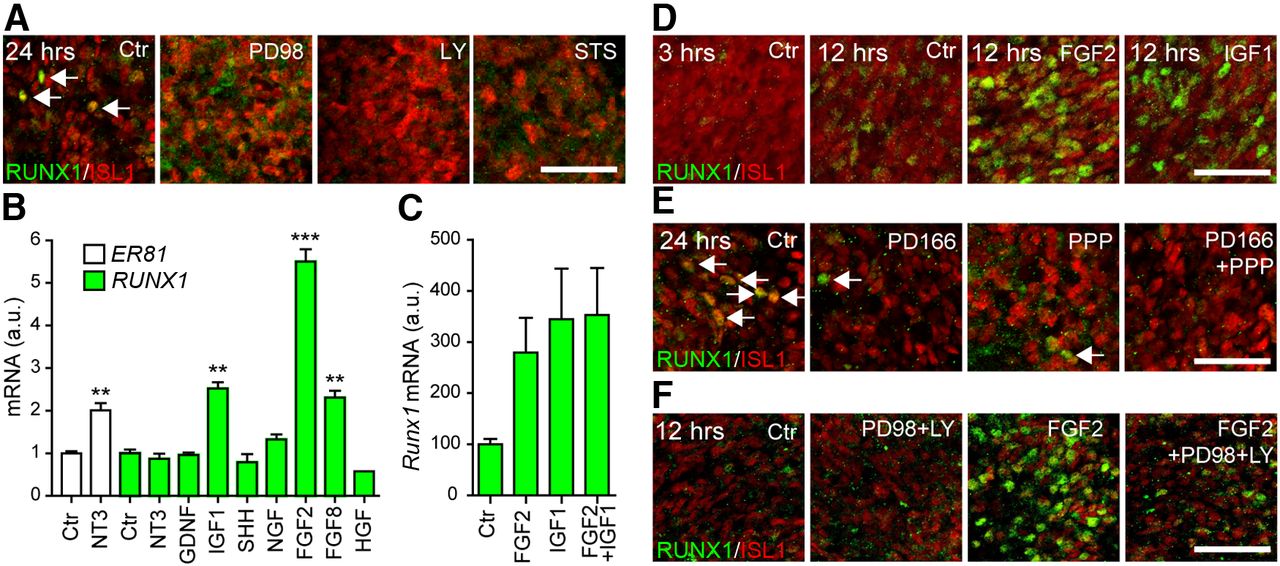
The fating of later-born DRG neurons by early neurons suggests the production of instructive molecules. Such molecules would be predicted to engage receptors resulting in intracellular signaling, which subsequently drives the acquisition of the RUNX1+ fate. To examine putative signaling pathways critical for development of RUNX1 expression, DRGs from E11.5 embryos were cultured for 24 h in the presence of various kinase inhibitors. Inhibition of MAPK pathway with PD98059 resulted in a reduction of RUNX1+ cells at 24 h of culture, whereas interfering with the PI3K pathway using LY294002 or with PKC activity using staurosporine led to a near complete block in development of RUNX1+ cells (24 h, Fig. 4A; similar observations at 48 h, data not shown).

FGF signaling induces RUNX1 in DRG cultures. A, Blocking of RUNX1 induction in E11.5 DRG cultures with inhibitor of MAPK/ERK and PI3K/Akt pathways (PD98059 and LY294002) indicates the involvement of a signaling factor for RUNX1 induction. B, FGF and IGF signaling increases RUNX1 mRNA in cultured E5 chicken DRG as measured by qPCR. ER81 upregulation by NT3 serves as a positive control. C, FGF and IGF signaling induces Runx1 mRNA expression also in mouse E11.5 brachial DRG cultures. D, Induction of RUNX1 protein in E11.5 brachial DRG 12 h after addition of FGF2 or IGF1. There is absence of RUNX1 expression in control condition (3 h in vitro). E, Inhibitors of FGF receptors (PD166866) and IGF receptors (PPP) block an endogenous induction of RUNX1 in 24 h cultures of E11.5 brachial DRG. F, FGF-induced expression of RUNX1 in E11.5 brachial DRG is reduced by PD98059 and inhibited by LY294002. Data are presented as mean ± SEM. **p < 0.005 (one-way ANOVA with Tukey's post hoc test, statistical comparison with control). ***p < 0.001 (one-way ANOVA with Tukey's post hoc test, statistical comparison with control). a.u., Arbitrary units; Ctr, control. Scale bars: A, D, E, and F, 50 μm.
We took a candidate approach to identify signaling molecules that induce RUNX1. For this purpose, we used E5 chick DRGs, which do not induce RUNX1 when cultured in a neutral medium within the first 12 h. In ganglia cultured for 6 h, among the many growth factors tested, only FGF ligands and IGF1 increased substantially the expression of RUNX1 mRNA transcripts (Fig. 4B). The rapid onset of RUNX1 expression suggests a direct inductive mechanism. In contrast, neither NT3 nor NGF had any effect on RUNX1, whereas NT3 significantly upregulated the expression of the ETS transcription factor ER81 (Fig. 4B), as previously shown (Patel et al., 2003). The effects of FGF2 and IGF1 on Runx1 mRNA expression were also observed using E11.5 mouse DRG (Fig. 4C). mRNA expression was significantly increased already after 6 h with either FGF2 or IGF1, and a combination of FGF2 and IGF1 did not show any additive effects (Fig. 4C). This increase of Runx1 mRNA represented an increase also of RUNX1 proteins because, in E11.5 DRG explant cultures grown for 12 h in the presence of either FGF2 or IGF1, a marked increase of RUNX1-immunoreactive neurons was observed compared with control conditions without any supplemented factors (Fig. 4D). Interestingly, PDBu, a phorbol ester that activates PKC, had similar effect on RUNX1 expression (data not shown).
To examine whether FGF and IGF produced within the DRG regulate the induction of RUNX1, ganglia were cultured for a period of 24 h, which allows for induction of RUNX1 independent of addition of growth factors. The FGF receptor inhibitor PD166866 as well as the IGF inhibitor PPP markedly attenuated the induction of RUNX1 (Fig. 4E). Remarkably, when applied together, the inhibitors completely abolished the induction of RUNX1 (Fig. 4E). Similar results were obtained with exogenously added growth factors where the effect of FGF2 on RUNX1 expression was blocked by PD166866 and the effect of IGF1, by PPP (data not shown). In accordance with the above results, PD98059 and LY294002 largely reduced expression of RUNX1 induced by FGF2 in culture (Fig. 4F), confirming the importance of the MAPK/ERK and PI3K/Akt pathways for FGF-induced RUNX1 expression.
The strong in vitro effects of FGF and IGF ligands and of FGF and IGF receptor inhibitors on RUNX1 induction prompted us to analyze the expression of the ligands in vivo. Whole-genome transcriptome analysis indicated mRNA expression of both Igf and Fgf ligands in the E11.5 mouse brachial DRG (data not shown). qPCR analysis from E11.5 brachial DRG confirmed these results, showing very high mRNA expression levels of Fgf13, Fgf11, and Fgf18 (Fig. 5A) and of Igf1 (14.4 arbitrary units) and Igf2 (504.1 arbitrary units) and mRNA expression of Fgf12, Fgf9, and Fgf2 (Fig. 5A). Other members of the FGF family showed lower or no expression (Fig. 5A). FGF ligands can be classified into different categories in regard to their mode of action or their capacity to bind to specific receptors (Reuss and von Bohlen und Halbach, 2003). Succinctly in regard to the present study, FGF11-FGF14 belong to the FGF homologous factor subfamily, are not secreted and do not activate any of the FGF receptors and have been shown to act as intracellular signaling proteins (Smallwood et al., 1996; Schoorlemmer and Goldfarb, 2001; Olsen et al., 2003), FGF9 and FGF18 do not bind Fgfr1, and FGF2 and FGF1 can bind all FGF receptors (Reuss and von Bohlen und Halbach, 2003). We therefore performed in situ hybridization for one member of each aforementioned category of ligands on DRG sections of E11.5 embryos and confirmed expression of Fgf13, Fgf18, and Fgf2 (and Fgf1, data not shown) within the DRG, whereas Igf2 (and Igf1, data not shown) was found located in the periphery of the DRG and in the mesenchyme surrounding the ganglion (Fig. 5B). These patterns of expression were also observed in E12.5 DRG (data not shown). Hence, several FGF ligands appear present at the time and place to act as potential inducers of RUNX1 in vivo. Interestingly, the strong expression of Fgf2 in E12.5 DRG was substantially reduced in TrkCCre;Isl2DTA embryos (Fig. 5C), suggesting that mostly early neurons highly express FGF2 at this stage.

FGFs and IGFs are expressed in the developing DRG. A, qPCR analysis for mRNA relative expression of FGF ligands in E11.5 brachial DRG (a.u., Arbitrary units, normalized to Fgf2). Blue represents FGF homologous factor subfamily members (FGF11-FGF14). Experiment performed in duplicate, each sample representing 80 brachial DRG from different embryos. B, In situ hybridization for Fgf and Igfligands in E11.5 brachial mouse DRG shows expression of Fgf13, Fgf18, and Fgf2 within the DRG. Igf2 is expressed in the DRG periphery and surrounding mesenchyme. C, Fgf2 expression is largely decreased in TrkCCre;Isl2DTA brachial DRG at E12.5 (n = 2 animals per genotype; experiment done in duplicate for each animal). D, A total of 20 ng/ml FGF1, but not 200 ng/ml FGF13 or FGF18, induces RUNX1 expression in neurons from DRG explants after 12 h (arrows). E, A time-dependent induction of RUNX1 by FGF2. At E11.5, brachial but not lumbar DRGs are competent to respond to FGF2 treatment within 12 h (arrows indicate positive neurons). However, lumbar DRG neurons upregulate RUNX1 within 20 h in vitro with FGF2. Scale bars: B, 50 μm; C, 100 μm; D, 20 μm; E, 50 μm.
The finding that several FGFs are expressed in the DRG at the critical developmental period when RUNX1 is induced in the DRG led us to test the identified FGFs on RUNX1 induction using mouse E11.5 DRG cultured for 20 h. Neither FGF13 nor FGF18 induced RUNX1 (Fig. 5D), even at high concentrations, whereas FGF1, similarly to FGF2 (Fig. 4B–D,F and 5E), was observed to efficiently upregulate RUNX1 (Fig. 5D). Because FGF13 act as intracellular modulators (Smallwood et al., 1996; Schoorlemmer and Goldfarb, 2001; Olsen et al., 2003) and FGF1 and FGF2 but not FGF18 (which does not bind Fgfr1) (Reuss and von Bohlen und Halbach, 2003) can induce RUNX1 (Fig. 5D), these results suggest a role for Fgfr1 activation in RUNX1 induction.
A time-dependent activation of RUNX1 expression by FGF2
We furthermore assessed whether the in vitro regulation of RUNX1 expression by FGF was temporally controlled, as seen in vivo (Fig. 1A–C). We therefore compared the induction of RUNX1 by FGF2 between E11.5 brachial and lumbar DRG neurons, which develop slightly later than brachial. After 12 h in culture, RUNX1 was strongly induced in brachial DRG treated with FGF2 (Fig. 5E). In contrast, RUNX1 was virtually absent in FGF2-treated lumbar DRG (Fig. 5E). However, lumbar DRG neurons initiated an FGF-dependent RUNX1 expression after 20 h (Fig. 5E). These results identify a critical window of competence for instructive signaling fating unmyelinated sensory subtypes.
Regulation of RUNX1 by FGFR1 in late-born DRG neurons
To identify whether the effects of FGFs on RUNX1 are mediated via any of the five known FGF receptors (FGFR1-FGFR5), we analyzed their expression by in situ hybridization at E12.5. Fgfr1 was abundantly present, consistent with the strong inductive effects of FGFs on RUNX1; but although Fgfr2, Fgfr3, and Fgfr5 were all detected in the proliferative zone of the spinal cord, they were not expressed in the DRG (Fig. 6A; and data not shown). Fgfr4 was not detected in the DRG or surrounding tissue at this stage (data not shown). Moreover, Fgfr1 expression was found in TrkA+ neurons at E12.5, and analysis of E10.5, E11.5, and E12.5 embryos revealed that Fgfr1 rapidly increased between E11.5 and E12.5 (Fig. 6B,C), thus coinciding with induction of Runx1 in the later-born TrkA+ neurons fated to differentiate into unmyelinated neurons of in the DRG. Together, these results identify onset of Fgfr1 in the DRG as a critical mediator of the competence to RUNX1 induction by FGFs.

Expression and function of FGF receptors in developing DRG. A, In situ hybridization on E12.5 brachial DRG sections shows that Fgfr1, but not Fgfr2, Fgfr3, or Fgfr5, is expressed in the early DRG. B, Fgfr1 mRNA expression in brachial DRG appears at E11.5 and reaches high levels by E12.5. C, Immunostaining for TrkA and ISL1 after in situ hybridization for Fgfr1 on E12.5 brachial DRG section shows expression of Fgfr1 in TrkA+ neurons. D, Immunostaining for βIII-tubulin and Tomato fluorescence on brachial DRG section from E12.5 Wnt1cre;Tomato (cross between the Wnt1cre transgenic line and the R26Tomato reporter mouse line). There is recombination in all neurons. E, In situ hybridization for Fgfr1 on E14.5 brachial DRG sections from Fgfr1lox/lox and Fgfr1cKO shows a nearly complete loss of Fgfr1 in Fgfr1cKO DRG. F, EdU staining (in green, 2 h after injection) on brachial DRG sections from E10.5 or E12 Fgfr1lox/lox and Fgfr1cKO embryos immunostained for SOX10 and ISL1. G, Quantification done within the DRGs depicted by dashed lines in F (n = 2 animals per genotype). H, Immunostaining for RUNX1, TrkA/ISL1, and TrkC/ISL1 on brachial DRG sections from E12.5 control Fgfr1lox/lox (n = 4) and Wnt1Cre;FGFR1lox/lox (FGFR1cKO) (n = 4) mice shows a decrease of RUNX1+ and TrkA+ cells at E12.5. There is lower intensity of RUNX1 in the mutant DRG (insets). I, Quantification of H. Data are presented as mean ± SEM. *p < 0.05 (two-tailed, unpaired t test, statistical comparison with Fgfr1lox/lox). **p < 0.01 (two-tailed, unpaired t test, statistical comparison with Fgfr1lox/lox). a.u., Arbitrary units. J, Whole-mount TrkA immunostaining of E14.5 Fgfr1lox/lox and Fgfr1cKO embryos reveals similar innervation pattern of the forelimb. K, Schematic illustration of the mechanism resulting in the birth of unmyelinated sensory neurons in the coalescing DRG (1) and intracellular signaling pathways participating (2). Scale bars: A, B, D–F, 50 μm; C, 10 μm; H, J, 100 μm.
Conditional mutant mice for Fgfr1 were analyzed to examine the requirement for Fgfr1 signaling during development of RUNX1-positive neurons in vivo. Because Fgfr1 full knock-out mice die before gastrulation (Deng et al., 1994), we used a conditional strategy using a Wnt1Cre strain (Danielian et al., 1998) that mediates recombination in dorsal neural tube and all neural crest derivatives (including DRG neurons) (Fig. 6D) at the premigration stage, clearly before the onset of Runx1 expression, together with mice carrying a loxP-based conditional Fgfr1 allele (Trokovic et al., 2003). Wnt1Cre effectively induced expression of TOMATO in DRG neurons of a reporter line, and Fgfr1 expression was confirmed to be efficiently eliminated in the DRG of Wnt1Cre;Fgfr1lox/lox mice (for simplicity hereafter referred to as Fgfr1cKO) by in situ hybridization (Fig. 6E). The mice survived to term, although they showed general weakness and all mutants pups died within the first postnatal days. In Fgfr1cKO embryos, the number of ISL1+ DRG neurons was decreased at E12.5, but this deficit was transient because E14.5 embryos displayed similar numbers of ISL1+ neurons as WT mice (Fig. 6H,I). Interestingly, the number of both RUNX1+ and TrkA+ DRG neurons was substantially reduced in the mutant embryos, whereas the earlier-born TrkC+ population was unaffected (Fig. 6H,I). Furthermore, the number of dividing cells (assessed by EdU incorporation) and SOX10+ population at E12.0 were unchanged (Fig. 6F,G), showing that the defects are restricted to neurons belonging to the small size unmyelinated class and does not involve effects on proliferation. In further support of this, the number of ISL1+ neurons and SOX10+ progenitor cells in the DRG was unaffected in the Fgfr1cKO embryos at E10.5 when many myelinated neurons are born (Fig. 6G). By quantifying the levels of RUNX1 expression in E12.5 Fgfr1cKO embryos, we observed that not only were the number of RUNX1+ DRG neurons decreased, but also the intensity of RUNX1 immunoreactivity in the remaining neurons (Fig. 6H,I). Further analysis revealed that the deficits of RUNX1 and TrkA expression was transient as it was recovered at E14.5 (Fig. 6I), indicating that other factors also participate in its expression and eventually becomes sufficient in the absence of Fgfr1. Furthermore, the peripheral innervation pattern was not affected by Fgfr1 ablation (Fig. 6J). Together with our in vitro analysis, these data suggest that FGF signaling via Fgfr1 participates in regulating the development of the RUNX1 lineage during early embryonic stages (Fig. 6K).
Discussion
In recent years, there has been a strong focus understanding the mechanisms that allow for the development of the many different kinds of sensory neurons with highly specialized features that have distinct response profiles to external stimuli and therefore provide the cellular fundament for modality-specific sensation. However, it has been unknown whether myelinated neurons and later-born unmyelinated neurons arise in a stochastic or controlled mechanism. Our results indicate that an initial commitment of a common pool of immature neurons, fated to the lineage of unmyelinated high-threshold sensory types that ultimately transduce pain, thermal, and itch modalities of sensation, arises in a controlled, reproducible process by means of inductive signals, FGFs, provided by a local organizing cell group, the early-born neurons.
Local induction of RUNX1 depends on activation of intracellular signaling pathways
During embryonic development, all presumptive unmyelinated neurons express TrkA and RUNX1 (Snider and McMahon, 1998; Kramer et al., 2006). Although TrkA appears before RUNX1 and maintenance of RUNX1 expression is under the control of TrkA signaling at late embryonic stages, NGF is not required for initiation of RUNX1 expression (Luo et al., 2007). RUNX1 plays a central role for the segregation of neuronal-type by regulating expression of many ion channels that are necessary for proper sensation of pain, itch, and temperature (Chen et al., 2006; Kramer et al., 2006; Luo et al., 2007; Abdel Samad et al., 2010; Gascon et al., 2010; Lopes et al., 2012). Therefore, the acquisition of RUNX1 is critical for diversification of this population into specialized types, although the exact mechanisms defining the birth of unmyelinated neurons have been unknown. Our results show that, over a period of 24 h, RUNX1 is induced when the DRG is cultured in a neutral medium and, hence, the neurons are either already fated for RUNX1 expression or local signals within the ganglia induce RUNX1 expression. Our results exclude a cell intrinsic programming because blocking PI3K and MAPK signaling pathways prevents the onset of RUNX1. We therefore conclude that local signaling within the DRG itself is sufficient to induce RUNX1 expression (Fig. 6K).
Timing of birth and the control of neuronal fate by local signals
In the retina (Masland, 2001), cerebral cortex (Angevine and Sidman, 1961; Rakic, 1974), and spinal cord (Hollyday and Hamburger, 1977), neuronal identity is coupled to the time of cell cycle exit and neurogenesis. The idea that early sensory neurons influence later-born neurons was proposed half a century ago because the later-born neural crest neurons of the trigeminal ganglion fail to differentiate in the form of peripheral projections when early-born placode neurons are ablated (Hamburger, 1961; Noden, 1978). We find that the genetic ablation of early DRG neurons leads to deficits of axon growth of later-born TrkA+ DRG neurons. The generation of a TrkCCre;Isl2DTA mouse depleting early neurons as soon as they are formed resulted in a loss of TrkA+/RUNX1+ neurons and a reduced RUNX1 expression in the remaining neurons. Hence, these data provide a causal link between birth date and fate in DRG by showing that early-born neurons contribute to the fating of later-born neurons.
FGF signaling and development of sensory neuron lineages
Our results suggest that neurogenesis associated with birth of neurons in the DRG leads to expression of soluble ligands, which instruct later-born neurons. Using a candidate strategy for factors inducing RUNX1, FGF2 and IGF1 were identified, and blocking FGF or IGF signaling in vitro significantly prevented RUNX1 initiation. Furthermore, a physiological role of Fgfr1 was confirmed using conditional null mice. These data clearly indicate a participation of FGFs, but the incomplete failure in generation of RUNX1+ neurons in Fgfr1cKO mice opens either for an incomplete recombination and elimination of Fgfr1 expression in the Fgfr1cKO mice or that other factors also are involved in vivo. The confirmation with in situ hybridization of an absence of Fgfr1 expression combined with the finding of a recovery of RUNX1 and TrkA expression at later stages in Fgfr1cKO mice support the conclusion that other factors, likely IGFs, also participate and eventually alone become sufficient. However, the recovery at E14.5 could also be explained by a compensatory reduction in cell death. This would indeed be expected because the TrkA ligand NGF that controls naturally occurring cell death at this stage is limiting, and hence proportionally more of the remaining neurons would be expected to successfully compete and survive in the Fgfr1cKO mice. An explanation for why early-born neurons are unresponsive to FGFs is that they are refractory to FGF signaling at the time Fgfr1 is first induced in DRG neurons. Consistently, large-size myelinated neurons, which RUNX3+/TrkC+ neurons as well as the small population of early-born TrkA+ neurons that develop into thinly myelinated neurons, are all born and committed largely before E11.5 (Lawson and Biscoe, 1979; Fariñas et al., 1998; Levanon et al., 2002). The coincidence of initiation of Fgfr1 expression with responsiveness of later-born neurons may explain the acquirement of FGF responsiveness of this population, which ultimately consolidates an unmyelinated phenotype by initiation of RUNX1 expression. Exogenously applied FGF1 and FGF2 can act as survival and mitogenic factors on NCCs (Kalcheim, 1989; Murphy et al., 1994). Thus, FGF1 and FGF2 may also be important at earlier stages in development on NCCs, possibly via other FGF receptors (e.g., FGFR2, FGFR3, FGFR4) because we did not observe any phenotype on proliferation in the Fgfr1cKO mice.
Along with RUNX1, the onset of TrkA expression in late-born neurons defines the development of unmyelinated neurons by gating for NGF responsiveness. In the absence of TrkA, essentially all aspects of development of unmyelinated neurons fail, including a failure of maintenance of RUNX1 expression, RET expression, and expression of both peptidergic and nonpeptidergic specific genes, which define the function of unmyelinated neurons (Patel et al., 2000; Luo et al., 2007). Interestingly, FGFR1 displayed a similar importance for TrkA expression as for RUNX1 in vivo. This effect was specific to unmyelinated cells because TrkC+ neurons and SOX10+ glial cells were unaffected. Consistently, in the absence of Fgfr1 signaling, we found an increase of ISL1+ neurons, which failed to acquire TrkA expression. Furthermore, the analysis of Fgfr1cKO mice shows that Fgfr1 signaling also participates in driving neurogenesis of the late-born neurons. We draw this conclusion based on our results showing a reduction of the total number of ISL1+ neurons at E12.5 without effects on EdU+ cells. We think that the effects on TrkA and RUNX1 can be ascribed to parallel functions of FGFR1 because Ngf null mutant mice display no deficit of RUNX1 induction, and Runx1 null mutant mice do not show any deficits of TrkA induction (Chen et al., 2006; Luo et al., 2007).
The underlying molecular mechanism for RUNX1 and TrkA transcriptional initiation in sensory neurons remains largely unknown. TrkA expression in the early DRG depends on the transcription factors Brn3a, KLF7, and ISL1 (Ma et al., 2003; Sun et al., 2008) with ISL1 playing a greater role (Dykes et al., 2011). The transcriptional mechanism for TrkA and RUNX1 expression might be shared because also RUNX1 expression depends on ISL1 (Sun et al., 2008; Dykes et al., 2011). Expression of KLF7, Brn3a, and ISL1 is coincident with neurogenesis (Fedtsova and Turner, 1995); before onset of TrkA and RUNX1, however, they are all coexpressed in most or all neurons (Ericson et al., 1992; Tsuchida et al., 1994; Fedtsova and Turner, 1995; Lei et al., 2001). The mechanism whereby specificity is conferred for generating unmyelinated neurons has therefore been unclear. Based on our data, we propose a mechanism by which inductive signals, including those arising from Fgfr1 activation, could provide cell-type specificity of KLF7 and ISL1 transcriptional activity.
FGF signaling can activate the ETS transcription factors ETV4/PEA3 and ETV5 to regulate cell identity (Brent and Tabin, 2004; Mao et al., 2009) downstream of several tyrosine kinase receptors (Kuure et al., 2010), consistent with multiple signals contributing to induction of RUNX1 and TrkA. Whereas ETV4 is present only in myelinated sensory neurons (Lin et al., 1998), ETV5 is abundantly expressed in TrkA+ neurons with expression coincident with onset of Fgfr1 (Chotteau-Lelièvre et al., 1997; Hagedorn et al., 2000); and similar to RUNX1 and TrkA, ETV5 depends on ISL1 for expression in sensory neurons (Sun et al., 2008).
Neurogenesis and the establishment of FGF signaling by sensory neurons
Among the FGF ligands FGF1-FGF23, FGF1 and FGF2 were the only FGF ligands among those expressed in the DRG that displayed a RUNX1-inductive activity. FGF2 seems to be expressed primarily in early-born neurons because, in TrkCCre;Isl1DTA mice lacking only the early neurons, Fgf2 expression was markedly attenuated at the time when RUNX1+/TrkA+ neurons are born. These results suggest that, as neurogenesis proceeds, increasing concentrations of FGFs define the functional lineage of late-born unmyelinated sensory neurons. The emergence of a role for FGF signaling in the specification of sensory neuron identity provides an insight into the relationship between neuronal birth date and fate of NCCs undergoing neurogenesis in the early coalescing DRG.
Footnotes
↵†P.E. and F.L. are co-senior authors.
This work was supported by the Swedish Medical Research Council, the Swedish Foundation for Strategic Research and Linné grants (DBRM grants), the Swedish Brain Foundation, Hållsten Foundation, Wallenberg Scholar, and ERC Advanced (232675) to P.E. and the Swedish Medical Research Council, Karolinska Institute, Ragnar Söderberg Foundation, Tysta Skolan, and Åke Wiberg Foundation to F.L. We thank Thomas Jessell, Silvia Arber, and Juha Partanen for generously providing us with the RUNX1 antibody, the Isl2DTA, and the Fgfr1lox/lox mouse lines, respectively, and Dinh Phong Nguyen for the Wnt1Cre;Tomato DRG picture.
The authors declare no competing financial interests.
- Correspondence should be addressed to either Dr. Patrik Ernfors, Division of Molecular Neurobiology, Department of Medical Biochemistry and Biophysics, 17177 Stockholm, Sweden, patrik.ernfors{at}ki.se; or Dr. Francois Lallemend, Department of Neuroscience, 17177 Stockholm, Sweden, francois.lallemend{at}ki.se





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}