

Abstract
Neuroplasticity is essential to prevent clinical worsening despite continuing neuronal loss in several brain diseases, including multiple sclerosis (MS). The precise nature of the adaptation mechanisms taking place in MS brains, ensuring protection from disability appearance and accumulation, is however unknown. Here, we explored the hypothesis that long-term synaptic potentiation (LTP), potentially able to minimize the effects of neuronal loss by providing extra excitation of denervated neurons, is the most relevant form of adaptive plasticity in stable MS patients, and it is disrupted in progressing MS patients. We found that LTP, explored by means of transcranial magnetic theta burst stimulation over the primary motor cortex, was still possible, and even favored, in stable relapsing-remitting (RR-MS) patients, whereas it was absent in individuals with primary progressive MS (PP-MS). We also provided evidence that platelet-derived growth factor (PDGF) plays a substantial role in favoring both LTP and brain reserve in MS patients, as this molecule: (1) was reduced in the CSF of PP-MS patients, (2) enhanced LTP emergence in hippocampal mouse brain slices, (3) was associated with more pronounced LTP in RR-MS patients, and (4) was associated with the clinical compensation of new brain lesion formation in RR-MS. Our results show that brain plasticity reserve, in the form of LTP, is crucial to contrast clinical deterioration in MS. Enhancing PDGF signaling might represent a valuable treatment option to maintain brain reserve and to attenuate the clinical consequences of neuronal damage in the progressive phases of MS and in other neurodegenerative disorders.
Introduction
Neuronal loss and gray matter atrophy progress since the early stages of multiple sclerosis (MS) (Tiberio et al., 2005; Compston and Coles, 2008), but their clinical consequences are initially well compensated, likely because of plastic adaptations of surviving neurons (Schirmer et al., 2013). The precise nature of the adaptation mechanisms taking place in MS brains, ensuring protection from disability appearance and accumulation, is however unknown, and its identification could be of great relevance for future treatments of primary and secondary progressive MS.
LTP of excitatory transmission is the most studied form of synaptic plasticity, and its occurrence in spared neurons might well compensate for neuronal loss occurring in acute and chronic neurological diseases. LTP, indeed, consists of the strengthening of synaptic communication between two connected neurons (Bliss and Lomo, 1973) and is virtually able therefore to restore membrane excitability of neurons that have lost part of their synaptic inputs (Singer et al., 2011; Zepeda et al., 2013). It can be hypothesized, therefore, that a certain degree of white and gray matter damage is tolerated in relapsing-remitting MS (RR-MS) as a consequence of LTP occurrence in unaffected neurons, although reversible or irreversible, clinical disability appears when the adaptive abilities of the brain fail. Instead, in primary progressive MS (PP-MS), accumulating disability might reflect the progression of neuronal damage without any compensation by adaptive LTP mechanisms exhausted during clinically silent inflammatory episodes.
LTP can be explored noninvasively in humans by means of transcranial magnetic stimulation (TMS) (Mariorenzi et al., 1991; Stefan et al., 2000), which offers therefore the possibility of addressing the hypothesis of synaptic plasticity involvement in the attenuation of MS clinical deficits. The synaptic plasticity hypothesis of clinical recovery in MS predicts that LTP is possible in RR-MS but not in PP-MS and that molecular factors regulating LTP induction also impact on MS disease clinical manifestation.
The present investigation was therefore specifically designed at investigating whether LTP is differentially expressed in nonprogressing and in progressing MS patients, and to try to uncover the role of inflammatory molecules in the regulation of LTP induction. According to the idea that synaptic plasticity is crucial to counterbalance clinical progression in MS subjects, we found that LTP was still possible, and even favored, in stable RR-MS patients, whereas it was absent in individuals with PP-MS. Recent studies reported that the PDGF can induce LTP in vitro (Peng et al., 2010), and that it may represent a key molecule for the recovery phase of MS because of its neuroprotective action (Vana et al., 2007). In the present study, we also identified PDGF as a crucial inflammatory molecule able to facilitate LTP induction and to promote the clinical compensation of brain damage associated with MS.
Materials and Methods
The study involving human subjects was approved by the Ethics Committee of the University Hospital Tor Vergata, Rome. All experiments in mice were performed in accordance with the Guide for the Care and Use of Laboratory Animals and the European Communities Council Directive of 24 November, 1986 (86/609/EEC).
Human subjects and CSF withdrawal.
A total of 194 central-southern Italian subjects (118 females, 76 males) were included in this study (Table 1). MS subjects were admitted to the neurological clinic of the University Hospital Tor Vergata of Rome and later diagnosed as suffering from RR-MS (n = 116), or PP-MS (n = 30). After their admittance, all patients underwent for diagnostic purposes, in sequence, brain (and in selected cases also spinal) MRI scan, and CSF withdrawal within 24 h. In all instances, patients underwent detection of oligoclonal banding in the CSF (positive in 88% of cases). Patients were drug-free before CSF withdrawal and neurophysiological assessment. Corticosteroids or other MS-specific immunoactive therapies were initiated later when appropriate.
Demographic and clinical characteristics of enrolled subjects
The diagnosis of RR-MS or PP-MS was established by clinical, laboratory, and MRI parameters, and matched published criteria (Polman et al., 2005, 2011). Demographic and clinical information was derived from medical records. MS disease onset was defined as the first episode of focal neurological dysfunction indicative of MS. Disease duration was estimated as the number of years from onset to the last assessment of disability. Disability was determined by a specially trained (Neurostatus training and documentation DVD for a standardized neurological examination and assessment of Kurtzke's functional systems and Expanded Disability Status Scale for MS patients. Basel, Switzerland: Neurostatus, 2006; available at http://www.neurostatus.net) and certified examining neurologist using Expanded Disability Status Scale (EDSS), a 10 point disease severity score derived from nine ratings for individual neurological domains (Kurtzke, 1983). Relapses were defined as the development of new or recurrent neurological symptoms not associated with fever or infection lasting at least 24 h. As controls, we used CSF from 48 age- and gender-matched healthy subjects (HSs) without inflammatory or degenerative diseases of the central or peripheral nervous system. These subjects underwent lumbar puncture because of a clinical suspect of acute peripheral neuropathy, meningitis, or subarachnoidal hemorrhage, which were not confirmed.
All the subjects gave their written informed consent to the study. Clinical and demographic data are presented as the mean ± SD. Differences between two groups were compared by univariate analysis using Student's t test or Mann–Whitney test for continuous variables and Fisher's exact test for categorical variables. Multiple comparisons were analyzed by performing ANOVA for independent measures followed by Tukey HSD.
MRI acquisition and analysis.
Three Tesla MRI scan consisted of dual-echo proton density, FLAIR, T2-weighted spin-echo images and precontrast and postcontrast T1-weighted spin-echo images. All images were acquired in the axial orientation with 3-mm-thick contiguous slices. The presence of gadolinium-enhancing (Gd+; 0.2 ml/kg e.v.) lesions was assessed by a neuroradiologist who was unaware of the patient's clinical details (Mori et al., 2011).
TMS.
TMS protocols were performed in a subgroup of 75 MS patients (63 RR-MS and 12 PP-MS), and in 13 healthy controls (Table 2). In a subgroup of 20 RR-MS patients, TMS measurements were performed during admission to the neurological clinic and within 24 h from CSF withdrawal. A subgroup of 62 MS subjects (50 RR-MS and 12 PP-MS) were evaluated at least 60 d since stabilization/resolution of a previous relapse in case of RR-MS (50 females, 12 males) (Table 2).
Demographic and clinical characteristics of the subgroup of subjects enrolled in TMS experiments
All subjects gave consent to the examination and were asymptomatic in the upper right limb. EMG traces were recorded from the right first dorsal interosseus muscle (FDI) with surface cup electrodes. The active electrode was placed over the muscle belly and the reference electrode over the metacarpophalangeal joint of the index finger. Responses were amplified with a Digitimer D360 amplifier (Digitimer) through filters set at 20 Hz and 2 kHz with a sampling rate of 5 kHz, then recorded by a computer with SIGNAL software (Cambridge Electronic Devices). Motor-evoked potentials (MEPs) were evoked through a figure-of-eight coil with external loop diameter of 70 mm connected to a Magstim 2002 magnetic stimulator (Magstim). Coil position was adjusted to find the optimal scalp site to evoke motor responses in the contralateral FDI, the motor “hot spot,” at the beginning of each experimental session and marked over the patients scalp with a pencil. The coil was held tangentially to the scalp surface with the handle pointing posteriorly and laterally at ∼45° with respect to the mid-sagittal axis of the head.
Intermittent theta burst stimulation (iTBS) or continuous theta burst stimulation (cTBS) was delivered over the primary motor cortex (M1) “hot spot” of the right FDI through a Magstim Rapid2 stimulator. The resting motor threshold (RMT) was defined as the minimum stimulation intensity required to evoke a liminal motor potential from the FDI at rest (∼50 μV in 50% of 10 trials). The active motor threshold (AMT) was defined as the minimum stimulation intensity required to evoke a liminal motor potential from the FDI during voluntary contraction (∼200 μV in 50% of 10 trials). Stimulation intensity was 80% of AMT. The iTBS protocol consisted of 10 bursts, each burst composed of three stimuli at 50 Hz, repeated at a theta frequency of 5 Hz every 10 s for a total of 600 stimuli (200 s). The cTBS protocol was delivered as a sequence of 200 bursts (600 stimuli) given at a rate of 5 Hz (total duration of 40 s).
The effect of iTBS or cTBS on corticospinal excitability was quantified by measuring the amplitude of MEPs evoked in the right FDI by a constant intensity TMS pulse given over the contralateral motor cortex. Twenty-five MEPs were collected before iTBS or cTBS (baseline) and at two different time points (0 and 15 min) after the end of stimulation procedure. Stimulation intensity was set to induce a stable MEP of ∼1 mV peak to peak amplitude in the relaxed right FDI at baseline and remained unchanged until end of recordings. MEP amplitudes were then averaged at each time point and normalized to the mean baseline amplitude.
Differences between groups for MEP latency, RMT, and AMT were evaluated through one-way ANOVA. For iTBS or cTBS aftereffects, we used a repeated-measures ANOVA with between-subjects GROUP (RR-MS, PP-MS, and controls) and within-subjects TIME (baseline, 0 and 15 min after iTBS or cTBS) main factors. Correlations between PDGF CSF levels and iTBS or cTBS effects on MEP size were analyzed through the Pearson correlation coefficient.
PDGF determination in the CSF.
For measurements of PDGF concentration, the CSF was centrifuged and immediately stored at −80°C until analyzed using Bio-Plex Multiplex Cytokine Assay (Bio-Rad), according to the manufacturer's instructions. Concentrations of PDGF were calculated according to a standard curve generated for the specific target and expressed as pictograms per milliliter. When the concentrations of PDGF were below the detection threshold, they were assumed to be 0 pg/ml. For data presented as the mean ± SEM, statistical analysis was performed using one-way ANOVA for independent measures followed by Tukey HSD. Differences among two groups were compared by univariate analysis using Student's t test.
In vitro electrophysiology.
Preparation of mouse brain slices was performed in accordance with the European Communities Council Directive (86/609/EEC). Parasagittal hippocampal slices (400 μm) were prepared from 3- to 4-week-old male C57BL/6J mice as previously described (Molinaro et al., 2011; Nisticò et al., 2013).
Slices were incubated for 1 h and then transferred to a recording chamber submerged in a continuously flowing artificial CSF (ACSF) (30°C, 2–3 ml/min), gassed with 95% O2 and 5% CO2 containing 124 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 2.5 mm CaCl2, 1.3 mm MgSO4, 26 mm NaHCO3, and 10 mm glucose. Hippocampal CA1 field EPSP (fEPSP) was evoked by Schaffer collateral stimulation (0.2 ms current pulses) using a bipolar tungsten-stimulating electrode. Synaptic responses were recorded with ACSF-filled microelectrodes (2–4 MΩ) positioned in the stratum radiatum and were quantified as the initial slope of fEPSP in CA1.
For slices in which the presynaptic fiber volley was distinguishable, input-output relations were examined by plotting the initial slope of the fEPSP against the amplitude of the presynaptic fiber volley. LTP was induced by conventional TBS applied to the Schaffer collateral-CA1 synapses TBS (4 trains of 5 pulses at a frequency of 100 Hz, with an intertrain interval of 200 ms) (Errico et al., 2008).
All data are presented as mean ± SEM. normalized to the preconditioning baseline (at least 30 min of stable responses) and assessed for significance using the Student's t test.
Results
Characteristics of enrolled subjects
The three groups (control, RR-MS, PP-MS) did not differ in terms of the demographic characteristics, and the two MS groups (RR-MS and PP-MS) did not differ in terms of the main clinical characteristics (Table 1). Of note, disease duration, reported to be inversely related to PDGF concentration in the CSF of MS patients (Harirchian et al., 2012), was similar in our samples. It was therefore not taken into account as confounding factor during subsequent analyses. EDSS was higher in PP-MS patients, according to the different grade of disability progression here investigated.
TMS was well tolerated from all subjects, and no adverse effect was recorded. One-way ANOVA revealed that MEP latency and AMT differed in the three groups. Post hoc comparisons revealed that MEP latency was significantly different between the three groups (PP-MS, 23.83 ± 3.2 ms; RR-MS, 22.02 ± 2.25 ms; HSs, 21.1 ± 0.9 ms; all p < 0.05). RMT differed significantly between the three groups (PP-MS, 41.5 ± 13.0%; RR-MS, 34.1 ± 7.2; HSs, 30.7 ± 4.96; all p < 0.05). AMT mean values were higher in the PP-MS (41.5 ± 13.0%) than in the RR-MS (34.1 ± 7.2) and HSs (30.7 ± 4.96) groups, but not statistically different between the three groups.
LTP induction in the motor cortex of RR-MS and of PP-MS
To assess plasticity reserve in nonprogressing RR-MS and in PP-MS, we delivered the iTBS and cTBS protocols over the M1 in 50 stable RR-MS patients, in 12 PP-MS subjects, and in 13 HSs (Table 2). Repeated-measures ANOVA showed a significant TIME per GROUP interaction (F = 3.80; p < 0.05). According to the idea that LTP reserve is greater in RR-MS than in PP-MS subjects, post hoc contrasts showed that iTBS-induced LTP was more pronounced in RR-MS than in PP-MS at 0 min (F = 3.50, p < 0.05) and 15 min (F = 3.60, p < 0.05) after the stimulation procedure. No differences emerged between RR-MS and healthy individuals, as iTBS caused the expected LTP-like phenomenon (Huang et al., 2005) in both groups (Fig. 1A).

Cortical excitability changes induced by (A) iTBS and (B) cTBS in RR-MS, PP-MS, and HSs. *p < 0.05.
We then explored the effects of the cTBS protocol, which also induces plasticity effects (LTP or LTD) in healthy individuals (Gentner et al., 2008; Iezzi et al., 2008; Huang et al., 2011). Repeated-measures ANOVA showed a significant effect of GROUP (F = 3.83, p < 0.05) and a significant TIME per GROUP interaction (F = 4.15; p < 0.05). Post hoc contrasts showed that, 15 min after cTBS, MEP amplitude was significantly higher in RR-MS subjects compared with PP-MS (F = 3.26, p < 0.05) and compared with HSs (F = 4.89, p < 0.05) at 0 min (F = 3.84, p < 0.05) and 15 min (F = 4.15, p < 0.05) and significantly lower in HSs compared with PPMS at 15 min (F = 3.31, p < 0.05), revealing that this alternative stimulation protocol caused a measurable LTP in RR-MS patients, although it failed to induce synaptic plasticity effects in PP-MS. In healthy individuals, cTBS caused a LTD-like phenomenon in the majority of the subjects (9 of 13), as already described (Huang et al., 2011) (Fig. 1B).
Together, these results indicate that LTP reserve is preserved in stable RR-MS and lost in PP-MS patients, and that LTP induction is favored in RR-MS patients compared with healthy individuals.
PDGF levels in the CSF of RR-MS and of PP-MS
Some inflammatory molecules released in the CSF of MS patients can alter excitatory synaptic transmission (Rossi et al., 2012) and could be therefore potentially implicated in the differential synaptic plasticity expression in RR-MS and in PP-MS. Because of its ability to enhance LTP induction and expression in vitro (Peng et al., 2010) and of its proposed role in limiting MS disease progression (Harirchian et al., 2012), we measured PDGF CSF levels in RR-MS, in PP-MS, and in a sample of control individuals.
PDGF levels were significantly affected by MS disease (F = 3.65, p = 0.03). Post hoc analysis showed that PDGF was significantly lower in PP-MS patients than in RR-MS, suggesting a contribution of this molecule in the differential expression of LTP seen in RR-MS and in PP-MS. PDGF levels were also lower in PP-MS patients than in controls, without reaching statistical significance. Of note, PDGF levels were higher, although not significantly, in the CSF of RR-MS subjects, in line with the TMS data showing a favored LTP induction compared with healthy individuals (Fig. 2).

PDGF levels in the CSF of MS subjects. The graph shows that PDGF was significantly lower in PP-MS patients than in RR-MS. *p < 0.05, versus RR-MS.
Role of PDGF in in vitro LTP
Having determined that PDGF CSF levels positively correlate to LTP induction in stable RR-MS, we next sought to test the effect of PDGF on synaptic transmission and LTP in mouse hippocampal slices. A scatter plot relating the initial slope of the fEPSP to the size of the presynaptic fiber volley showed that slices incubated with PDGF (20 ng/ml for 1 h) displayed normal input-output characteristics in the CA1 area (Fig. 3A). Next, to determine whether synaptic plasticity is modulated by PDGF, we studied hippocampal LTP induced by TBS. Similarly to previous results (Peng et al., 2010), CA1-LTP was always facilitated when slices were preincubated with PDGF (Fig. 3B). Indeed, the magnitude of potentiation measured between 50 and 60 min after TBS was significantly higher in PDGF-treated (162 ± 9%, n = 6) compared with control slices (134 ± 9%, n = 6) (p < 0.05; Fig. 3B).

Effect of PDGF on hippocampal synaptic function. A, Input/output curves are shown as plots of the fEPSP slopes against the corresponding presynaptic fiber volley amplitudes. Each treatment group is the average (±SEM) of at least six recordings performed on separate slices. No significant differences between treatment groups were detected. B, Time plot of fEPSP responses showing that pretreatment with PDGF enhances the magnitude of CA1-LTP. Representative fEPSP recordings from time points 1 and 2 are shown for each condition. Calibration: 0.5 mV, 10 ms.
Thus, our in vitro data indicate that PDGF modulates LTP induction at excitatory synapses and are consistent with the idea that this molecule could contribute to the maintenance of plasticity reserve seen in nonprogressing MS patients.
Association between PDGF levels and clinical relapses in RR-MS patients
Based on the assumption that PDGF-favored plasticity reserve could be relevant for the compensation of evolving brain damage in RR-MS, we also examined the impact of PDGF on the clinical correlates of disease evolution in RR-MS subjects. To this aim, RR-MS subjects with active MS lesions (n = 49), as evidenced by Gd enhancement at the MRI, were divided into two groups (relapsing, n = 26 vs silent, n = 23), based on the presence or absence of concomitant clinical symptoms. Gd+-relapsing patients had significantly lower PDGF CSF concentrations than Gd+ clinically silent patients (p = 0.01), a finding that is in agreement with the idea that PDGF plays a role in plasticity reserve and clinical compensation of brain damage in MS, by preventing symptom appearance despite new lesion formation (Fig. 4A). In line with this, relapsing patients were significantly more frequent among subjects with undetectable PDGF at the time of MRI activation (undetectable PDGF, n = 23; detectable PDGF, n = 26; 78% vs 30%, p = 0.001) (Fig. 4B).

Association between PDGF levels and clinical compensation in RR-MS. A, The histogram shows that PDGF CSF levels were lower in Gd+ relapsing patients than in Gd+ clinically silent patients. B, Relapsing patients were more frequent among RR-MS subjects with undetectable PDGF levels at the time of MRI activation. *p < 0.05.
Correlation between PDGF levels and LTP amplitude in RR-MS patients
To further address the proposed involvement of PDGF in LTP reserve in RR-MS patients, the correlation between CSF concentrations of this molecule and the amplitude of this form of synaptic plasticity was explored in a subgroup of 20 RR-MS patients (5 males and 15 females, aged 17–47 years). The results of this investigation were in agreement with the conclusion that PDGF has a role in the maintenance of brain plasticity potential in MS subjects because those individuals with higher CSF PDGF levels showed larger LTPs in response to cTBS than those with low PDGF levels (r = 0.57, p < 0.05) (Fig. 5). Conversely, no significant correlation emerged between CSF PDGF levels and MEP latency, AMT, or RMT (data not shown).
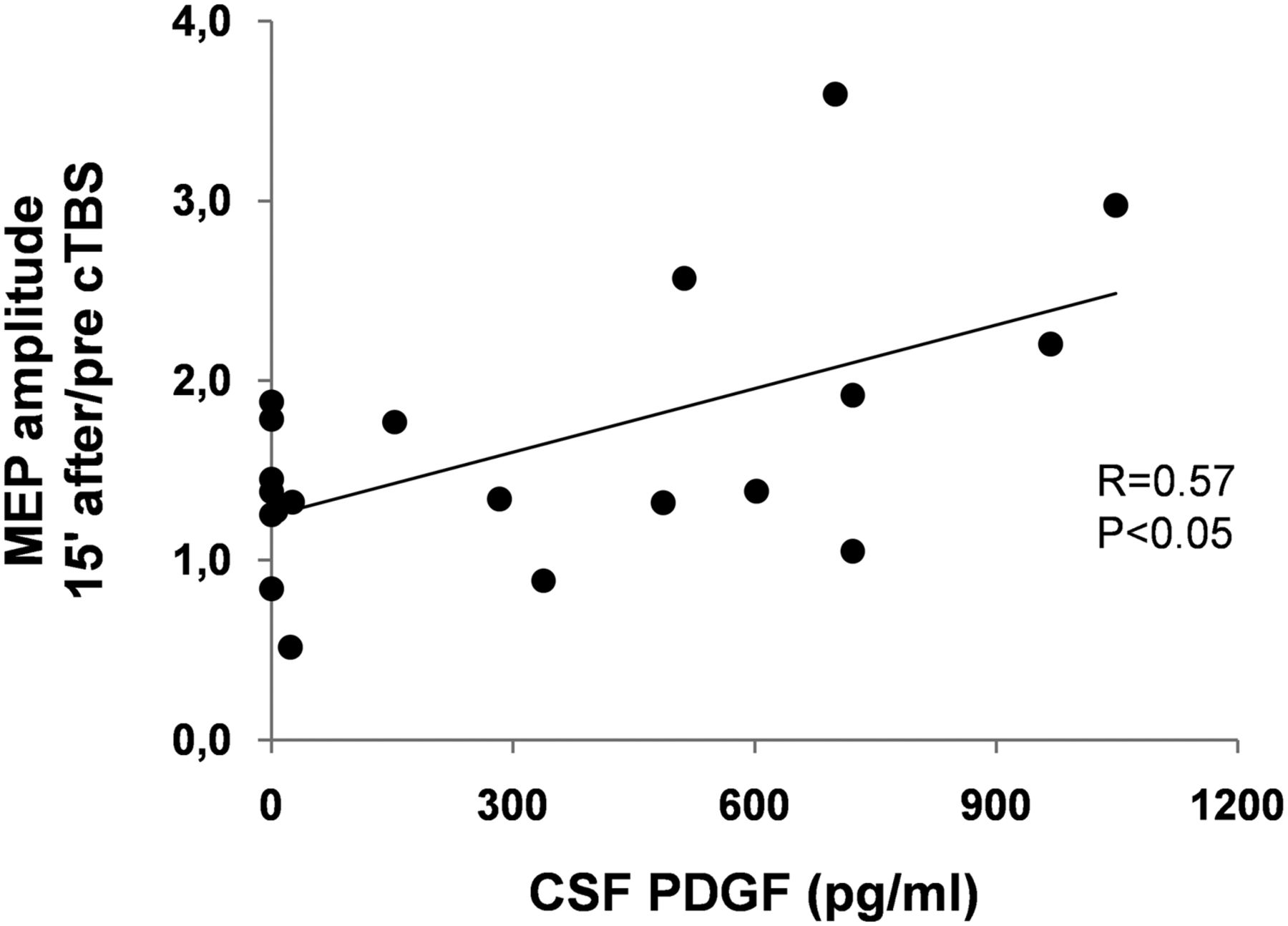
PDGF favors LTP over LTD in RR-MS patients. Correlation plot shows that PDGF levels in the CSF correlate with the magnitude of LTP-like changes induced 15 min after cTBS in RR-MS patients.
Discussion
LTP is a form of use-dependent synaptic plasticity that strengthens the communication between two connected neurons, able to minimize the effects of neuronal loss in a network by providing extra excitation of denervated neurons (Bliss and Lomo, 1973).
The present study showed defective LTP in patients with progressive MS course, supporting the idea that altered synaptic plasticity reserve plays a substantial role in the manifestation and progression of clinical deficits in MS. In stable RR-MS patients, in contrast, LTP was still possible and even favored, as evidenced by the finding that not only iTBS but also cTBS resulted in significant and long-lasting enhancement of cortical excitability. In healthy individuals, conversely, cTBS generally results in LTD, but LTP has been found to emerge in response to cTBS under specific circumstances altering the excitability of the motor cortex (Gentner et al., 2008; Iezzi et al., 2008).
NMDA receptor-dependent LTP induction is greatly facilitated in response to ischemic brain damage in rodents (Benveniste et al., 1984; Picconi et al., 2006), and its occurrence in the peri-infarct area is associated with better clinical outcome (Centonze et al., 2007). Also, in humans, LTP reserve explored with iTBS after focal brain ischemia is associated with better functional recovery (Di Lazzaro et al., 2010), a finding that is in good agreement with our results, and extends to other acute and chronic pathological conditions the concept that this form of synaptic plasticity is indeed able to attenuate the clinical consequences of brain tissue damage.
Immune cells infiltrating the CNS and causing tissue damage contribute to neuronal and oligodendroglial cell survival and tissue repair by secreting different growth factors (Schwartz et al., 1999; Kerschensteiner et al., 2003) and, among these, PDGF acts as a key molecule for the recovery phase. PDGF, indeed, promotes neuronal differentiation (Williams et al., 1997; Erlandsson et al., 2001), improves significantly remyelination and oligodendrocyte density during acute demyelination, and reduces apoptosis during the recovery period after chronic demyelination (Vana et al., 2007). Furthermore, strong upregulation of PDGF occurs in peripheral lymphocytes of experimental MS, with the highest expression after the disease maximum (Koehler et al., 2008). PDGF has also been implicated in neuroprotection against energy deprivation and oxidative injury (Cheng and Mattson, 1995), against human immunodeficiency virus protein toxicity (Peng et al., 2008), and after injurious events, such as focal brain ischemia (Egawa-Tsuzuki et al., 2004). Importantly, PDGF concentration in the CSF of MS patients decreases with disease duration, and its serum and CSF levels have been proposed as markers of disease severity (Harirchian et al., 2012). Here, we also provided evidence that PDGF plays a substantial role in favoring both LTP and brain reserve in MS patients, as this molecule: (1) enhanced LTP emergence in hippocampal slices, (2) was associated with more pronounced LTP in RR-MS patients, (3) was associated with clinically stable disease course, and (4) was associated with the clinical compensation of new brain lesion formation in RR-MS. The ability of PDGF to favor LTP, and its association with clinically stable MS course, is indeed consistent with its prosurvival effects because a striking convergence between the intracellular signaling pathway mediating LTP and neuronal survival exists (Bartlett and Wang, 2013).
As expected, PP-MS patients had higher EDSS values and more pronounced MEP alterations (higher MEP latency and RMT). PP-MS patients may thus have lost the ability to express LTP resulting from increased axonal damage and thus to a lower number of synaptic connections. It must be considered, however, that activity-dependent LTP is essential for synaptogenesis (Collin et al., 1997) and that axonal damage occurs also in RR-MS patients since the early stages (Schirmer et al., 2013). However, LTP was higher in RR-MS than in HSs in our study. At the present stage, this point remains controversial and needs to be elucidated in further studies.
The mechanism by which PDGF enhances LTP is only marginally understood, but animal studies showed that PDGF and its receptors are widely expressed in the CNS (Sasahara et al., 1991; Gozal et al., 2000), where they modulate the expression of Arc/Arg3.1 and give rise to LTP in hippocampal slices (Peng et al., 2010; present study). While reduced PDGF activity seems to contribute to exhausted LTP in PP-MS patients, LTP facilitation in RR-MS, as evidenced by the induction of this form of synaptic plasticity not only after iTBS but also after cTBS, does not seem to be directly dependent upon PDGF. Although higher than in PP-MS, indeed, PDGF CSF levels were similar in RR-MS and in control subjects, who showed LTD rather than LTP in response to cTBS.
Facilitated LTP is likely to be important for prompt compensation of brain damage in RR-MS and could instead be favored by inflammatory molecules released during MS but not in nondiseased brains. Among them, IL-1β has already been found to lower the threshold for LTP induction and to favor LTD to LTP switch in MS brains, possibly through the inhibition of GABA synapses (Nisticò et al., 2013). cTBS, indeed, induces a mixture of excitatory and inhibitory long-lasting effects (Stagg et al., 2009), and the direction of the post-cTBS synaptic plasticity is determined in physiological conditions by the excitation/inhibition balance (Huang et al., 2011). Accordingly, LTP is attenuated by enhancing GABA transmission (Wigström and Gustafsson, 1983; Chapman et al., 1998; Grover and Yan, 1999; Levkovitz et al., 1999; Lu et al., 2000; Yoshiike et al., 2008; Martin et al., 2010) and potentiated by reducing it (Hess and Donoghue, 1996; Stäubli et al., 1999).
During a relapse, MS patients show decreased short interval intracortical inhibition (Caramia et al., 2004). These findings could reflect changes of both NMDA receptors (Schwenkreis et al., 1999) or GABAA receptor activities (Ziemann et al., 1996). Thus, the increased LTP responses observed in RR-MS patients may possibly be secondary to alterations in both NMDA or GABA receptor activity. In this respect, reduction of cortical inhibition has been proposed as a mechanism to augment plastic properties (Baroncelli et al., 2011; Imbrosci and Mittmann, 2011), as synaptic inhibition limits the plastic properties of the cortex (Teo et al., 2009).
In our study, the excitability of the motor cortex was evaluated indirectly, through the amplitude of the MEP, which is considered reflective of corticospinal excitability. This may represent a limitation, as the plastic modulation of the MEP amplitude may have occurred at noncortical sites. Indeed, plastic changes in the spinal H reflex after 5 consecutive days of iTBS over the motor cortex have been reported in MS patients with lower limb spasticity (Mori et al., 2010). However, H reflex remained unchanged after only one single session of iTBS (Huang et al., 2005; Mori et al., 2010; Conte et al., 2012) or cTBS (Huang et al., 2005; Zapallow et al., 2012).
Also, spinal recordings through epidural electrodes at the cervical site showed that, after a TMS pulse at intensities 3% below the AMT over the motor cortex, no corticospinal volleys could be recorded (Di Lazzaro et al., 1998). The lack of change in the H reflex after TBS and the low intensity used by TBS suggest that there is no effect of one single session of motor TBS on excitability of spinal motor neurons and the inhibitory circuits around these neurons. Moreover, EEG (Vernet et al., 2013), MEG (McAllister et al., 2013), and cervical epidural (Di Lazzaro et al., 2005) recordings showed that TBS induces excitability changes at the cortical level. Investigating the relationship between cortical plasticity through measures of corticocortical connectivity (i.e., by means of paired pulse TMS, EEG, MRI), and clinical and biochemical variables in MS may provide further and more direct evidence of the role of neuronal plasticity and its regulators in MS.
LTP can be influenced by a number of different factors, among these, proinflammatory cytokines, with detrimental effects on MS progression (Pickering et al., 2005; Haji et al., 2012; Rossi et al., 2012; Mori et al., 2013). It thus appears reasonable that the net effect on plasticity is the result of a very complex interaction between proinflammatory and anti-inflammatory cytokines and genetic factors. Future studies designed to evaluate the impact of multiple variables and of their interaction on MS clinical severity are thus warranted.
In conclusion, despite brain damage progresses overtime, maintenance of LTP-like, brain plasticity reserve is crucial to contrast clinical deterioration in MS, and possibly in other acute or chronic neurological diseases. Enhancing PDGF signaling might represent a valuable treatment option to preserve brain reserve and to attenuate the clinical consequences of neuronal damage in the progressive phases of MS. This possibility should be carefully considered as plasticity can also lead to maladaptive clinical manifestations as reported in dystonia (Quartarone et al., 2003), dyskinesias (Cenci and Konradi, 2010) or spasticity itself (Tan et al., 2012).
Footnotes
This work was supported by Fondazione Italiana Sclerosi Multipla Special Project Grant to D.C.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Diego Centonze, Clinica Neurologica, Dipartimento di Medicina dei Sistemi, Università Tor Vergata, Via Montpellier 1, 00133 Rome, Italy. centonze{at}uniroma2.it





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}