

Abstract
Integrins have been implicated in various processes of nervous system development, including proliferation, migration, and differentiation of neuronal cells. In this study, we show that the serine/threonine kinase Ndr2 controls integrin-dependent dendritic and axonal growth in mouse hippocampal neurons. We further demonstrate that Ndr2 is able to induce phosphorylation at the activity- and trafficking-relevant site Thr788/789 of β1-integrin to stimulate the PKC- and CaMKII-dependent activation of β1-integrins, as well as their exocytosis. Accordingly, Ndr2 associates with integrin-positive early and recycling endosomes in primary hippocampal neurons and the surface expression of activated β1-integrins is reduced on dendrites of Ndr2-deficient neurons. The role of Ndr2 in dendritic differentiation is also evident in vivo, because Ndr2-null mutant mice show arbor-specific alterations of dendritic complexity in the hippocampus. This indicates a role of Ndr2 in the fine regulation of dendritic growth; in fact, treatment of primary neurons with Semaphorin 3A rescues Ndr2 knock-down-induced dendritic growth deficits but fails to enhance growth beyond control level. Correspondingly, Ndr2-null mutant mice show a Semaphorin 3A−/−-like phenotype of premature dendritic branching in the hippocampus. The results of this study show that Ndr2-mediated integrin trafficking and activation are crucial for neurite growth and guidance signals during neuronal development.
- dendritic and axonal growth
- dendritic branching
- integrin activation
- integrin trafficking
- mouse
- serine/threonine kinase Ndr2
Introduction
Kinases of the Ndr (nuclear Dbf2-related) family are abundantly expressed in neural tissue and play an important role in neuronal differentiation (Emoto, 2011). Current evidence suggests that the Ndr1/2 homologs SAX-1 in Caenorhabditis elegans and tricornered in Drosophila melanogaster are required for neurite initiation and for the control of dendritic branching and tiling (Zallen et al., 1999; Emoto et al., 2004; Gallegos and Bargmann, 2004; Emoto et al., 2006). Consistent with that, expression of dominant-negative Ndr1 and a combined Ndr1/2 knock-down in rat hippocampal neurons enhances, whereas constitutively active Ndr1 reduces, dendritic growth and spine density. Potential Ndr1 substrates involved in control of dendritic growth and spine formation are the endosome-associated Rabin-8 and AAK (Ultanir et al., 2012), but it is rather likely that further targets downstream of Ndr1/2 will contribute to the complex process of neurite outgrowth and dendrite branching.
Integrins are heterodimeric cell and matrix receptors that provide sites of adhesion and signals for the dynamic rearrangement of cytoskeletal elements in various stages of neuronal differentiation, including outgrowth and branching of axons and dendrites (Moresco et al., 2005; Marrs et al., 2006). Eighteen α-subunits and eight β-subunits have been identified that combine into at least 24 different receptors. β1-integrin-containing dimers appear to be particularly relevant for neuronal differentiation because they provide adhesive functions and a versatile signaling platform mediating adhesion contact signaling (Moresco et al., 2005; Marrs et al., 2006).
Integrin binding can be triggered either through an initial low-affinity contact with a suitable substrate (“outside-in”) or through the activation of alternative intracellular pathways (“inside-out”), which enhances adhesion by increasing the affinity and avidity of integrin surface receptors (Clegg et al., 2003; Harburger and Calderwood, 2009). This activation involves endosomal trafficking, a coordinated assembly of one α-subunit and one β-subunit at the membrane, the exposure of their extracellular binding domains, and the intracellular association of signaling factors and linkers to the actin cytoskeleton, such as filamins, talins, and kindlins (Larjava et al., 2008; Abram and Lowell, 2009; Huveneers and Danen, 2009; Moser et al., 2009a). In developing and mature neurons, both the adhesive function and the integrin-mediated signaling to the actin cytoskeleton are well documented (Clegg et al., 2003; Bernard-Trifilo et al., 2005). Intriguingly, Ndr2 and tricornered associate with the actin cytoskeleton, suggesting a role in cytoskeletal reorganization during cell polarization and neurite formation (Stork et al., 2004; Fang and Adler, 2010). However, whereas integrin activation mechanisms have been investigated in detail in non-neuronal cells (Gahmberg et al., 2009; Margadant et al., 2011), they are still not sufficiently understood in neurons.
In this study, we provide evidence that Ndr2 contributes to dendrite outgrowth and branching by stimulating the phosphorylation and endosomal trafficking of β1-integrins, thereby controlling integrin-dependent differentiation of hippocampal neurons both in vitro and in vivo.
Materials and Methods
DNA constructs and antibodies.
Cloning of Ndr2 and generation of EGFP-Ndr2 fusion proteins has been described previously (Stork et al., 2004) and mCherry fusion constructs were generated in the same way using SalI and BglI restriction endonuclease digestion (New England Biolabs). Plasmids pCMV-tdTomato and pEGFP-C1 were purchased from Clontech. To generate a constitutive active Ndr2 (Stork et al., 2004) and Mst1 (purchased from the Harvard Plasmid Bank), full-length cDNA clones were amplified by PCR and cloned either into the pEFBOS-LAT vector or the pEFBOS-Flag vector as described previously (Boerth et al., 2000). Specific shRNA against murine Ndr2 (5′-CCT-CAT-CTG-CCA-ATC-CCT-C-3′), murine β1-integrin (according to #2 shITGB1 from Lei et al. (2012) and commercial anti-luciferase shRNA from pMIR-mU6-Luc (Mirus Bio), was used as a control and cloned into pLL.3.7 vector (Addgene). RNAi rescue experiment where achieved by cloning the sequence of Ndr2 shRNA together with an shRNA-resistant EGFP-Ndr2 gene (with five point mutations, the sequence was changed from 5′-GAA-GGA-TTG-GCA-GAT-GAG-3′ to 5′-GAG-GGT-TTG-GCC-GAC-GAA-3′, causing no change of encoded amino acids but mismatching of Ndr2 shRNA) into the pLL3.7 vector backbone. Generation and purification of a GST fusion protein of the cytoplasmic domain of β1-integrin was done as described previously (Moser et al., 2009b). The GST-Filamin-A 19–24 pGEX clone was generously provided by Dr. Martin Humphries (Manchester University). The construct pECFP-SARA-FYVE was a gift from S. Corvera (Hayakawa et al., 2004).
The primary antibodies anti-caveolin1 (Santa Cruz Biotechnology), anti-Mst1 and anti-pT183 Mst1 (Cell Signaling Technologies), anti-Ndr1 (MyBioSource), anti-actin (Millipore), anti-β-actin (Abcam), and anti β-galactosidase (Promega) were used for immunoblotting. Anti-GFP (Abcam), anti-pT788/789 β1-integrin (Abcam), anti-Rab11 (BD Bioscience PharMingen), and anti-Rab5 (Synaptic Systems) primary antibodies were used for immunoblotting and immunocytochemistry. For detecting Ndr2 kinase, we used our c-terminal-binding antibody abNdr2 described by Stork et al. (2004). For flow cytometric analysis, FITC-labeled or nonconjugated anti-CD29 (β1-integrin, clone MEM101A) were purchased from Immunotools; FITC-conjugated anti-mouse IgG were from Dianova. Primary antibodies anti-MAP2 (Abcam), anti-β1-integrin (BioLegend) and 9EG7 antibody against activated β1-integrin were used for immunocytochemistry. Alexa Fluor dye and Cy-coupled secondary antibodies (Invitrogen and Dianova) were used for immunofluorescence analysis. HRP-coupled secondary antibodies for ECL Western blot detection were purchased from Dianova and GE Healthcare.
NIH 3T3 and HEK-293T culture.
Cell lines were maintained in DMEM containing 10% fetal bovine serum and penicillin/streptomycin (100 U/ml/100 μg/ml; all from Invitrogen). HEK-293T cells were transiently transfected using the standard calcium phosphate precipitation method. When indicated, the transfected cells were treated with 1 μm okadaic acid (Merck) for 1 h before harvesting. To analyze potential dependency of β1-integrin phosphorylation at Thr788/789 on the calcium-calmodulin-dependent kinase II (CaMKII) and the protein kinase Cε (PKCε) kinase activity, cells were treated with autocamtide-2 related inhibitory peptide II (0.1 μm, cell-permeable; Tocris Bioscience) or Ro-31–7549 (0.35 μm; Merck) according to the manufacturers' specifications.
Isolation of lipid rafts and non-lipid-raft fractions.
Preparation of lipid rafts by sucrose gradient centrifugation has been described previously (Boerth et al., 2000). HEK 293T cells (2 × 106 cells) were lysed in morpholine-propane-sulponic acid (MES)-buffered saline (25 mm MES, pH 6.5, 150 mm NaCl) containing 1% Triton X-100 and 1 mm PMSF. The lysates were mixed with 1 ml of 80% sucrose and overlaid with 2 ml of 30% sucrose and 1 ml of 5% sucrose all solved in MES-buffered saline. After centrifugation (200,000 × g for 18 h at 4°C), fractions were removed sequentially starting from the top of the gradient. Anti-caveolin1 antibody was used to identify lipid rafts fractions on a dot blot assay. Rafts fraction (2–4) and non-raft fractions (8–10) were pooled and analyzed by Western blotting.
Isolation of endosome fractions.
Endosome fractionation experiments were done with neural differentiated rat pheochromocytoma (PC-12) cells using a sucrose gradient centrifugation adopted from Waugh et al. (2011). Cells were grown in RPMI-1640 with 2 mm l-glutamine, 10% horse serum, 5% fetal bovine serum (all reagents from Invitrogen). Then, 30 × 106 PC-12 cells were harvested and lysed using a manual homogenizer in a buffer containing 250 mm sucrose, 100 mm triethanolamine, 100 mm acetic acid, 1 mm EDTA, 1 mm PMSF, and complete proteinase inhibitor (Roche). Lysate was overlaid on a continuous 10–40% sucrose gradient. After 20 h of centrifugation (at 75,000 × g), 10 fractions were aliquoted from the top and analyzed by Western blotting and immunodetection of intracellular membrane organelles. Specificity of fraction was tested with markers for endosomes, mitochondria, endoplasmic reticulum, and the Golgi apparatus (data not shown).
Western blot analysis, immunoprecipitation, and in vitro kinase assay.
For phosphoprotein isolation, cells were washed once with ice-cold PBS, lysed in lysis buffer containing 1% lauryl maltoside N-dodycyl-β-d-maltoside (Merck), 1% NP-40 (Sigma-Aldrich), 1 mm Na-orthovanadate, 1 mm PMSF, 50 mm Tris-HCl, pH 7.4, 10 mm NaF, 10 mm EDTA, and 160 mm NaCl and incubated for 20 min on ice. After centrifugation, the protein concentration of the postnuclear supernatant was determined using the Roti-Nanoquant reagent (Roth) according to the manufacturer's instructions. Equivalent amounts of protein were used in precipitation studies (500 μg of total protein). Ndr2 was immunoprecipitated using anti-Ndr2 antibody in combination with 30 μl of protein-A agarose beads (Santa Cruz Biotechnology) for 2 h at 4°C. Precipitates were washed 3 times in NP-40 washing buffer (0.1% NP-40, 1 mm PMSF, 50 mm Tris-HCl pH 7.4, 10 mm NaF, and 160 mm NaCl). Cell lysates (50 μg of total protein) or immune complexes were separated by SDS-PAGE and transferred to nitrocellulose and PVDF (Millipore) membrane, respectively. Western blots were conducted with the indicated primary antibodies and the appropriated HRP-conjugated secondary antibodies and analyzed with a Luminol detection system from Roth and Millipore, respectively. For in vitro kinase assays, precipitates were washed three times in NP-40 washing buffer; for some experiments, samples were divided into two equal aliquots. Both samples were pelleted and one used for Western blot analysis; the other was washed 3 times and resuspended in kinase buffer (25 mm HEPES, 10 mm MgCl2, 1 mm dithiothreitol, and 100 μm [γ-32P]ATP, ∼1 μCi/ μl; PerkinElmer) supplemented with 5 μg of myelin basic protein (Merck), GST, and GST-cytβ1 and incubated for 30 min at 30°C. Reactions were stopped by addition of 5× sample buffer and proteins were resolved by SDS-PAGE. After drying, the gels were exposed to autoradiography films. The generation and purification of the GST fusion protein for the cytoplasmic domain of β1-integrin has been described previously (Moser et al., 2009b).
Analysis of β1-integrin recycling.
Twenty-four hours after transfection, HEK-293T cells were resuspended in PBS and 0.5 × 106 cells were stained for 30 min at 4°C with FITC-labeled CD29 antibodies (or anti-mouse IgG conjugated with FITC as a negative control). After labeling, cells were incubated for various time points at 37°C and the remaining cell-surface-bound FITC-labeled CD29 was removed by incubating the cells for 10 min in stripping buffer (0.2 m glycine, pH 7.4). The stripping procedure was controlled for each experiment. For exocytosis, cells were stained for 30 min at 4°C with unconjugated CD29 antibodies and incubated for 45 min at 37°C. Upon endocytosis, remaining cell-surface-bound antibodies were removed in the same manner, cells were incubated for various time points at 37°C, and recycled antibody-bound integrins at the surface were stained with anti-mouse IgG conjugated with FITC. Samples were analyzed using FACSCalibur flow cytometer and CellQuestPro software (Invitrogen).
Primary cell culture.
Dissociated primary hippocampal cultures were prepared using a modified protocol from Banker and Goslin (1988). Briefly, hippocampi from embryonic day 18 (E18) to E19 C57 BL/6JBomTac mice (Taconic) were dissected, dissociated in 0.25% trypsin (Invitrogen), and treated with 0.1% DNase (Roche). Dissociated cells were plated at a density of 30,000–50,000 cells/cm2 (high density for transfection) or 10,000–20,0000 cells/cm2 (low density for immunocytochemistry) on poly-d-lysin-coated (Sigma-Aldrich) or on fibronectin-/poly-d-lysin-coated coverslips (fibronectin was purchased from Roche). Three to 4 h after plating, DMEM containing 10% FBS (v/v), l-GlutaMAX (2 mm), and penicillin/streptomycin (100 U/ ml/100 μg/ ml) was changed to neurobasal medium containing B27 (2% v/v), l-GlutaMAX (0.5 mm), and penicillin/streptomycin (100 U/ml and 100 μg/ml, respectively; all from Invitrogen) that had been glia conditioned for 72 h. For low-density cultures and echistatin treatment, coverslips were transferred to 30 mm dishes containing a 10- to 14-d-old glial feeder layer and maintained in neurobasal medium. After 2 d in vitro (DIV), cells were treated with 3 μm AraC (Sigma-Aldrich). Neuronal cultures were transfected at DIV 3 with the calcium phosphate precipitation method according to Köhrmann et al. (1999). After transfection, coverslips were returned to the glia-containing dish or glia-conditioned medium and cultured further for four additional days. To analyze β1-integrin-dependent growth, echistatin (Sigma-Aldrich) was applied at a concentration of 200 nm 24 h after transfection or Semaphorin 3A (R&D Systems) was applied at a concentration of 60 ng/ml for 16 h.
RNA isolation real-time PCR.
RNA isolation of primary hippocampal neurons was performed at different time points of culturing (DIV 3, 5, 7, 14, and 21) and first-strand synthesis was done with MMRV reverse transcriptase according to the protocol of the “Cells-to-cDNA II” kit. Quantitative PCR was performed in a StepOnePlus Real-time PCR System using TaqMan reagents with a custom-made expression assay for Ndr2 (5′-TGCCCGTCTCTGAGAAAGC-3′; 3′-CCACCATTCCCAATTCTGTTTTCAG-5′ and FAM ACTTGATTCTCAGATTTTG NFQ) and a predesigned expression assay for GAPDH (4352923E), β1-integrin (Mm01253230), and β3-integrin (Mm00443980) (all Invitrogen).
Immunocytochemistry and neurite growth analysis.
Primary neurons at DIV 7 were fixed with 4% paraformaldehyde and 4% sucrose in 0.1 m PBS, pH 7.4, washed in PBS, and embedded with Immuno-Mount (Thermo Scientific). Axons and dendrites of GFP- or tdTomato-filled neurons were differentiated and tracked according to their morphological features (Kaech and Banker, 2006) and immune staining for Tau and MAP2, respectively. Outgrowth and branching was evaluated according to the method of Sholl (1953) with the first radius of 20 μm from the soma's barycenter, using a DMI 6000 microscope and QWin software (Leica Microsystems).
For immunocytochemistry, after fixation, cells were permeabilized for 10 min with 0.3% PBS/Triton X-100 and unspecific binding was blocked with 5% secondary antibody serum in PBS, followed by primary antibody incubation in blocking solution at 4°C overnight. Cells were washed in PBS, incubated for 1 h at room temperature in secondary antibodies in 2% bovine serum albumin in PBS, and again washed in PBS. Nuclear staining was achieved with 5 min of incubation in 600 nm DAPI before embedding. Cells were finally examined using the DMI 6000 epifluorescence microscope and LAS AF software for deconvolution (both Leica Microsystems).
Live immunocytochemistry of activated β1-integrin was done according to the method of Tan et al. (2012). 9EG7 antibody was added 1:50 in the culture medium of hippocampal primary cells at DIV 3 and incubated for 15 min at 37°C. Before fixation with PFA/sucrose, cells were washed with warm culture medium. Cells were costained for Map2, followed by application of Alexa Fluor 488- and Cy3-conjugated secondary antibodies and washing and mounting of slides. For quantification, randomly selected cells (n = 10) were imaged using LAS AF software with identical light intensity. Each cell was tracked as a stack of 30 pictures with a width of 0.2 μm each. Blind deconvolution was performed, dendrites and soma of each neuron were traced, and fluorescence intensity of immune staining was analyzed with the histogram tool of LAS AF software.
Generation of Ndr2−/− mice.
The mouse Ndr2 (Stk38l) gene is located on chromosome 6 forward strand between 146724995bp and 146778812bp, with no apparent overlap of other genes or noncoding RNA sequences (Eppig et al., 2012). Ndr2−/− mice were generated from a gene-trap clone of the ES cell line E14TG2a (Stk38lGt(RRT116)Byg; Bay Genomics) supplied through the NIH Mutant Mouse Regional Resources Center (Stryke et al., 2003). The position of the gene trap was determined by the supplier, who identified with a 5′ rapid amplification of cDNA ends (5′RACE) a sequence tag (Table 1) matching exon 9–7 of the Ndr2 gene and thus indicating gene trap location in intron 9 (see Fig. 7G).
Primers for sequence analysis and genotyping of Ndr2−/− mice
Upon rederivation of ES cells according to the supplier's instructions and injection into C57BL/6 blastocyst (Polygene), one mouse line with stable gene-trap insertion was obtained and backcrossed to C57BL/6OlaHsd genetic background. The genotype of Ndr2 offspring was determined on tail biopsies taken shortly after weaning using multiplex PCR with the common forward primer wt/ko and each one primer for the wt and the ko allele (Fig. 7H).
Histological analysis of Ndr2−/− mice.
Male Ndr2−/− mice and their Ndr2+/+ littermates were used (n = 8). For analysis of overall brain structure and cell lamination, 8- to 10-week-old animals were killed by cervical dislocation and brains were removed and quickly frozen in cold isopentane. Next, 20-μm-thick coronal sections were cut at the level of the dorsal hippocampus, mounted on poly-l-lysine-coated glass slides, and stained using a 0.5% solution of cresyl violet in acetic acid buffer, pH 4.0. Sections were washed with acetic acid buffer, dehydrated in ethanol, cleared with xylol, and finally embedded with Permount.
For LacZ staining, brain sections after mounting were treated with a solution of 1 mg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactoside in 0.1 m phosphate buffer, pH 7.2, containing 2 mm MgCl2, 5 mm K4Fe(CN)6, and 5 mm K3Fe(CN)6. After washing with tap water, sections were embedded with Immumount.
Golgi impregnation was done as described previously (Mylius et al., 2013). Brains were removed and impregnated in the dark for 14 d at room temperature in a Golgi-Cox solution (Glaser and Van der Loos, 1981), dehydrated, and embedded with celloidin. After polymerization, 150-μm-thick horizontal sections were cut on a sliding microtome (Microm), collected in 70% ethanol, and rinsed in aqua dest before being treated with 50% alkaline ammonia followed by 0.5% phenylene-diamine (Sigma-Aldrich). Staining was developed in 1% Dectol (Kodak) and fixated in 5% Tetenal (Calbe Fotochemie). Finally, sections were rapidly dehydrated in a graded series of ethanol and xylol (Roth) and mounted between two coverslips using Merckoglas (Merck). The brain morphology of wild-type and Ndr2−/− mice was inspected and analyzed using a light microscope system (DMRX; Leica) with a motorized stage (Märzhäuser). Representative pyramidal neurons of the left and right CA3 at the level of the root region of the fimbria were analyzed for initial dendrite segment length. Some of them were further reconstructed by means of the Neurolucida software (Microbrightfield). For the quantitative analysis of their branching patterns, we used Neuroexplorer software (Microbrightfield) and the Excel spreadsheet application.
Statistical analysis.
Statistical analysis on multiple groups was performed with multivariate ANOVA followed by Fischer's protected least significant difference (PLSD) test. For direct pairwise comparisons, Student's t test or the nonparametric Mann–Whitney U test was applied. A statistical value of p < 0.05 was considered significant.
Results
Ndr2 is involved in dendrite and axon development in mouse hippocampal neurons
In primary hippocampal neurons, Ndr2 is localized in the soma, dendrites, axons, and presynaptic and postsynaptic sites (Stork et al., 2004). When analyzing the expression of Ndr2 mRNA in hippocampal cultures, we found that levels correlated with the formation and maturation of dendrites (ANOVA F(6,25) = 3.9, p = 0.011), increasing 1.54-fold (±0.24) from DIV 3 to a maximum at DIV 7 and decreasing on DIV 14 (0.89 ± 0.20) and on DIV 21 (0.70 ± 0.12). Ndr1 mRNA was >100-fold lower than Ndr2 at all time points and was hardly detectable. In fact, Western analysis revealed that Ndr2, but not Ndr1, is expressed in the mouse hippocampus and frontal cortex during development (Fig. 1I). In contrast, in rats, both Ndr kinases could be observed, as described previously (Ultanir et al., 2012). The increase in Ndr2 mRNA expression during the period of extensive dendritic and axonal growth between DIV 3 and DIV 7 (Goslin and Banker, 1990) prompted us to investigate the relevance of the kinase in neurite outgrowth of mouse hippocampal neurons.

Ndr2 is required for neurite growth in mouse hippocampal neurons. Cells were transfected with tdTomato and shLuc as control shRNA (control; A), with tdTomato and shRNA directed against Ndr2 (shNdr2; B), or with tdTomato, shRNA directed against Ndr2 (shNdr2; C) together with an shNdr2-insensitive form of EGFP-Ndr2. Neurons (DIV 7) with Ndr2 knock-down show a drastic reduction of both the dendritic tree and the axon. This reduction was detained by a coexpression of EGFP-labeled Ndr2. Scale bars, 100 μm. Quantitative analysis of dendrites (D,G) and axons (E,H) using the Sholl analysis (n = 12 cells each; N = 3) confirms their profoundly reduced density upon Ndr2 knock-down and the recovery by the shNdr2-insensitive EGFP-Ndr2. Data are presented as mean ± SEM. *p < 0.05; ***p < 0.001 (Fischer's PLSD). F, Efficiency of the shNdr2 is revealed by the suppression of cotransfected GFP-Ndr2 in HEK-293T. Further transfection with the shNdr2/EGFP-Ndr2* reconstitution construct prove the shNdr2 insensitivity of EGFP-Ndr2*. Double transfection with the control shRNA shLuc (control) does not alter GFP-Ndr2 levels. I, Ndr2, but not Ndr1, is prominently expressed in mouse frontal cortex (FC) and hippocampus (Hip) during development. In contrast, both kinases are found in rat tissue at P0, P10, and P20.
First, we found that shRNA-mediated knock-down of Ndr2 led to a significant reduction in the growth of both dendrites (repeated-measures ANOVA with the between-subject factor gene and the within-subject factor distance: F(1,38) = 49.816, p < 0.001) and axons (F(1,38) = 23.184, p < 0.001) as determined by Sholl analysis (n = 20 cells each; N = 3, data not shown). In a second set of experiments (Fig. 1A–H), we also used a reconstitution vector coexpressing shRNA-resistant Ndr2, with which we confirmed the Ndr2 knock-down-induced reduction and, moreover, demonstrated its recovery in both dendrites (repeated-measures ANOVA with the between-subject factor gene and the within-subject factor distance: F(2,33) = 33.075, p < 0.001, and axons: F(2,33) = 20.745, p < 0.001). Comparing the total number of intersections, Ndr2 shRNA expression led to a significant reduction in the growth of both dendrites (Fischer's PLSD, p < 0.001) and axons (p < 0.001) compared with control-transfected neurons. The reconstitution of Ndr2 expression resulted in an increase of dendritic (p < 0.001) and axonal branching (p < 0.001) compared with shNdr2-transfected neurons, reaching levels comparable to those of controls.
Ndr2-sensitive neurite development is integrin dependent
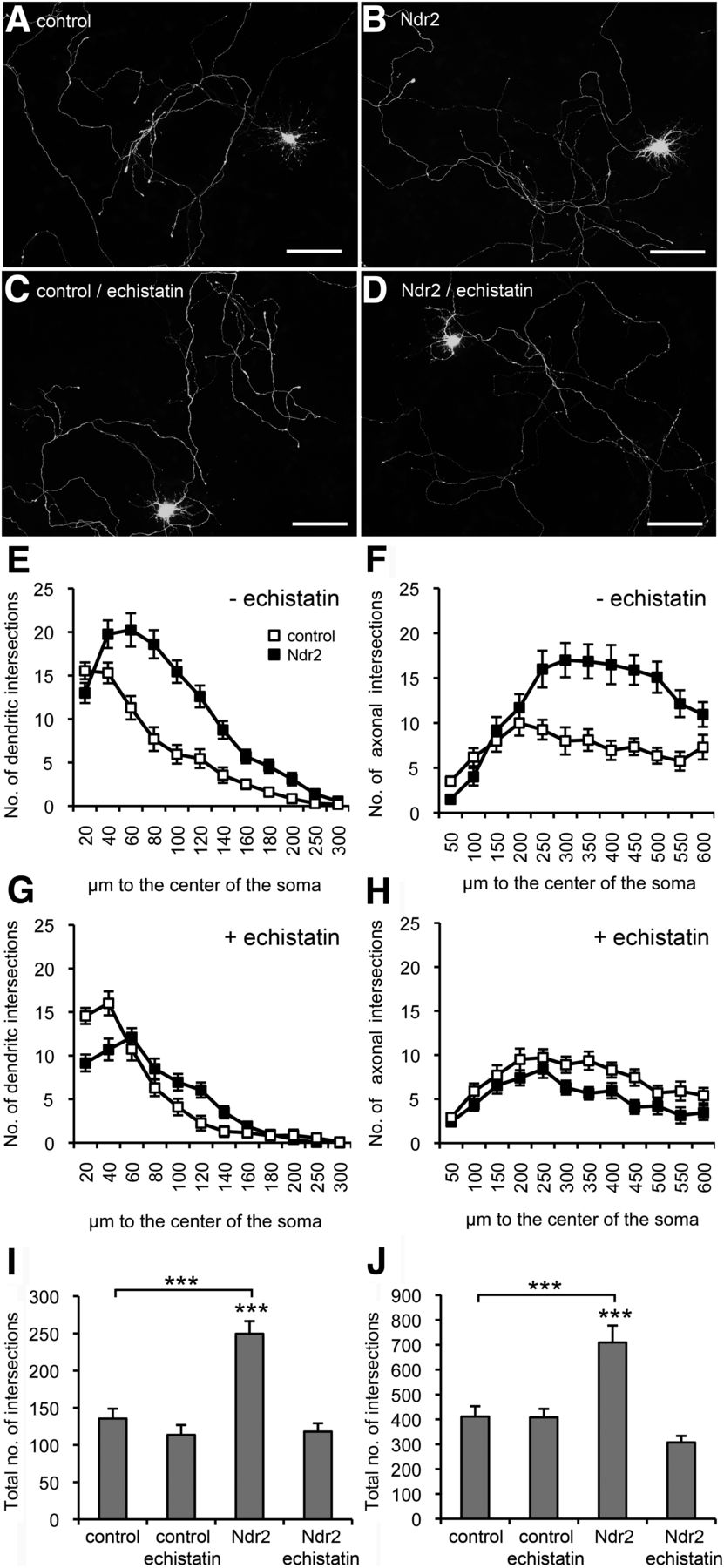
Overexpression of Ndr2 leads to significant increase in dendritic growth (repeated-measures ANOVA with the between-subject factor gene and the within-subject factor distance: F(1,39) = 28.013, p < 0.001) without any change in the number of primary dendrites evading the soma. Moreover, axonal growth of Ndr2 overexpression neurons was strongly enhanced (F(1,39) = 13.991, p = 0.001), thus supporting the growth-stimulating function of Ndr2 in our experiments (Fig. 2A,B,E,F). In primary cell culture, astrocytic deposition of extracellular matrix components strongly activates integrin-dependent neuronal growth (Nishio et al., 2003) and the RGD-receptors α5β1-integrin and αvβ3-integrin appear to be critical for dendrite development (Gupton and Gertler, 2010; McGeachie et al., 2011). With application of the RGD disintegrin echistatin, we were indeed able to entirely prevent the Ndr2-enhanced dendritic growth (repeated-measures ANOVA: F(1,39) = 0.064, p = 0.802 compared with echistatin-treated control cells) and furthermore to slightly reduce axonal growth (F(1,39) = 5.419, p = 0.025) in Ndr2-overexpressing cells (Fig. 2C,D,G,H). Comparison with untreated Ndr2-overexpressing cells confirmed the blockage of Ndr2-induced overgrowth of both types of neurites through echistatin (Fischer's PLSD, p < 0.001; Fig. 2I,J).
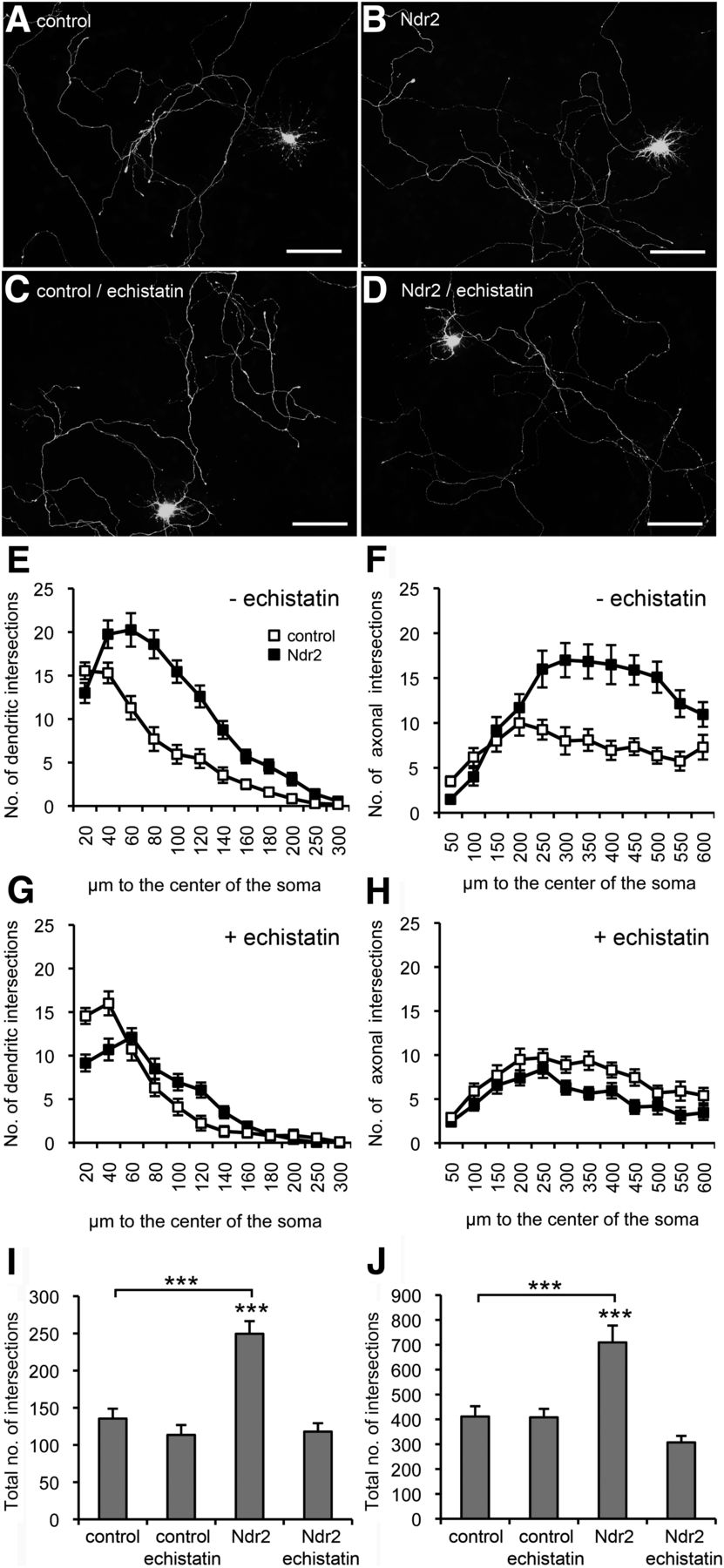
Ndr2-mediated dendritic and axonal growth effects are integrin dependent. A–D, Primary hippocampal neurons were transfected with EGFP (control; A,C) or with both EGFP and mCherry-tagged Ndr2 (Ndr2; B,D). A, B, Overexpression of Ndr2 leads to an increased growth of both dendrites and axons compared with control cells (DIV 7). C, D, Under treatment with echistatin, no difference can be observed between Ndr2-transfected primary neurons and controls. Scale bars, 100 μm. E, F, Quantitative analysis (n = 20 cells each; N = 3) of dendritic and axonal arbors confirms the significant increase upon Ndr2 overexpression. G, H, Under echistatin treatment, no enhancement of growth and branching is evident in the Ndr2-overexpressing cells. In fact, axons show a slight reduction of arborization. I, J, Cross-comparison of the total number of intersections with untreated neurons demonstrates that echistatin normalizes the number of dendritic and axonal branches in Ndr2-overexpressing cells to control level, whereas EGFP-transfected control cells are not affected by the disintegrin. Data are presented as mean ± SEM. ***p < 0.001 (Fischer's PLSD).
Because echistatin blocks both β1- and β3-integrin-containing dimers, we tested the mRNA levels of these two β isoforms at early developmental stages of hippocampal neurons. We found that mRNA levels of β3-integrin were <0.5% of β1-integrin mRNA expression levels at DIV 3, DIV 5, and DIV 7. Therefore, we focused on testing the function of β1-integrin in neurite growth and arborization by expressing a shRNA that partially knocked down the endogenous β1-integrin protein (Fig. 3G). We found both dendritic (repeated-measures ANOVA with the between-subject factor gene and the within-subject factor distance: F(1,22) = 45.013, p < 0.001) and axonal (F(1,22) = 32.363, p < 0.001) branches to be drastically reduced (Fig. 3A–F), akin to the shNdr2 phenotype.

Reduced β1-integrin expression leads to a reduced dendritic and axonal growth in mouse hippocampal neurons. Cells were transfected with tdTomato and shLuc as control shRNA (control; A), or with tdTomato and shRNA directed against β1-integrin (shITGB1; B). Knock-down of β1-integrin at DIV 7 exhibits a drastically reduced growth and branching of either dendrites (C,E) or axons (D,F) using the Sholl analysis (n = 12 cells each; N = 3). Data are presented as mean ± SEM. ***p < 0.001 (Fischer's PLSD). Scale bars, 100 μm. (G) Efficiency of shITGB1 was proven in the murine cell line NIH3T3.
Ndr2 controls β1-integrin phosphorylation at Thr788/789
We next tested whether Ndr2 may phosphorylate the cytoplasmic domain of the β1-integrin subunit. Expression of a membrane targeted, constitutively active form of Ndr2 (LAT-Ndr2) in fact did increase β1-integrin phosphorylation (3.91 ± 0.94-fold, p = 0.001, Student's t test) at Thr788/789 in HEK-293T cells (Fig. 4A,B). CaMKII and PKCε have been reported previously to mediate β1-integrin phosphorylation at this site (Suzuki and Takahashi, 2003; Stawowy et al., 2005). In fact, LAT-Ndr2 induced phosphorylation at Thr788/789 was sensitive to Ant-AIP-II and Ro-317549, which are specific inhibitors of CaMKII (Ishida et al., 1995) and PKC activity (Turner et al., 1994), respectively. Ndr2 did not directly phosphorylate β1-integrin because we were not able to detect phosphorylated cytoplasmic domain of β1-integrin in an in vitro kinase assay using either LAT-Ndr2 (Fig. 4C) or ocadaic-acid-stimulated native Ndr2 (data not shown). At the same time, autophosphorylation of Ndr2 and phosphorylation of the myelin-binding protein were clearly evident (Fig. 4D) and an association of activated Ndr2 with the lipid rafts fraction of the plasma membrane was verified (Fig. 4E). Finally, expression of constitutively active Mst1 kinase did not stimulate Thr788/789 phosphorylation of β1-integrin in HEK-293T cells (Fig. 4F).

Ndr2 stimulates phosphorylation of β1-integrin. A, β1-integrin phosphorylation at Thr788/789 is increased in HEK-293T cells upon overexpression of LAT-Ndr2. The increase is prevented through application of the CaMKII inhibitor Ant-AIP-II or the PKC inhibitor Ro-317549. B, Quantification of Western blot signals confirms a significant increase of β1-integrin Thr788/789 phosphorylation upon LAT-Ndr2 expression and its blockage by Ant-AIP-II and Ro-317549, respectively (N = 3). Data are presented as mean ± SEM. **p < 0.01 compared with control transfection (pEFBOS). C, In an in vitro kinase assay, however, constitutively active LAT-Ndr2 fails to phosphorylate the intracellular domain of β1-integrin. Autophosphorylation of LAT-Ndr2 and transphosphorylation of MBP confirm the activity of the kinase. D, Scheme of membrane-anchored, constitutively active LAT-Ndr2 and activity of precipitated Flag-Ndr2 (FNdr2) or LAT-Ndr2 in an in vitro kinase assay. Autophosphorylation of FNdr2 can be induced with ocadaic acid (OA) treatment of transfected cells, whereas LAT-Ndr2 is constitutively autophosphorylated. Myelin basic protein (MBP) serves as a positive control. E, Western blot analysis of lipid rafts (3–5) and nonrafts (8–10) membrane fractions reveals a localization of both ocadaic-acid-stimulated FNdr2 and LAT-Ndr2 in lipid rafts of transfected HEK-293T cells. Caveolin1 serves as a marker for the lipid rafts fractions. F, LAT-Mst1 overexpression in HEK293 cells results in increased phosphorylation of Mst1 at Thr183 compared with control conditions (pEFBOS transfected). However, constitutively active LAT-Mst1, in contrast to LAT-Ndr2, fails to increase β1-integrin phosphorylation at Thr788/789 of β1-integrin.
Ndr2 modulates integrin trafficking
The phosphorylation of β1-integrin at Thr788/789 has been shown to protect it from lysosomal degradation and to enhance its cell surface expression (Böttcher et al., 2012). We, therefore, next investigated in HEK-293T cells whether Ndr2 may alter β1-integrin endo- or exocytosis. Through FACS analysis with an antibody that detects the extracellular domain of native β1-integrin we found that overexpression of the constitutively active LAT-Ndr2 enhances the exocytosis of β1-integrins, whereas Ndr2 shRNA-mediated knock-down suppresses their recycling to the plasma membrane. In contrast, neither of these manipulations affected the endocytosis rate or total expression levels of β1-integrin (Fig. 5).

Ndr2 stimulates β1-integrin exocytosis. Flow cytometric analysis of β1-integrin (CD29) surface expression was performed in HEK-293T cells. A, B, Expression of constitutively active LAT-Ndr2 (A) and Ndr2 knock-down with shNdr2 (B) do not affect total β1-integrin expression or surface expression in unstimulated cells (top). Moreover, no effect is observed on the stimulated endocytosis of β1-integrin (“internalized CD29,” middle). However, LAT-Ndr2 increases and shNdr2 reduces recycling of β1-integrin 15 to 45 min after induction of exocytosis “surface expression of recycled CD29,” bottom). A and B each show one representative experiment of three.
Ndr2 localizes at integrin-recycling endosomes
Next, we examined whether Ndr2 may be associated with integrin-containing recycling vesicles in primary neurons. Indeed, we observed a colocalization of endogenous and transfected Ndr2 with various markers of the endosomal pathway, including EEA1 (data not shown), Rab5 (Fig. 6A–H), and Rab11 (Fig. 6I–P), as well as β1-integrin in dendrites and somata of hippocampal neurons. Western blot analysis of isolated subcellular fractions from neural differentiated PC12 cells confirmed the expression of endogenous Ndr2 in early and recycling endosome containing fractions, again using Rab5 and Rab11 as markers (Fig. 6Q).

Ndr2 associates with β1-integrin-positive early and recycling endosomes. Colocalization of endogenous Ndr2 (green) and β1-integrin (red) can be observed with the early endosome marker Rab5 (A–H) and with Rab11 (I–P) (both blue), a marker for recycling endosomes in somata, dendrites, and axons. Arrowheads depict sites of costaining in proximal dendrites. Scale bars: A–D, I–L, 25 μm; E–H and M–P, 5 μm. Q, Western blot analysis of a subcellular fractionation of neural differentiated PC12 cells indicates that Ndr2 is selectively detected only in Rab5- and Rab11-positive endosome fractions.
Reduced β1-integrin activation in outgrowing dendrites of Ndr2-deficient neurons
Primary hippocampal neurons from Ndr2−/− mice (see Successful gene targeting in Ndr2−/− mice, below) were used to determine whether Ndr2 deficiency also alters the amount of activated β1-integrin on the surface of outgrowing dendrites. Using an antibody that detects β1-integrins in their active conformation, we indeed found decrease in dendritic β1-integrin labeling on DIV 3 hippocampal neurons derived from Ndr2−/− mice (p < 0.001, Student's t test; Fig. 7A–F).

Ndr2 deficiency in hippocampal neurons leads to a reduced surface expression of activated β1-integrin. Map2 and activated β1-integrin were immune labeled in wild-type (A,B) and Ndr2−/− (C,D) hippocampal neurons at DIV 3. Scale bars, 100 μm. E, The number of dendrites was not significantly affected by Ndr2 deficiency, but tend to be reduced. F, Ndr2 knock-down impairs integrin activation in dendrites, as indicated by lower level of 9EG7 labeled β1-integrin. n = 19–22, N = 3, ***p < 0.001. G, Ndr2 gene ablation was achieved with a gene trap insertion of vector pGT0lxf between exons 9 and 10 of the Ndr2 gene (KOMP). Almost the entire cassette (bp 212- 8379), including a slice acceptor (SA), full β-galactosidase (β-geo) coding sequence, and polyadenylation site, has been inserted. Arrows indicate the location of primers geof/r for confirmation of the ES cells, seqf/r for sequencing of critical sites, and wt/kof, wtr,kor for genotyping. H, Multiplex PCR detects specific signals for homozygous and heterozygous mutant mice, with the ko allele at 638 bp and the wt allele at 1098 bp fragment size. I, Western blot analysis with our c-terminal binding antibody for Ndr2 (abNdr2) confirms the loss of full-length Ndr2 protein from the hippocampus of Ndr2−/− mice and a reduced expression in Ndr2±mice. Beta-actin is used as a loading control. J, Ndr21-282:: β-geo fusion protein was detected in Ndr2−/− and Ndr2±mice with a 33 kDa molecular weight increase to β-galactosidase alone (control). K, Staining of tissue sections with X-Gal substrate detects prominent expression of the inserted cassette in the hippocampus, particularly in area CA3, reflecting the high expression levels of Ndr2 mRNA in this region (Stork et al., 2004). Scale bar, 1 mm. L, Scheme of Ndr21-282:: β-geo fusion protein indicating the lack of most parts of the activating sequence, the kinase subdomains VIII-XII including the substrate recognition site, the c-terminal hydrophobic motif of the Ndr2 gene, as well as the Ndr2 antibody (abNdr2)-binding site (Stegert et al., 2004).
Successful gene targeting in Ndr2−/− mice
Ndr2-null mutant mice were generated by an insertion of a functional gene-trap cassette covering bp 212–8379 of vector pGT0lxF, including the splice acceptor site, full β-galactosidase-neomycin-phosphotransferase II (β-geo) coding sequence, and polyadenylation site as confirmed with serial PCR and sequence analyses (see Fig. 7G, Table 1 for primers and their locations). The precise location in intron 9 was determined in the same manner: sequencing of a 638 bp fragment generated with common forward primer wt/ko (position bp 596–615 of intron 9) and reverse primer ko (position bp 451–431 of the gene trap vector) unambiguously showed that the gene trap vector integrated at position bp 995 of intron 9 of the mouse Ndr2 gene (Fig. 7G).
Homozygous mutants were viable and Ndr2± × Ndr2± breeding produced offspring with genotype distributions at Mendelian frequency. Western analysis confirmed the complete loss of full-length Ndr2 from the null mutant hippocampus (Fig. 7I). The gene trap insertion resulted in the generation of an Ndr21-282::β-geo fusion protein (Fig. 7L) with a predicted molecular weight of 140 kDa, deleting most of the activating sequence and kinase subdomains VIII-XII, including the substrate recognition site and the c-terminal hydrophobic motif of the Ndr2 gene (Stegert et al., 2004). Western analysis confirmed the expected 33 kDa molecular weight increase of the Ndr21-282::β-geo fusion protein in Ndr2±and Ndr2−/− mice compared with β-geo (Fig. 7J). A complete loss of the 54 kDa band in Ndr2−/− mice and a reduced signal in Ndr2± mice were found with an Ndr2-specific antibody (Stork et al., 2004) targeting the c-terminal region of the kinase.
Ndr2-null mutant mice display premature branching phenotype
To investigate the role of Ndr2 on neuronal differentiation in the hippocampus in vivo, we applied histological analysis of brain sections of Ndr2-null mice. An overall view of Nissl-stained tissue revealed (Fig. 8A,B) no overt alteration in hippocampal structure and lamination. However, an analysis of dendritic arborization with the Golgi impregnation method (Fig. 8C,D) showed a distinct morphological change of CA3 pyramidal neurons: whereas no significant differences in the basal dendrite branching was observed in the mature hippocampus of adult Ndr2−/− mice (repeated-measures ANOVA with the between-subject factor gene and the within-subject factor distance: F(1,30) = 0.065, p = 0.301), the apical dendritic tree was found to be altered in its complexity compared with wild-type littermates (Fig. 8E). In the proximal part, apical dendrites of Ndr2−/− mice (Fig. 8E,J) branch earlier and show an increased complexity (repeated-measures ANOVA: F(1,25) = 5.295, p = 0.03), whereas a more distal part of the dendritic tree (Fig. 8E,K) shows less branching (F(1,25) = 5.389, p = 0.029). This shift of arborization toward the soma is associated with a significant shortening in the length of the initial apical dendritic segment of adult Ndr2−/− mice (Fig. 8F) compared with wild types (Mann–Whitney U test: U = 172.5, p < 0.05). In young, Ndr2-deficient mice (P21), no reduction is evident in the length of the initial segment (Fig. 8H, Mann–Whitney U test: U = 104, p > 0.05) or in the complexity of the proximal (repeated-measures ANOVA: F(1,13) = 0.269, p = 0.768) and more distal (repeated-measures ANOVA: F(1,13) = 0.106, p = 0.9) part of their apical dendrites, respectively (Fig. 8G,J,K). However, an increased branching of basal dendrites is evident in Ndr2−/− mice on P21 (F(1,13) = 6.328, p = 0.026) that cannot be observed in the adult knock-out mouse (Fig. 8G,I).

Adult Ndr2−/− mice show premature dendritic branching. Morphology of CA3 pyramidal cells was investigated in Golgi-stained tissue of Ndr2−/− mice and their wild-type littermates. A, B, No major change in hippocampus structure and lamination is observed in Ndr2-deficient mice (A) compared with their wild-type littermates (B). Scale bar, 500 μm. C, D, However, Golgi staining of control animals (C) and Ndr2−/− mice (D) demonstrates a shortening in the initial segment and a premature branching (arrow) in hippocampal CA3 pyramidal neurons lacking the expression of full-length Ndr2. E, Sholl analysis of traced CA3 pyramidal neurons reveals that proximal apical dendrites in adult Ndr2−/− mice (n = 14 cells each, N = 4–5 mice per group) branch earlier and thus have more dendritic branches. Due to this early branching, there is a shift in the length of more distal dendrites and reduction of arborization in Ndr2−/− pyramidal neurons. Whether further distal parts of the dendritic arbor are equally affected (Warren et al., 2012) remains to be determined. There is no difference in the basal dendrite complexity. F, The length of the initial segment of CA3 pyramidal neurons is shorter in the Ndr2−/− mice compared with their wild-type littermates (n = 22–27 cells each, N = 6–7 mice per group). *p < 0.05. G, Sholl analysis of traced CA3 pyramidal neurons at P21 indicates no difference in the apical dendrite complexity but does show overbranching in basal dendrites in the Ndr2−/− mice (n = 7–8 cells each, N = 4–7 mice per group). H, At P21, the length of the primary segment of apical dendrites does not differ between Ndr2+/+ and Ndr2−/− mice (n = 12–18 cells each, N = 5–6 mice per group). I, Mean total dendrite length of basal dendrites shows difference at P21 as the length of dendrites is increased in Ndr2−/− mice. J, K, Mean total length of the proximal apical dendrites is increased in adult Ndr2−/− mice (J) and in more distal parts of apical dendrites (K) there is a reduction compared with their wild-type littermates. Scale bars, 11 μm. Data are presented as mean ± SEM. *p < 0.05.
Ndr2-dependent dendritic growth is responsive to Semaphorin 3A
Semaphorin 3A (Sema3A) has been shown to stimulate dendritic growth in primary hippocampal neurons in a β1-integrin-dependent manner (Schlomann et al., 2009). We tested whether Sema3A can rescue the knock-down-induced growth deficit or if the Sema3A effects themselves are sensitive to the Ndr2 knock-down (Fig. 9). In fact, we observed a general stimulation of dendritic growth by Sema3A (repeated-measures ANOVA: F(1,28) = 14.196, p = 0.01) and recovery of Ndr2 knock-down induced dendritic growth deficits indicating compensation through the Sema3A stimulation (Fig. 9A–F). However, Sema3A induced dendrite growth in shNdr2-transfected neurons only to nonstimulated control levels (Fig. 9G). Ndr2 knock-down cells failed to reach the level of dendritic growth seen in control shRNA-transfected cells with Sema3A treatment (Fishers PLSD, p = 0.016; Fig. 9), suggesting a contribution of Ndr2 to Sema3A growth signaling.

Sema3A partially recovers dendritic growth after Ndr2 knock-down. A, B, Ndr2 knock-down-induced reduction of dendritic growth is evident in the absence of Sema3A treatment. C, D, Dendritic growth is stimulated in both controls and in shNdr2-treated neurons by Sema3A. However, a reduced level of differentiation is still evident in shNdr2 cells compared with the Sema3A-treated controls. E, F, Comparison of the number of dendritic intersections with the Sholl analysis confirms that application of Sema3A partially rescues dendritic growth in Ndr2 knock-down neurons (n = 20 cells each, N = 3). G, The total number of dendritic intersections is increased in shNdr2 cells, but does not reach levels of Sema3A-stimulated growth in control cells. Data are presented as mean ± SEM. *p < 0.05, **p < 0.01 compared with untreated controls (Fischer's PLSD).
Discussion
Ndr kinases are targets of the Hippo signaling pathway and control centrosome duplication and alignment, cell-cycle exit, apoptosis, and cell polarity (Hergovich et al., 2006; Emoto, 2011). The mammalian Ndr kinases Lats1/2 and their invertebrate homologs have been implicated in the proliferation and differentiation of neural progenitors (Cao et al., 2008; Fernandez et al., 2009). Ndr1/2 and their homologs, in contrast, appear to be more relevant for neurite development (Emoto, 2011; Ultanir et al., 2012).
Here, we report the selective expression of Ndr2, but not Ndr1, in the developing mouse hippocampus and frontal cortex and its importance as a stimulator of β1-integrin-dependent dendritic growth in mouse hippocampal neurons. This is in contrast to the rat hippocampus, in which both Ndr1 and Ndr2 kinase are expressed and Ndr1 acts as a negative regulator of dendritic branching (Ultanir et al., 2012). However, rat Ndr1 function appears to be differentially regulated along the dendritic tree, because constitutively active Ndr1 also enhances distal dendritic branching. In the same study, Ndr2 was found to compensate for the loss of Ndr1, but only a trend toward reduced dendrite branching was observed upon single Ndr2 knock-down.
In our experiments with primary mouse neurons, the effect of Ndr2 on neurite growth was more clearly evident, because its knock-down significantly reduced and its overexpression profoundly increased dendritic growth. Consistent with findings in C. elegans, we further observed that Ndr kinase function is not confined to dendritic, but is also involved in the control of axonal, growth (Zallen et al., 2000).
Considering the generally larger dendritic arborization in our cultures than those reported by Ultanir et al. (2012), we hypothesized that experimental conditions may account for the observed differences between studies. For example, in primary cell culture, astrocytic deposition of extracellular matrix components powerfully activates integrin-dependent neuronal growth (Nishio et al., 2003) and the Hippo kinase Mst1 activates integrins in hematopoietic cells (Kliche et al., 2012). In fact, in our hippocampal cultures, growth deficits were also evident in shNdr2-treated cells plated on fibronectin instead of a glia-derived matrix and the overgrowth of dendrites and axons in Ndr2-overexpressing cells was abolished through the application of the β1-/β3-disintegrin echistatin. We found that β1-integrin is the predominant of these two integrin isoforms in our neuronal cultures and that knock-down of β1-integrin interferes with dendritic and axonal growth similar to Ndr2 knock-down. Such involvement of integrins in dendritic differentiation has been reported previously: the integrin ligand laminin-1 was found to enhance branching of dendrites and axons in mouse cortical primary neurons in an echistatin-sensitive manner (Moresco et al., 2005) and inactivation of β1-integrins induced rapid retraction of dendrites in retinal ganglion cells (Marrs et al., 2006). More recently, β1-integrins have been shown to facilitate both dendritic and axonal growth in hippocampal primary neurons (Schlomann et al., 2009; Tan et al., 2012).
It is firmly established that, during endosomal trafficking and activation, β1-integrin becomes phosphorylated at the cytoplasmic residues Thr788/789 (Nilsson et al., 2006). This phosphorylation site is involved in the binding of sorting nexin 17 and the protection of β1-integrin from degradation; its inactivation through mutagenesis disturbs recycling to the cell surface without an alteration of internalization rates (Böttcher et al., 2012). Indeed, we observed that Ndr2 is able to stimulate both the phosphorylation of Thr788/789 and the exocytosis rate of β1-integrin in HEK-293T cells. However, increased Thr788/789 phosphorylation upon Ndr2 activation apparently was not direct, but instead was dependent on PKC and CaMKII, which have also been reported to phosphorylate this site in other cellular systems (Suzuki and Takahashi, 2003; Stawowy et al., 2005). It is unlikely that these kinases act upstream of Ndr2, because Thr788/789 phosphorylation was stimulated by constitutively active Ndr2, but not by a constitutively active form of its upstream activator Mst1; rather, they appear to act as intermediate kinases and synergistically with Ndr2 in the activation of β1-integrin. PKCε, through phosphorylation of vimentin, is able to control the surface expression of β1-integrin (Ivaska et al., 2005; Kim et al., 2010). CaMKII-mediated Ca2+ signaling is known as critical regulator of dendritic differentiation (Fink et al., 2003; Konur and Ghosh, 2005) and Ca2+-dependent β1-integrin recycling has been reported during axonal growth cone repulsion (Hines et al., 2010).
We further found that Ndr2 is associated with β1-integrin-positive early and recycling endosomes in neuronal cells. Dynamin-dependent endocytosis, endosomal trafficking, and exocytosis are important steps in the control of integrin-mediated growth. For example, integrins are internalized at the rear end of migrating cells and form new sites of adhesion at their leading edge (Ng et al., 1999; Zerial and McBride, 2001; Pellinen and Ivaska, 2006; Caswell et al., 2009). It is conceivable that Ndr2 may be recruited to integrin-containing endosomes to support the phosphorylation of β1-integrins and their recycling. Achieving high-affinity conformation, however, further requires the assembly of intracellular binding partners such as talin and kindlin. Interestingly, the conserved TT (i.e., Thr788/789 of β1-integrin) phosphorylation motif of β-integrins also plays a role in regulating the competitive association of these intracellular binding partners (Gahmberg et al., 2009).
Evidence suggests that recycling and activation of integrins at the plasma membrane are critically involved in neurite outgrowth: nerve growth factor induces accumulation of β1-integrins in filopodial tips at axonal growth cones (Grabham and Goldberg, 1997) and stimulation of AMPA type glutamate receptors can lead to an increase of α5- and β1-integrins in the cell (Lin et al., 2005). α5β1-integrin relocalizes from somata to dendrites during dendrite formation and maturation in the course of the first two postnatal weeks in vivo (Bi et al., 2001) and β1-integrin associates with integrin-linked kinase, which is required for the balance between axon and dendrite formation, at the dendritic tips of differentiating hippocampal neurons (Guo et al., 2007). Tan et al. (2012) have recently shown that kindlin-1 stimulates growth through associating with β1-integrins in outgrowing axons. We found that the expression of activated β1-integrin is reduced on dendrites of Ndr2 deficient primary neurons. Therefore, the ability of Ndr2 to control the phosphorylation of the cytoplasmic domain of β1-integrin may well serve to regulate integrin activity during neural differentiation.
The relevance of these processes for hippocampal development is evident in Ndr2−/− mice. A gene trap insertion to intron 9 of the Ndr2 gene of these animals resulted in generation of a Ndr21-282::β-geo fusion protein with disrupted protein kinase domain. Because of the activity of the β-geo marker indicating pronounced expression in the hippocampal CA3 pyramidal cell layer of Ndr2−/− mice, we analyzed the dendritic structure of these neurons in mutant and wild types using the Golgi impregnation method. Our data indicate arbor- and layer-specific alterations of dendritic complexity in these cells during postnatal development and maturation. These observations are in agreement with a previous report on the role of β1-integrin in the maturation and stabilization of hippocampal dendrites (Warren et al., 2012), although further work will be required to examine potential Ndr2 effects on distal parts of the apical dendrite. Ndr2-mediated β1-integrin activation may be used by locally acting growth signals for the fine regulation of dendritic structure. For example, CA3 hippocampal neurons express neuropilins and plexin-A1, plexin-A2, and plexin-A3, which can act as receptors for Sema 3A and Sema 3F (Chédotal et al., 1998; Murakami et al., 2001; Pozas et al., 2001). Sema3A has been shown to stimulate dendritic growth in hippocampal primary neurons by activating β1-integrins (Schlomann et al., 2009). Strikingly, the phenotype of Ndr2−/− mice resembles that of Sema3A−/− mice in that ∼1/3 of adult pyramidal cells showed a premature branching (Nakamura et al., 2009).
We therefore finally tested the effect of Sema3A on hippocampal primary neurons and found that it recovers dendritic growth in neurons with Ndr2 knock-down to levels that are comparable to those of untreated control cells. However, shNdr2-treated cells did not reach the level of dendritic growth of control neurons under Sema3A stimulation. Therefore, Sema3A-induced, integrin-dependent dendritic growth appears to interact with the expression of Ndr2. Semaphorins are known to control neuronal differentiation through various signaling pathways (Pasterkamp, 2012) and, intriguingly, both Ndr1 and Ndr2 have been shown to interact with MICAL-1 (molecule interacting with CasL-1), an enzyme that links semaphorins to F-actin rearrangement during axonal and dendritic growth. MICAL-1 inhibits Ndr1/2 by competing with Mst kinases for binding at the hydrophobic motif and it has been suggested that semaphorins may disinhibit the kinase by sequestering MICAL (Hung et al., 2010; Zhou et al., 2011). Ndr kinases and semaphorins thus seem to interact in the intracellular signaling pathways that lead to β1-integrin activation in differentiating neuronal cells.
Conclusions
The control of neurite formation and branching is fundamental to the development of neuronal circuits. Ndr family kinases play multiple roles in these processes and do so, at least in part, by regulating the intracellular trafficking and the activation of β-integrins. The activation of integrins appears to be an important general function of the Hippo signaling pathway, which has been described previously for T cells (Katagiri et al., 2009; Ueda et al., 2012).
Footnotes
This work was supported by the German Research Foundation (DFG; Grant SFB854 to O.S., S.K., and C.F. and Grant STO488/3 to O.S.) and the European Union (ERA-Net on Rare Diseases Project CURE-FXS to O.S.). K.R. was a scholar of the DFG graduate school (Grant GRK1167). We thank D. Wolters, A. Koffi von Hoff, A. Ramonat, S. Stork, and F. Webers for expert technical assistance; A. Deter and T. Porcuzek for excellent animal care; Y.E. Demiray for experimental support; D. Lang for helpful discussions; and M. Kreutz for comments on the manuscript.
The authors declare no competing financial interests.
- Correspondence should be addressed to Oliver Stork, Department of Genetic and Molecular Neurobiology, Institute of Biology, Otto-von-Guericke University, Magdeburg, Leipziger Strasse 44, 39120 Magdeburg, Germany. oliver.stork{at}ovgu.de





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}