

Abstract
The elevation of kynurenic acid (KYNA) observed in schizophrenic patients may contribute to core symptoms arising from glutamate hypofunction, including cognitive impairments. Although increased KYNA levels reduce excitatory neurotransmission, KYNA has been proposed to act as an endogenous antagonist at the glycine site of the glutamate NMDA receptor (NMDAR) and as a negative allosteric modulator at the α7 nicotinic acetylcholine receptor. Levels of KYNA are elevated in CSF and the postmortem brain of schizophrenia patients, and these elevated levels of KYNA could contribute to NMDAR hypofunction and the cognitive deficits and negative symptoms associated with this disease. However, the impact of endogenously produced KYNA on brain function and behavior is less well understood due to a paucity of pharmacological tools. To address this issue, we identified PF-04859989, a brain-penetrable inhibitor of kynurenine aminotransferase II (KAT II), the enzyme responsible for most brain KYNA synthesis. In rats, systemic administration of PF-04859989 dose-dependently reduced brain KYNA to as little as 28% of basal levels, and prevented amphetamine- and ketamine-induced disruption of auditory gating and improved performance in a sustained attention task. It also prevented ketamine-induced disruption of performance in a working memory task and a spatial memory task in rodents and nonhuman primates, respectively. Together, these findings support the hypotheses that endogenous KYNA impacts cognitive function and that inhibition of KAT II, and consequent lowering of endogenous brain KYNA levels, improves cognitive performance under conditions considered relevant for schizophrenia.
Introduction
Kynurenic acid (KYNA) is a naturally occurring product of tryptophan metabolism that is reported to inhibit NMDA and neuronal nicotinic α7 receptors (Hilmas et al., 2001), both of which contribute to spatial working memory processes (Castner et al., 2011). Though measured brain levels of KYNA are low (nanomolar), chronic increases, such as those observed in the brains and CSF of patients with schizophrenia (Erhardt et al., 2001; Schwarcz et al., 2001; Nilsson et al., 2005; Linderholm et al., 2012), are hypothesized to contribute to cortical hypofrontality and, thus, impairments in cognitive function. In animal models, increased KYNA levels have been reported to produce effects consistent with a role in symptom etiology in schizophrenia, including deficits in spatial and working memory (Chess et al., 2007, 2009), impairments in sensory gating (Shepard et al., 2003), and reduction in dopaminergic and glutamatergic neurotransmission (Rassoulpour et al., 2005; Amori et al., 2009; Konradsson-Geuken et al., 2010). It is important to highlight the role of kynurenine aminotransferase II (KAT II) in the modulation of dopaminergic tone and function in the rodent striatum, which is critical for motivated behaviors, including addiction (Justinova et al., 2013). Further, in some neurodegenerative diseases such as Parkinson's disease, Huntington's disease, and epilepsy, it is believed that elevated levels of KYNA or its analog provide neuroprotection potential (Wu et al., 2000; Carrillo-Mora et al., 2010). Therefore, both activation and downregulation of KAT II activity can be envisioned to play a role in phenomena ranging from motor control to cognition.
Despite the fact that behavioral impairments can be induced by exogenously administered KYNA, establishing the role of endogenous brain KYNA has been hampered by the limited availability of pharmacological agents, none of which appear to cross the blood–brain barrier. The identification of PF-04859989 (Fig. 1A), a potent and selective KAT II inhibitor with excellent brain penetration after systemic administration (Dounay et al., 2012), addresses this limitation and provides an opportunity to better understand the physiological role of KYNA in the brain.
In this article, the pharmacological characterization of PF-04859989 is described; specifically, its receptor affinity, its effects on the KAT II enzyme, its ability to penetrate the brain, and, most significantly, its ability to reduce brain KYNA levels. Next, the effect of reducing brain KYNA synthesis on neural function is described in studies that evaluated the impact of PF-04859989 in animal models where elevated KYNA had previously been shown to induce impairments. Thus, auditory gating was used as an indicator of preattentive processing measured by an attenuation of auditory evoked potentials in response to pairs of acoustic stimuli (Hajós, 2006). A previous study showed that systemic administration of l-kynurenine led to increased brain KYNA and disruption of hippocampal auditory gating in anesthetized rats (Shepard et al., 2003). However, it was unknown whether lowering KYNA would impact auditory gating processes or could prevent gating deficits induced by drugs such as amphetamine or ketamine, which mimic impairments measured in schizophrenic patients (Javitt et al., 2008). In the present study, auditory gating was evaluated in the hippocampus (CA3) and entorhinal cortex of anesthetized rats.
In addition to preattentive processing abnormalities, schizophrenia is also marked by deficits in several cognitive domains, particularly impaired attentional and working memory processes. To investigate the consequences of decreased KYNA on attentional processes, the effects of PF-04859989 administration were measured in an operant sustained attention task (SAT) in rats. In addition, since NMDA antagonists have been shown to profoundly impair spatial working memory in rodents (Baddeley, 1992), nonhuman primates, and humans (Goldman-Rakic, 1994; Levin et al., 1998), and are commonly used to model the hypoglutamatergia hypothesized to underlie cognitive deficits in schizophrenia, the ability of PF-04859989 to antagonize the disruptive effects of ketamine was tested in a radial arm maze (RAM) working memory task in rats, and in a spatial delayed response (SDR) working memory task in nonhuman primates.
Materials and Methods
Chemicals and drugs
PF-04859989 was synthesized by the Pfizer Neuroscience Medicinal Chemistry group. [3H]MDL-105,519 was purchased from GE Healthcare Bio-Sciences Corp. All other chemicals were purchased from Sigma-Aldrich.
Production of enzymes
Human KAT I, KAT III, KAT IV, KMO, and rat KAT II cDNAs were ordered from DNA2.0. Human KAT II cDNA was ordered from GeneCopoeia. Rhesus monkey KAT II cDNA was cloned in-house directly from rhesus brain RNA obtained from BioChain using reverse transcription-PCR with SuperScript III RT enzyme, gene-specific primer, custom 5′ and 3′ PCR primers incorporating a C-terminal HIS6 tag followed by a stop codon to facilitate protein purification, and Accuprime Pfx DNA Polymerase according to vendor protocol. All cDNAs were cloned into pFastBac1 vector (Invitrogen), propagated in Escherichia coli DH5α, and used to infect SF21 insect cells according to the manufacturer's directions for large-scale expression of recombinant enzymes. Cleared cell lysate was batch bound to pre-equilibrated Ni-NTA resin and washed with buffer. The enzyme/resin complex was transferred to a column, washed with 20 and 60 mM imidazole/lysis buffer, and eluted with 60–400 mM imidazole gradient (five column volumes). Enzyme fractions were pooled based on purity by SDS-PAGE analysis and dialyzed into an appropriate storage buffer.
Enzyme assays
Kynurenine metabolic enzymes used in this study are described in Table 1. KAT activity was measured using an absorbance assay, based on the observation that the substrate l-kynurenine absorbed light at 370 nM, while the product KYNA did not. Activity was measured by monitoring decreases in absorbance at 370 nM as the substrate was consumed. KAT II activity was measured in a 384-well format using 150 mM Tris acetate buffer, pH 7.0; 1 mM l-kynurenine; 1 mM α-ketoglutaric acid; 70 μm pyridoxal 5-phosphate (PLP); 0.1% bovine gamma globulin (BGG); and 30 nM human, 5 nM rat, or 20 nM rhesus monkey recombinant KAT II enzyme. KAT I activity was measured in a 384-well format using 200 mM potassium phosphate buffer, pH 7.4, 1 mM l-kynurenine, 1 mM pyruvate, 70 μM PLP, 0.1% BGG, and 50 nM human recombinant KAT I enzyme. KAT III activity was measured in a 384-well format using 100 mM K2HPO4/100 mm boric acid buffer, pH 9.0, 1 mM l-kynurenine, 1 mM phenylpyruvate, 70 μM PLP, 0.1% BGG, and 200 nM human recombinant KAT III enzyme. To determine its IC50 values versus that of the KAT enzymes, PF-04859989 was dissolved in DMSO and diluted by a half-log scheme with a final dilution into aqueous buffer for a final concentration of 0.36% DMSO per well. Plates were sealed and incubated at 37°C for 16 h (KAT I, KAT II) or 42°C for 22 h (KAT III), and A370 nm was read on a SpectroMax plate reader. The IC50 values were calculated by comparing percents inhibition at each inhibitor concentration to that of the DMSO control.
Kynurenine metabolic enzymes used to evaluate potency and specificity of PF-04859989
KAT IV activity was measured in an NMR assay using 50 mm deuterated Tris buffer, pH 7.5, 100 mM NaCl and 10% deuterated water, 2 mM l-aspartate, 500 μm 2-oxoglutarate, 10 μm PLP, and 80 nm human recombinant KAT IV enzyme. To determine the percentage of inhibition, PF-04859989 was dissolved in deuterated DMSO and added at a final concentration of 50 μM. Samples were incubated at room temperature for 45 min, reaction was quenched with 1N HCl, and NMR data were collected at 298K on a Bruker DRX500 NMR spectrometer. The percentage of inhibition was calculated by comparing the integral of the glutamate signal at 2.49 ppm to that of the DMSO control.
Kynurenine monooxygenase (KMO) activity was measured in a kinetic absorbance assay in 384-well format using 50 mM potassium phosphate buffer, pH 7.5, 2.5 mM MgCl2, 0.3 mm l-kynurenine, 0.8 mM NADPH, and 159 nM human recombinant KMO enzyme. To determine the IC50 value, PF-04859989 was dissolved in DMSO and diluted by a half-log scheme with a final dilution into aqueous buffer for a final concentration of 0.36% DMSO per well. Plates were sealed and incubated at 37°C for 45 min, and read on a SpectraMax absorbance microplate reader at 340 nm every 5 min for 1 h at 37°C. Slope (as a measure of enzyme activity) was plotted versus drug concentration, and IC50 values were calculated using linear regression analysis.
Binding at the glycine site of the NMDA receptor (NMDAR) was determined using a competitive radioligand receptor binding assay, as previously described (Baron et al., 1996).
Animal care and handling
Rodents.
All animals were housed two per cage under a 12 h light/dark cycle in a temperature- and humidity-controlled, Association for Assessment and Accreditation of Laboratory Animal Care International-accredited vivarium, for at least 7 d before testing, except where noted otherwise. Food and water were available ad libitum, except as noted in individual sections below. Animals were handled and cared for according to the Guide for the Care and Use of Laboratory Animals, and all procedures were performed with the approval of the Institutional Animal Care and Use Committee at the Pfizer Global Research and Development site in Groton, CT.
Nonhuman primates.
Ten rhesus monkeys (Macaca mulatta; 6 females and 4 males; 10–25 years of age) were housed at the Yale School of Medicine and maintained in accordance with the Yale Animal Care and Use Committee procedures as well as federal guidelines for the care and use of nonhuman primates. Animals were provided water ad libitum, were fed their standard fruit and monkey chow immediately after testing, and were supplied with environmental enrichment.
Determination of the unbound fraction in rat plasma and brain and rat pharmacokinetic studies
The rat plasma and brain unbound fractions for PF-04859989 were determined using 96-well equilibrium dialysis using an established methodology (Kalvass and Maurer, 2002).
Male Sprague Dawley rats weighing 200–250 g were obtained from Charles River Laboratories. Upon arrival, the rats were maintained for at least 2 d on a 12 h light/dark cycle. Rats were administered a single subcutaneous dose of PF-04859989 (3.2, 10, or 32 mg/kg; N = 3/dose/time point). Dosing solutions were prepared in sterile water and administered at a volume of 2 ml/kg in the mid-back region. Rats were euthanized in a CO2 chamber at 0.25, 0.5, 1, 2, 4, or 7 h after dosing (0.5, 1, and 3 h time points after administration at 10 mg/kg only). Whole blood was collected by cardiac puncture into Vacutainer tubes containing heparin and were stored on ice until centrifuged for the preparation of plasma. Whole brains were collected by decapitation, rinsed with PBS, weighed, and immediately frozen on dry ice upon collection. The concentration of PF-04859989 in brain and plasma samples was determined via HPLC-tandem mass spectrometry. The total plasma and total brain concentrations were subsequently converted to unbound molar concentrations through use of the unbound fraction in the respective matrix and the molecular weight of the compound.
Microdialysis in freely moving rats
Male Sprague Dawley rats (280–340 g) with surgically implanted guide cannulae (Bioanalytical Systems) in the prefrontal cortex (distance from bregma: anteroposterior, +3.7 mm; mediolateral, −0.7 mm; dorsoventral −2.0 mm; Paxinos and Watson, 1997) were ordered from Charles River Laboratories. The surgery was performed under isoflurane anesthesia, and cannulae were affixed to the skull using bone screws and dental acrylic. Approximately 18 h before testing, guide stylets were removed, and probes (BR-4; Bioanalytical Systems) with a polyacrylonitrile membrane (outer diameter, 340 mm; molecular weight cutoff, 30,000 Da) and a 4 mm dialysis tip were inserted. The inlet and outlet of the probe were connected with flexible polyether ether ketone tubing (inner diameter, 0.005 inches; Upchurch Scientific), and the probes were perfused at 1.5 μl/min with artificial CSF (147 mM NaCl, 2.7 mM KCl, 1.0 mM MgCl2 hexahydrate, 1.3 mM CaCl2 dihydrate) with a CMA/100 microperfusion pump (CMA Microdialysis). On the test day, microdialysate samples (30 μl) were continuously collected and automatically injected every 20 min for analysis by on-line HPLC/fluorimetry detection.
Rats were dosed subcutaneously with vehicle (2 ml/kg) or PF-04859989 (3.2, 10, and 32 mg/kg) dissolved in vehicle [1% acetic acid in distilled water (v/v)]. Test compound or vehicle was administered ∼2 h after basal KYNA levels had stabilized, and the effects on KYNA levels were monitored for a period of at least 4 h.
Determination of KYNA in dialysate
KYNA was analyzed on a 150 × 3 mm, 3 μM BDS Hypersil C18 column, (Thermo Electron Corporation) set at 30°C. The analytes were separated with a mobile phase containing 50 mM sodium acetate (pH to 6.3 with glacial acetic acid), 250 mM zinc acetate, 2–6% acetonitrile, delivered at 0.3–0.350 ml/min by a LC-10ADvp pump (Shimadzu) and detected fluorometrically at excitation 344/emission 398 nm (RFXL-10 detector, Shimadzu). Extracellular levels of KYNA were measured and reported as nanomolar concentrations and the percentage of baseline level, respectively. Data were collected by EZChrom Elite Client/Server software version 3.2.1 (Agilent Technologies).
In vivo electrophysiology
Experiments were performed on male Sprague Dawley rats (Harlan; weighing 260–310 g) under chloral hydrate anesthesia (400 mg/kg, i.p.). Simultaneous field potentials were recorded from the CA3 region of the left hippocampus and the ipsilateral entorhinal cortex; and were amplified, filtered (0.1–100 Hz), displayed, and recorded for analysis, as described previously (Hajós et al., 2008). Quantitative EEG analysis was performed using fast Fourier transforms. Auditory stimulation consisted of two consecutive tone bursts of 10 ms duration at a frequency of 5 kHz; the sound pressure level was 95 dB. The delay between the first “conditioning” stimulus and the second “test” stimulus was 0.5 s, and the time interval between tone pairs was 10 s. Auditory evoked potentials were determined by measuring the potential difference between the positive and the negative deflection, 15–30 and 40–50 ms after stimulation (P20 and N40), respectively. For quantification, sweeps were averaged, the amplitude was determined, and the ratio of the response after the second stimulus [test (T)] and the first stimulus [conditioning (C)] was calculated. The percentage of gating was determined by the following formula: (1 − T amplitude/C amplitude) × 100. Individual waveform averages consisted of 25 gating stimuli with an interstimulus interval of 10 s. Individual averages were separated by 50 s.
Animals were administered either PF-04859989 (32 mg/kg, s.c.) or saline vehicle (1.0 ml/kg, s.c.) and returned to their home cages. After ∼45 min, animals were anesthetized and experimental procedures were initiated. Baseline auditory gating was determined by an average of 50 sequential evoked potentials (two blocks of 25, separated by 50 s) in response to conditioning and test stimuli over a 10 min interval. After baseline auditory gating was determined, either amphetamine (1 mg/kg, d-amphetamine sulfate, dissolved in H2O) or ketamine (3 mg/kg, dissolved in saline solution) was administered intravenously to all animals (∼60–120 min after initial subcutaneous injection) to disrupt sensory gating. Recordings of evoked potentials commenced 5 min after amphetamine or ketamine administration. Six additional blocks of 25 auditory evoked potentials were computed over a 30 min period. The amphetamine and ketamine effects were determined by combining all six blocks into a single 150 sweep average. Animals pretreated with saline vehicle were subsequently administered haloperidol 0.1 mg/kg, i.v., as a positive control; disrupted auditory gating was significantly restored by administration of haloperidol (0.1 mg/kg; data not shown).
Averages for each experiment were normalized and printed out. Means and SEs were tabulated for response amplitudes and the percentage of response gating. Significant differences between saline and PF-04859989-pretreated rats were determined by means of two-tailed paired Student's t tests.
Sustained attention task
The effects of PF-04859989 in the SAT were determined in male Wistar rats (Charles River) performing the standard SAT and a distractor SAT (dSAT) version of the task. Animals initially weighing between 250 and 300 g were individually housed, were handled extensively before the beginning of training, and were food deprived by restricting food access (8 g rodent chow; Lab Diet 5053, Purina) after each operant training or practice session. Food was also provided as a reward during task performance (see below). On nontesting days, food access was increased to 16 g/d. Water was available ad libitum.
Behavioral training and testing were conducted in 18 operant chambers located inside sound-attenuating chambers (Med Associates Inc.). This operant procedure consists of a random sequence of signal and nonsignal events followed by a variable intertrial interval (ITI), as described in detail by McGaughy et al. (1996). Briefly, after a signal (center panel light illumination for 500, 50, or 25 ms) or a nonsignal event (no light illumination), levers were extended into the chambers 2 s later and remained active for 4 s. Correct responses (lever presses) were either hits (signal trial) or correct rejections (nonsignal trial), for which animals were rewarded. Incorrect responses were misses (signal trial) or false alarms (nonsignal trial), which were unrewarded and triggered the ITI (9 ± 3 s). An error of omission was defined as the failure to operate a lever within 4 s after a trial initiation. To obtain an overall measure of performance that reflects the animals' accuracy in signal and nonsignal trials, an overall vigilance index (SAT/dSAT score) was calculated as follows: (hits − false alarms/[2(hits − false alarms) − (hit + false alarms)2]. SAT/dSAT values range from −1 (total inaccuracy) to +1 (perfect response accuracy), with 0 indicating random lever selection.
The task duration was 40 min, and data were separated into five 8 min blocks. In the dSAT, a distractor (chamber houselights flashing on/off at 0.5 Hz) was presented during the second and third blocks, which reduces the discriminability of the signal and increases the demand on top-down control to maintain performance (St Peters et al., 2011). The animals were exposed to “distractor” sessions two to three times within a 2 week period before drug administration. PF-04859989 (3.2, 10, and 32 mg/kg, s.c.) or saline solution was administered 60 min before SAT or dSAT was assessed.
Statistical analysis
The relative number of hits was calculated as [hits/(hits + misses)] for each signal length, the relative number of correct rejections was calculated as [correct rejections/(correct rejections + false alarms)], and errors of omission were recorded. Angular transformations were used to adjust the percentage of hits and the percentage of correct rejections using the formula (x′ = 2 × Arsinx1/2). Repeated-measures ANOVA was used to assess the effects of dose, block, and signal duration on dSAT score and hits. For correct rejections and omissions, the factor “signal duration” was not included. Post hoc comparisons were conducted using a Fisher's PLSD test.
Radial arm maze task
Administration of the NMDA antagonist ketamine to well-trained rats consistently produces significant disruption of performance in the RAM task, resulting in a significant increase in the number of working memory errors. This study was designed to test the hypothesis that KAT II inhibition would provide significant protection versus the ketamine-induced working memory deficits in well-trained rats. Since these experiments were conducted in highly trained animals that were performing the task with very few errors, the cognition-enhancing effects of PF-04859989 alone could not be evaluated in this paradigm, and additional experiments in an acquisition version of the task would be needed to evaluate the potential working memory-enhancing effects of the compound on its own.
Subjects.
Sixty male Long–Evans rats (Charles River Laboratories) were used in the RAM experiments. Body weights ranged between 200 and 225 g upon arrival and between 400 and 450 g during testing.
Drug administration.
For the RAM experiment, the KAT II inhibitor PF-04859989 HCl was formulated as a solution in saline. Ketamine was diluted from a commercially available veterinary injection solution of 100 mg/ml for a 10 mg/kg dose with saline. All compounds were administered subcutaneously at a volume of 1 ml/kg.
Ketamine-disrupted radial arm maze experiment.
Two cohorts of Long–Evans rats (N = 30 each) were trained in a spatial working memory task on an eight-arm RAM (Pathfinder Maze System, Lafayette Instruments), using a procedure adapted from Ward et al. (1990) and described in detail in Strick et al. (2011). Briefly, beginning 1 week after arrival, rats were food-restricted to provide motivation to perform the RAM task. Maze habituation (2 d) and then training were initiated 1 week later. During training, one reinforcement pellet was placed in the food cup at the end of each arm. The doors to the arms of the maze were not used and were always kept open to simulate the “free-foraging” that rodents encounter in the wild. The task requires that the animals enter each arm to retrieve the pellets, using spatial cues in the room to remember which arms of the maze they have previously entered. Rats were individually placed on the maze and allowed to navigate until all eight arms were entered and the pellets were consumed, until 30 choices were made, or until 5 min had elapsed. Entry into an arm previously entered was counted as an error. If an animal failed to choose all eight arms in 5 min, the arms not chosen were also counted as errors. Animals were trained once a day, 5 d a week (Monday to Friday), and the learning criterion was defined as two or fewer errors on 2 consecutive days. Training continued until all animals had reached the training criterion, typically after ∼15 d of training.
On test days, animals were given vehicle or various doses of PF-04859989, followed 90 min later by administration of ketamine (10 mg/kg, s.c.), and performance on the maze was evaluated 30 min later by an observer that was blinded to animal treatments. Subjects were randomly assigned to treatment groups and tested as follows: on Mondays and Thursdays, all animals were tested on the RAM to be “qualified” for drug testing on the following day by committing two or fewer errors. On test days, qualifying animals were randomly assigned to treatment groups as follows: (1) vehicle plus vehicle; (2) vehicle plus ketamine; (3) PF-04859989 (1.0 mg/kg) plus ketamine; (4) PF-04859989 (3.2 mg/kg) plus ketamine; (5) PF-04859989 (10.0 mg/kg) plus ketamine; and (6) PF-04859989 (32.0 mg/kg) plus ketamine. The dependent measure was the number of errors committed during the 5 min test period. Due to the extensive amount of time necessary for animal training and testing, the randomization and testing procedure was repeated several times until the group numbers equaled 10–11 animals for each of the two cohorts.
Statistical analyses.
Data from each of the two cohorts (i.e., replications) were combined, resulting in final group numbers of 21–23 animals per group. Data were analyzed by the Pfizer Biostatistics Group using a generalized linear mixed model, assuming an underlying Poisson distribution, followed by Hochberg's test for multiple post hoc comparisons using the vehicle-plus-ketamine group as the control (SAS software, SAS Institute).
Spatial delayed response in the nonhuman primate
Administration of the NMDA antagonist ketamine consistently produces significant disruption of performance in the SDR task in nonhuman primates, resulting in a significant increase in the number of working memory errors. This study was designed to test the hypothesis that KAT II inhibition would provide significant protection versus the ketamine-induced working memory deficits in nonhuman primates.
Drug administration.
Subjects received pretreatment with vehicle (sterile saline solution) or PF-04859989 (0.0032, 0.32, or 3.2 mg/kg, s.c.) 4 h before cognitive testing. They then received an intramuscular injection of either placebo (sterile saline) or ketamine (0.7–1.7 mg/kg; Fort Dodge Co.) 0.25 h before cognitive testing. The dose of ketamine for each animal was predetermined such that all animals achieved a comparable magnitude of cognitive impairment (e.g., a score of less than ∼50% correct) relative to their pretreatment baseline performance of ∼70% correct. Thus, for the present study, one monkey received 0.7 mg/kg, seven monkeys received 1.0 mg/kg, and two monkeys received 1.7 mg/kg. The vehicle/PF-04859989 and placebo/ketamine treatments were assigned using a randomized Latin square design with a total of eight conditions. With the exception of the vehicle/placebo condition, there was a minimum 1 week washout period between all acute challenges during which time animals were required to “restabilize,” which was defined as at least three consecutive testing sessions wherein cognitive performance ranged between 65% and 75% correct.
Behavioral observations.
Behavioral observations were performed in the homecage of the animal 5–10 min after vehicle/ketamine treatment using SimTrack, a computer program developed for this purpose by S.A.C. and G.V.W. The behaviors were separated into various categories, including gross motor and hallucinatory-like behaviors. The gross motor category encompassed behaviors such as circling, pacing, and cage crossing. Hallucinatory-like behaviors included tracking nothing, staring at nothing, abnormal sniffing of fingers or hands, hypervigilance, excessive grooming of self, checking, and manipulation of thin air (for further details of categorization of behaviors, see Castner and Goldman-Rakic, 1999; Roberts et al., 2010). Other behaviors of interest included orobuccal dyskinesias as well as bradykinesia.
Cognitive testing.
Animals were trained to stability on a variable spatial delayed response task in a sound-attenuated Wisconsin General Testing Apparatus. Briefly, the monkey watches while the investigator baits one of two to seven wells with a highly preferred food reward and then covers all of the wells with identical square plaques. An opaque screen is then lowered for one of five variable delays, which are pseudorandomized across trials within a session. Thus, delays are defined as 0–4 N, where “N” is a value that is animal dependent and ranges from 1 to 10 depending upon the difficulty level of the task at which an animal reaches the criterion of stable performance. At the end of the delay period, the opaque screen is raised and the animal must select the well that had been baited to obtain a reward. Each test session consists of 20 trials wherein both the baited well and delay length are pseudorandomized across trials. Before study initiation, all subjects were required to reach stability over a period of ≥10 consecutive test sessions, where stability was defined as an average of 65–75 ± ≤2.5% correct. Stability was attained by varying the number of wells and the delay value for each animal. Subjects were originally trained on a two-well board with an N value of 1. The N value and the number of wells were gradually increased until the animal consistently scored within stability range. Once stability was attained for a given number of wells and N value, that combination was kept constant throughout the course of the study (Roberts et al., 2010). The range of stable performance for the 10 subjects was two to five wells and an N value of 1–7 s (median, four-well testing board, N = 5 s).
Statistical analysis and results.
Working memory performance was first assessed by two-way ANOVA, and then one-way ANOVAs were performed as appropriate on placebo and ketamine conditions separately, as described in the Results. Post hoc comparisons were performed using a Dunnett's t-test (two-tailed) for these data. A three-way ANOVA was performed to examine the impact of PF-04859989 on ketamine-induced, delay-dependent errors in the task. Behavioral data were analyzed by one- and two-way ANOVA.
Results
Characterization of PF-04859989
PF-04859989 (Fig. 1A) was discovered during high-throughput screening using a functional enzyme assay. At 23 nm, PF-04859989 inhibited human KAT II activity by 50% (Fig. 1B). Further characterization showed ∼10-fold weaker activity at the rat KAT II enzyme (263 nm) and >460-fold selectivity over all other KAT -related enzymes (Fig. 1C). In addition, PF-04859989 showed limited or negligible activity at other KATs; at KMO, another enzyme that shares l-kynurenine as a substrate; and at serine racemase, an enzyme that also contains PLP cofactor and had negligible affinity for the NMDAR glyB site, to which KYNA itself binds. These results are presented in Figure 1C. Furthermore, PF-04859989 had no affinity for other receptors or channels, and failed to inhibit enzymes in a broad-panel screen BioPrint (CEREP) when tested at 10 μm (data not shown).

PF-04859989, a potent, selective, and brain-penetrant inhibitor of KAT II, reduced extracellular KYNA in rat brain. A, Structure of PF-04859989. B, A representative plot of % Effect (inhibition) of KAT II enzyme activity vs inhibitor concentration. C, Potency of PF-04859989 at KAT II and related enzymes. Activity was determined using recombinant human and rat proteins or rat brain tissue. Results shown are the mean of N > 3 determinations.
Determination of the unbound fraction in rat plasma and brain and in rat pharmacokinetic studies
The mean rat plasma and brain unbound fractions were 0.893 and 0.393, respectively. The plasma and brain concentrations of PF-04859989 increased with increasing dose. PF-04859989 exhibited intercompartmental equilibrium, and the concentration–time profiles are displayed in Figure 2A. The mean total brain-to-plasma area under the curve (AUC) ratio and the mean unbound brain-to-unbound plasma AUC ratio were 1.4 and 0.6, respectively.

PF-04859989 readily enters the brain after systemic administration and inhibits KAT II to reduce brain KYNA. A, Unbound plasma and brain exposure of PF-04859989 after subcutaneous administration of 3.2 mg/kg drug (n = 18, error bars indicate SD), 10 mg/kg drug (n = 12, error bars indicate SEM), or 32 mg/kg drug (n = 18; error bars indicate SEM). B, Endogenous extracellular levels of KYNA are reduced in rat prefrontal cortex in response to the inhibition of KAT II by PF-04859989 administration of 3.2 mg/kg drug (n = 4, error bars indicate SEM), 10 mg/kg drug (n = 17, error bars indicate SEM), or 32 mg/kg drug (n = 9; error bars indicate SEM). C, Endogenous extracellular levels of KYNA are reduced to a similar extent in rat brain hippocampus, striatum, and prefrontal cortex. (n = 3, 5, and 17, respectively, error bars indicate SEM).
PF-04859989 decreased KYNA in rat prefrontal cortex extracellular fluid
Systemic subcutaneous dosing of PF-04859989 resulted in a dose-responsive decrease in KYNA as measured by microdialysis in PFC of freely moving rats. A maximum decrease in KYNA, by ∼80% of basal level, was observed after administration of the 32 mg/kg dose. This decrease was achieved by ∼1 h after dosing and was sustained for at least 4 h. No further decrease was seen at a dose of 100 mg/kg (data not shown). A lower dose of 10 mg/kg decreased basal KYNA levels by ∼40%. At the lowest dose tested, 3.2 mg/kg, KYNA decreased by ∼20% by 3 h postdose (Fig. 2B). PF-04859989 caused comparable decreases across other brain regions tested, including hippocampus and striatum (Fig. 2C).
PF-04859989 reversed amphetamine- or ketamine-induced deficits in the entorhino-hippocampal system as measured by in vivo electrophysiology
The effect of reducing brain KYNA synthesis on neural function was examined by evaluating the impact of PF-04859989 in a rat model of auditory gating, an indicator of preattentive processing. The attenuation of auditory evoked potentials in response to pairs of acoustic stimuli was measured (Hajós, 2006) in the hippocampal CA3 region and in the entorhinal cortex. Auditory gating values, as assessed by calculating the difference between the P20 and N40 deflections of the local field potential (LFP) were as follows: 85 ± 4 μV (CA3) and 79 ± 5 μV (entorhinal cortex) after saline injection (n = 11); and 74 ± 4 μV (CA3) and 72 ± 8 μV (entorhinal cortex) after PF-04859989 injection (n = 11). Preinjection with PF-04859989 (32 mg/kg, s.c.) alone had no effect on auditory gating parameters. Interestingly, however, PF-04859989 pretreatment prevented auditory gating deficits induced by either amphetamine (n = 5) or ketamine (n = 6) administered ∼1 h after pretreatment in both hippocampal CA3 area and the entorhinal cortex (Fig. 3). Furthermore, spontaneous LFP oscillations measured concomitantly with gating were evaluated after pretreatment with PF-04859989 but before amphetamine or ketamine challenge, and a significant (p < 0.05, paired t tests, n = 11) reduction of low-frequency delta oscillation power was observed.

Inhibition of KAT II reversed entorhino-hippocampal deficits induced by amphetamine or ketamine. A, Baseline low-frequency LFP delta oscillation is significantly reduced (horizontal bar and asterisk denote significance at p < 0.05, paired t tests; n = 11) by PF-04859989 (32 mg/kg, s.c.; light gray traces) in both hippocampus (CA3) and entorhinal cortex (ENT) compared with saline-treated rats (dark gray traces). Insets show representative LFP traces exhibiting fast, regular oscillations in animals pretreated with PF-04859989 before amphetamine or ketamine challenge. Note the large-amplitude, slow, irregular activity recorded from saline-treated rats. Shaded areas indicate SEM. B, PF-04859989 pretreatment prevented or attenuated disruptive effects of amphetamine (n = 5) and ketamine (n = 6) in both regions examined, as expressed as the percentage decrease in auditory gating relative to control. *p < 0.05, **p < 0.001. Error bars indicate SEM.
PF-04859989 improved performance in the sustained attention task
The presentation of a distractor resulted in a deficit in performance on the dSAT score with interactions observed between distractor × block (F(4,28) = −12.249, p < 0.001), block × signal duration (F(8,56) = 2.213, p < 0.05), and distractor × signal duration × block (F(8,56) = −2.820, p < 0.05). The distractor also resulted in a deficit in the percentage of hits (F(1,7) = −18.978, p < 0.001; data not shown), but interactions were observed between distractor × block (F(4,8) = −6.106, p < 0.001), signal duration × block (F(8,56) = −2.083, p < 0.01), and distractor × signal duration × block (F(8,56) = −3.226, p < 0.01). Finally, there was a significant deficit in the percentage of correct rejections caused by the distractor with a strong distractor × block interaction (F(4,28) = −12.205, p < 0.001).
Statistical analysis of the effects of PF-04859989 on the dSAT score (Fig. 4) showed no effect of dose (F(3,21) = 0.367, p > 0.05), while the expected significant effects of both signal duration and block were observed (F(1.179,8.255) = −87.500, p < 0.0001; and F(3.186,22.304) = −22.306, p < 0.01, respectively). No interactions were observed for signal duration × block (F(8,56) = −0.745, p > 0.05), signal duration × dose (F(4.025,28.172) = −1.349, p > 0.05), or dose × block (F(12,84) = −1.219, p > 0.05); however, there was a strong interaction among signal duration, dose, and block (F(21.023,147.162) = −2.093, p < 0.01). Post hoc comparisons showed significant attenuation of the distractor effect at the medium signal (50 ms) duration at 10 and 32 mg/kg (p < 0.01 and p < 0.01, respectively) during the first postdistractor block. The 10 mg/kg dose also improved the dSAT score during the presence of the distractor at the long signal duration (500 ms; p < 0.05).

KAT II inhibition improved disrupted performance in sustained attention task in rats. PF-04859989 attenuated the decline in performance in the presence of distractor (blocks 2 and 3) in the sustained attention task (n = 8) and improved significantly the vigilance index for long and medium signal durations (500 ms and 50 ms, respectively). This positive effect was due to an increase in correct rejections during distractor presentation and hits in response to the 500 ms stimulus (data not shown) and to the 50 ms stimulus, mainly during postdistractor performance (blocks 4 and 5). Error bars indicate SEM.
Significant effects on percentage hits were again found for both signal duration and block in animals dosed with PF-04859989 (F(1.307,9.149) = −144.752, p < 0.001; and F(4,28) = −8.240, p < 0.001, respectively), but not for dose (F(3,21) = −0.091, p = −0.964). There were no observed interactions for dose × signal duration (F(4.086,28.601) = −1.024, p > 0.05) or dose × block (F(8.959,62.710) = −0.656, p > 0.05), but strong interactions occurred for both signal duration × block (F(8,56) = −3.042, p < 0.01) and signal duration × dose × block (F(24,168) = −1.64, p < 0.05). Post hoc comparisons showed an improvement in the percentage of hits (data not shown) in the medium signal (50 ms) at 10 mg/kg (p < 0.05).
For percentage correct rejections, a significant effect was observed for block (F1.826,12.784) = −38.229, p < 0.001), but not for dose (F(3,21) = −0.400, p > 0.05) or dose × block interaction (F(4.916,34.41) = 1.252, p > 0.05; data not shown). Overall, these data showed that PF-04859989 enhanced cognitive effects as measured in the sustained attention task, partially reversing the performance impairment produced by the presence of the distractor.
PF-04859989 improved ketamine-disrupted working memory in the radial arm maze task
In rats that were well-trained on the RAM task, the administration of ketamine produced a significant deficit in performance on the RAM task, as evidenced by a significant increase in mean errors from 0.82 ± 0.34 errors in the vehicle-plus-vehicle group to 5.68 ± 0.69 in the vehicle-plus-ketamine group (p < 0.0001, as determined by post hoc analysis; Fig. 5). Administration of the KAT II inhibitor PF-04859989 at the 32.0 mg/kg dose before ketamine administration significantly attenuated the mean errors produced by ketamine (mean errors = 2.10 ± 0.22, p < 0.0001). These data show that PF-04859989 afforded significant protection from the ketamine-induced deficits in working memory in rats on the RAM.

KAT II inhibition attenuated the working memory deficits produced by ketamine administration in rats performing a radial arm maze task. When administered before ketamine, PF-04859989 (32.0 mg/kg) significantly attenuated the ketamine-induced increase in mean working memory errors on the maze (p < 0.0001; n = 21–23 animals per group; error bars indicate SEM).
PF-04859989 improved ketamine-disrupted spatial memory in nonhuman primates
To further demonstrate relevance to cognitive impairments in schizophrenia, the impact of lowering endogenous KYNA levels on working memory in the nonhuman primate was examined using the acute ketamine model. While there was no effect of pretreatment with PF-04859989 on spatial delayed response performance under vehicle conditions, PF-04859989 prevented the working memory deficits induced by ketamine when administered at 3.2 mg/kg. A two-way ANOVA for spatial delayed response performance revealed a significant effect of PF-04859989 (F(3,27) = 12.943, p < 0.001) and ketamine (F(1,9) = 27.684, p = 0.001), but no interaction (F(3,27) = 1.797, p > 0.05). As can be seen in Figure 6A, a one-way ANOVA across vehicle conditions yielded no significant effects on working memory performance (F(3,36) = 1.437, p > 0.05). By contrast, a one-way ANOVA across ketamine conditions (F(3,36) = 3.845, p < 0.05) revealed significant protection of working memory at the highest dose of 3.2 mg/kg compared with the vehicle/ketamine condition as determined by a two-tailed Dunnett's t-test (p < 0.05; Fig. 6B). Spatial working memory performance was further analyzed by examining delay-dependent errors across vehicle/placebo, vehicle/ketamine, and 3.2 mg/kg PF-04859989/ketamine conditions (Fig. 7). An overall three-way ANOVA revealed a main effect of PF-04859989 (F(3,27) = 8.641, p < 0.001) and a main effect of ketamine (F(1,9) = 37.149, p < 0.05) with no significant interaction (F(3,24) = 0.057, p = 0.982). Furthermore, there was a main effect of delay (F(4,36) = 5.588, p < 0.001), but no significant interactions for PF-04859989 × ketamine (F(3,27) = 1.039, p > 0.05), PF-04859989 × delay (F(12,108) = 0.476, p > 0.05), or ketamine × delay (F(4,36) = 1.315; p = 0.283). There was, however, a significant three-way interaction of PF-04859989 × ketamine × delay (F(12,108) = 2.106, p < 0.05). Thus, the improvement in working memory in the presence of ketamine after pretreatment with the KAT II inhibitor PF-04859989 at the highest dose was manifested by a decreased number of errors, especially at the shorter delay lengths (Fig. 7). This cognitive remediation is consistent with a restoration of attention and encoding of spatial information in working memory buffers and concurs with the demonstrated support for improved preattentive and attentional processes.

Impact of pretreatment with PF-04859989 on spatial delayed response performance. A, Pretreatment with PF-04859989 had no effect on working memory performance under normal placebo conditions. B, In the presence of ketamine, which normally impairs cognitive function, the high dose of PF-04859989 provided substantial protection against the deficit induced. *p = 0.024, significantly different from vehicle/ketamine; n = 10; error bars indicate SEM).
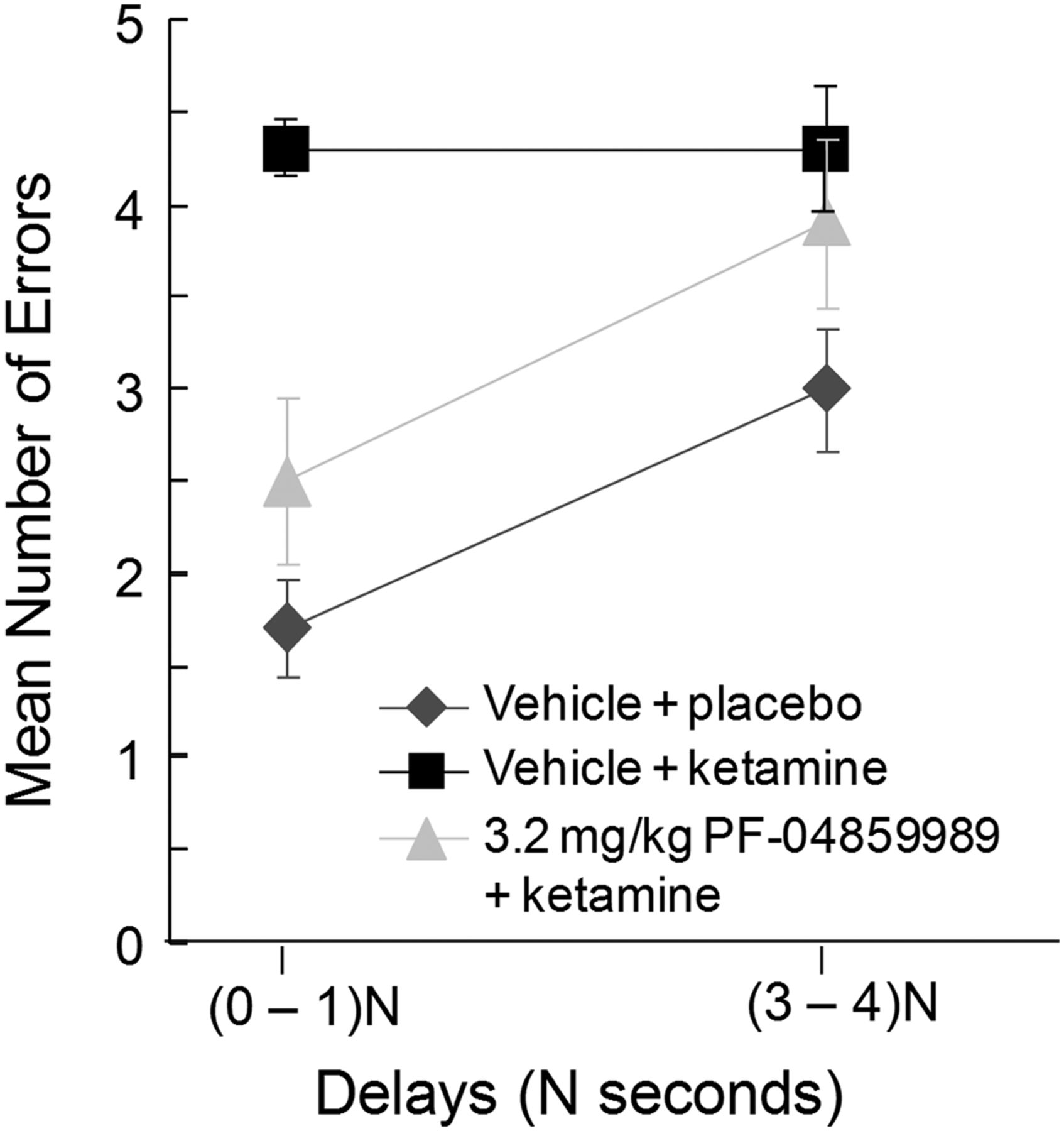
Effect of PF-04859989 on delay-dependent errors. Ketamine induced a substantial increase in errors on the task at both short and long delays. Pretreatment with PF-04859989 at 3.2 mg/kg provided protection against this effect, which was clearly evident at short delays. n = 10; error bars indicate SEM.
Behavioral observations in the nonhuman primate
Ketamine also produced a robust enhancement of positive-like symptoms compared with the vehicle/placebo condition where such symptoms are not typically evident, but this effect was partially attenuated after pretreatment with PF-04859989 at 3.2 mg/kg (Fig. 8A). A one-way ANOVA across treatment conditions (F(4,45) = 3.040, p < 0.05) revealed significance for all ketamine conditions except for the highest dose of 3.2 mg/kg compared with vehicle/placebo, as determined by a Dunnett's t test (vehicle/ketamine, p < 0.05; 0.032 mg/kg, p < 0.05; 0.32 mg/kg, p < 0.01; 3.2 mg/kg, p = 0.108). The induction of negative-like symptoms, or psychomotor retardation, by ketamine was unaffected by pretreatment with PF-04859989 at any of the doses tested (Fig. 8B). A two-way ANOVA revealed no significant main effect of PF-04859989 pretreatment (F(3,24) = 0.233, p > 0.05), a significant main effect of ketamine treatment (F(1,8) = 7.972, p < 0.05), but no interaction between pretreatment and treatment (F(3,24) = 0.057, p > 0.05) on the expression of gross motor behaviors. Together, these findings suggest that substantial reduction of endogenous kynurenic acid levels significantly attenuated the ketamine-induced disruption of mechanisms involved in working memory and, possibly, behavioral organization, but had no effect on the mechanisms involved in internally generated behaviors that are characteristic of the normal behavioral repertoire of the animal.

Effect of PF-04859989 on positive- and negative-like behaviors. A, A substantial incidence of hallucinatory-like behaviors emerged in the presence of ketamine. Pretreatment with PF-04859989 did not prevent this occurrence, but it was not significant at the highest dose in comparison with placebo. B, Likewise, PF-04859989 did not protect against the diminution in gross motor behaviors induced by ketamine. *p = 0.022, significant main effect of ketamine treatment; n = 10; error bars indicate SEM.
Discussion
The physiological function of KYNA continues to be a subject of debate. Though circulating levels are lower than the reported affinity at NMDA and nicotinic α7 receptors, studies in which KYNA is elevated have demonstrated a role in regulating glutamate and/or dopamine signaling within the brain and altered performance in cognitive processing (Shepard et al., 2003, Koshy Cherian et al., 2014; Rassoulpour et al., 2005; Chess et al., 2007, 2009; Amori et al., 2009; Konradsson-Geuken et al., 2010). While these studies often introduce KYNA concentrations that are greatly elevated, it can be hypothesized that even small chronic increases during critical development stages such as those associated with schizophrenia may lead to disturbances in normal brain function throughout life (Pocivavsek et al., 2012). However, this hypothesis relies on the prediction that KYNA is present in synapses at concentrations sufficient to innately impact brain function, an assertion difficult to demonstrate in vivo without appropriate pharmacological agents. The discovery of PF-04859989 as the first potent, selective, and brain-penetrable KAT II inhibitor is an important advance in the study of endogenous KYNA function in the brain. The present findings demonstrate that acute reduction in brain KYNA after KAT II inhibition is an effective mechanism to prevent sensory gating deficits induced by either excessive dopamine neurotransmission or blockade of NMDA receptors. Since both enhanced dopamine activity and NMDA receptor dysfunction are hypothesized to be involved in the pathophysiology of schizophrenia, and elevated KYNA levels are reported in schizophrenia patients, it is predicted that KAT II inhibitors may normalize auditory gating deficits, and consequently clinical symptoms, in schizophrenia patients. Although PF-04859989 alone did not change the degree of auditory gating, it significantly reduced the power of low delta oscillations in both the hippocampus and entorhinal cortex, independent of pharmacological challenges with ketamine or amphetamine. Interestingly, recent findings demonstrate an opposite change in the power of low delta activity after NMDA receptor antagonism under a similar recording condition (Kiss et al., 2011), warranting further electrophysiological studies to evaluate how KYNA might directly or indirectly regulate low delta frequency.
In addition to the effects on auditory gating, the present findings demonstrate that acute reduction in brain KYNA levels also effectively improved performance in a sustained attention task compromised by an environmental disruptor, and prevented ketamine-induced disruption of performance in rodent and nonhuman primate working memory tasks. As seen in previous studies in the nonhuman primate, ketamine appeared to result in a disruption of encoding that was apparent even at the shortest delays (Roberts et al., 2010). This finding is consistent with that of Gold et al. (2010), who demonstrated that patients with schizophrenia primarily exhibit a deficit in the capacity of working memory rather than in maintenance itself. Together, these findings support the hypotheses that endogenous KYNA impacts cognitive function, and that inhibition of KAT II and consequent lowering of endogenous brain KYNA levels improves cognitive performance under conditions considered relevant for schizophrenia.
From the current experiments, we can infer that basal KYNA tone is sufficient to regulate key neuronal circuitry involved in attention and spatial working memory in agreement with prior studies in juvenile KAT II knock-out mice (Potter et al., 2010), and may also impact preattentive processing. Data from the present studies support the hypothesis that endogenously produced KYNA negatively modulates NMDA receptor function (Watson et al., 1988; Hilmas et al., 2001; Mok et al., 2009) or possibly may regulate glutamate and/or acetylcholine release through an interaction at the nicotinic α7 receptor (Hilmas et al., 2001; Rassoulpour et al., 2005; Lopes et al., 2007; Wu et al., 2010), although it is worth noting that direct changes in dopamine release after treatment with PF-04859989 were not observed (data not shown). The effects of KAT II inhibition on glutamate neurotransmission may be particularly important in schizophrenia where the KYNA level is not only elevated, but the number of endogenous NMDA coagonist ligands, including glycine and d-serine, are also reduced (for review, see Millan, 2002, 2005). Under these conditions, one might expect that signaling through NMDA receptors would be impaired from both a reduction in stimulatory tone and an increase in inhibitory tone, thus contributing to glutamate receptor hypofunction. If true, then lowering KYNA levels through KAT II inhibition, and thereby facilitating activity-dependent NMDA receptor signaling, may indeed be a promising means to treat symptoms arising from disrupted cortical networks in schizophrenia.
Footnotes
All aspects of this research were funded by Pfizer Inc. G.V.W. and S.A.C. received research support from Pfizer Inc., the CHDI Foundation, and Forest Research Institute Inc.
R.K., C.A.S., W.E.H., T.K., D.S.C., D.M., K.F., D.A.Y., and A.D. are full-time employees of Pfizer Inc., and own and/or hold options for/restricted stock units of publicly traded shares of the company. B.M.C., W.H., V.G., P.A.S., and M.H. were full-time employees of Pfizer at the time our research was conducted but is no longer affiliated with the company.
During these studies, T.K. was on sabbatical leave from the Wigner Research Centre for Physics of the Hungarian Academy of Sciences, H-1121 Budapest, Hungary.
- Correspondence should be addressed to Dr. Rouba Kozak, Pfizer Inc., 610 Main Street, Cambridge, MA 02139. rouba.kozak{at}pfizer.com





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}