

Abstract
New granule cells are continuously integrated into hippocampal circuits throughout adulthood, and the fine-tuning of this process is likely important for efficient hippocampal function. During development, this integration process is critically regulated by the α-calcium/calmodulin-dependent protein kinase II (α-CaMKII), and here we ask whether this role is conserved in the adult brain. To do this, we developed a transgenic strategy to conditionally delete α-CaMKII from neural progenitor cells and their progeny in adult mice. First, we found that the selective deletion of α-CaMKII from newly generated dentate granule cells led to an increase in dendritic complexity. Second, α-CaMKII deletion led to a reduction in number of mature synapses and cell survival. Third, consistent with altered morphological and synaptic development, acquisition of one-trial contextual fear conditioning was impaired after deletion of α-CaMKII from newly generated dentate granule cells. Previous work in Xenopus identified α-CaMKII as playing a key role in the stabilization of dendritic and synaptic structure during development. The current study indicates that α-CaMKII plays a plays a similar, cell-autonomous role in the adult hippocampus and, in addition, reveals that the loss of α-CaMKII from adult-generated granule cells is associated with impaired hippocampus-dependent learning.
Introduction
During development, neuronal structure is dynamic. Initially dendritic growth is rapid, with frequent addition and retraction of dendritic branches. However, as dendritic arbors mature and become more complex, growth slows and structure stabilizes. Calcium/calmodulin-dependent protein kinase II (CaMKII) is thought to play a key role in this stabilization process. For example, in the developing Xenopus, the onset of CaMKII expression coincides with a reduction in dendritic growth rate and stabilization of dendritic arborization (Wu and Cline, 1998). Overexpression of CaMKII prematurely initiates stabilization of dendritic arborization by reducing rates of branch addition and retraction (Wu and Cline, 1998). Conversely, inhibition of CaMKII activity increases neurite complexity and branch dynamics (Wu and Cline, 1998; Zou and Cline, 1999). Consistent with this role in dendritic and axonal maturation, CaMKII regulates synaptic maturation (Wu et al., 1999). The role of α-CaMKII in activity-dependent structural stabilization appears to be conserved in the mammalian brain. For example, in the developing rodent brain, CaMKII expression peaks postnatally (Sugiura and Yamauchi, 1992; Tang et al., 2004; Nakashiba et al., 2012), and genetic inhibition of the α isoform of CaMKII increases dendritic complexity of mouse dorsal horn neurons during development (Pattinson et al., 2006). Similarly, CaMKII regulates structural stabilization in neuronal cell lines (Goshima et al., 1993; Tashima et al., 1996; Massé and Kelly, 1997) and primary neuronal cultures (Redmond et al., 2002).
These data indicate that during development initially dynamic neuronal structure is stabilized by α-CaMKII-dependent processes (see also Wang et al., 1994). Neurogenesis persists beyond development in the subgranular zone of the hippocampus; therefore, here we test whether this role for α-CaMKII in stabilization of neuronal structure is conserved when new neurons integrate into established circuits in adult mice. To address this, we used a transgenic strategy to delete α-CaMKII in a Cre recombinase inducible manner (α-CaMKIIf/f mice). To restrict α-CaMKII deletion to neural progenitor cells, we crossed α-CaMKIIf/f mice with nestin-CreERT2 mice, in which a tamoxifen (TAM)-inducible Cre recombinase is expressed under the control of a nestin promoter (Imayoshi et al., 2008). In adult offspring from this cross, TAM administration deletes α-CaMKII in neural progenitor cells (NPCs) and their progeny. We found that deletion of α-CaMKII led to a an increase in complexity of adult-generated granule cells in the dentate gyrus (DG), suggesting that α-CaMKII plays a cell-autonomous role in stabilization of dendritic structure as new neurons integrate into established hippocampal circuits. Further, we found a reduction in the number of mature synapses onto α-CaMKII-deficient newly generated granule cells, and, consistent with this, impaired learning in a hippocampus-dependent task. These results suggest that α-CaMKII regulates integration of adult-generated granule cells into the dentate gyrus by arresting dendritic growth and stabilizing input connections.
Materials and Methods
Mice
Nestin-CreERT2 mice.
Nestin-CreERT2+ mice express a TAM-inducible form of Cre recombinase driven by a progenitor specific (nestin) promoter, as previously described (Imayoshi et al., 2008; Arruda-Carvalho et al., 2011). In all experiments, we used line 4 from Imayoshi et al. (2008) as recombination efficiency is highest in the subgranular zone of the hippocampus in this line (Imayoshi et al., 2008; Arruda-Carvalho et al., 2011).
α-CaMKIIf/f mice.
In α-CaMKII floxed mice, α-CaMKII f/f exon 2 of the α-CaMKII gene is flanked by loxP sequences (Achterberg et al., 2014). In all the experiments, homozygote mice were used, so that Cre recombinase-mediated excision of exon 2 led to complete knock-out of α-CaMKII in cells in which Cre recombinase is active.
Rosa26-LacZ mice.
The Rosa26-LacZ reporter mice have been previously described (Zambrowicz et al., 1997). In this reporter line, LacZ expression is under the control of the ubiquitous Rosa26 locus promoter, and expression of the LacZ transgene is dependent on the Cre recombinase-mediated removal of a transcriptional STOP cassette. We crossed this reporter line with nestin-CreERT2 and α-CaMKII f/f mice to characterize recombination and α-CaMKII deletion.
All lines were maintained on a C57BL/6 background (Taconic Farms). Genotypes were determined by PCR analysis of tail DNA samples as previously described (Zambrowicz et al., 1997; Buch et al., 2005; Imayoshi et al., 2008). Two breeding schemes were used. For all behavioral and morphological analyses, nestin-CreERT2+/α-CaMKIIf/f mice were bred with α-CaMKIIf/f/Rosa26-LacZ+/+ mice, resulting in nestin-CreERT2+/α-CaMKIIf/f/Rosa26-LacZ+/− and nestin-CreERT2−/α-CaMKII f/f/Rosa26-LacZ+/− offspring. For all remaining analyses (proliferation, survival, maturation), nestin-CreERT2+/α-CaMKIIf/+ mice were bred with α-CaMKIIf/+/Rosa26-LacZ+/+ mice, resulting in nestin-CreERT2+/α-CaMKIIf/f/Rosa26-LacZ+/−, nestin-CreERT2+/α-CaMKIIf/+/Rosa26-LacZ+/−, nestin-CreERT2+/α-CaMKII+/+/Rosa26-LacZ+/−, nestin-CreERT2−/α-CaMKIIf/f/Rosa26-LacZ+/−, nestin-CreERT2−/α-CaMKIIf/+/Rosa26-LacZ+/−, and nestin-CreERT2−/α-CaMKII+/+/Rosa26-LacZ+/− offspring, of which only the nestin-CreERT2+ pups were used. In the morphological and behavioral experiments, we compared nestin-CreERT2−/α-CaMKIIf/f/Rosa26-LacZ+/− (referred to as α-CaMKIIf/f for simplicity) and nestin-CreERT2+/α-CaMKIIf/f/Rosa26-LacZ+/− (referred to as α-CaMKII inducible knock-out or α-CaMKII-iKO) mice. For the proliferation, survival, and maturation analyses, we compared LacZ+ dentate granule cells in nestin-CreERT2+/α-CaMKII+/+/Rosa26-LacZ+/− (referred to as α-CaMKII+/+ mice for simplicity) to LacZ+ dentate granule cells in nestin-CreERT2+/α-CaMKIIf/f/Rosa26-LacZ+/− (α-CaMKII-iKO) mice. All mice were treated with TAM. Importantly, this design ensures that group effects cannot be attributed to nonspecific effects of TAM.
Mice were bred in our colony at The Hospital for Sick Children and maintained on a 12 h light/dark cycle with free access to food and water. Before all behavioral experiments, mice were handled for 2 min twice a day for 4 d. Male and female offspring were used in all experiments. All experiments were performed in the light cycle and conducted according to protocols approved by The Hospital for Sick Children Animal Care and Use Committee.
Drugs
TAM treatment.
TAM (Sigma) was dissolved in ethanol (10%) and suspended in sunflower seed oil (Lagace et al., 2007). Mice received daily injections (180 mg/kg; i.p.) for 5 d. Mice were treated with TAM either at 4–5 or at 12 weeks of age.
Immunohistochemistry
Mice were perfused transcardially with PBS (0.1 m) and 4% PFA. Brains were removed, fixed overnight in PFA, and transferred to 0.1 m PBS. Coronal sections (40 μm) were cut using a vibratome (Leica VT1200S). The following primary antibodies were used: mouse monoclonal anti-α-CaMKII (1:2000; Millipore), goat polyclonal anti-doublecortin (DCX) (1:4500; Santa Cruz Biotechnology), rabbit polyclonal anti-Egr1 (Zif268) (1:10,000; Santa Cruz Biotechnology), mouse monoclonal anti-NeuN (1:1000; Millipore), rabbit polyclonal anti-LacZ (1:6000; Invitrogen), mouse monoclonal anti-nestin (1:150; BD Biosciences PharMingen), rabbit monoclonal anti-calbindin (D28K; 1:600; Cell Signaling Technology), mouse monoclonal anti-calretinin (1:1500; Swant), rat monoclonal anti-BrdU (1:1000; Axyll), rabbit polyclonal anti-Ki67 (1:10,000; Abcam), anti-GFP (1:500; Invitrogen), and anti-VGlut1 (1:1000; Millipore). All sections were treated with 1% hydrogen peroxidase. Sections were then incubated overnight with the primary antibody and then for 60 min at room temperature with HRP-conjugated or biotinylated secondary antibodies (1:750; Jackson ImmunoResearch Laboratories). Signals were amplified and visualized using Vectastain Elite ABC kit (Vector Laboratories), tyramide signal amplification, Alexa-Fluor-conjugated streptavidin (Invitrogen). Sections were mounted on slides with Permafluor anti-fade medium.
Imaging and quantification
All images were acquired using a confocal (LSM 710 Zeiss) microscope. To calculate cell number, cell density, or proportion of double-positive cells, we used 1/5 systematic section sampling fractions covering the entire anterior–posterior extent of the DG. To calculate the proportion of double-labeled cells, confocal 1 μm Z-stack images were obtained using ZEN software (Zeiss) with a minimal interval of 15 μm to prevent duplicate counts of the same cell. We estimated the total number of LacZ+ cells after TAM treatment using the optical fractionator method on an Olympus BX61 epifluorescence microscope using a 60×, 1.45 NA objective and a motorized XYZ stage attached to a computer with Stereoinvestigator 9.1 (MBF Bioscience) (Chen et al., 2004). A random systematic sampling was used for these stereological analyses (section interval of 1/5, grid size of 250 × 250 μm, 2D counting frame of 90 × 90 μm using fractionators of 30 μm in thickness). Tissue thickness measured in each counting frame was used to estimate the total number of LacZ+ cells in the entire DG. Conditions were optimized to obtain a Gundersen coefficient of error <0.05 (Gundersen et al., 1999). We quantified BrdU+ cells throughout the entire anterior–posterior extent of the DG (11–13 sections per mouse, with every fifth section analyzed) using the Nikon Eclipse 80i microscope. Granule cell layer volume was estimated in α-CaMKII+/+ (n = 7) and α-CaMKII-iKO (n = 7) mice from 10 hippocampal slices per mouse (collected every sixth section) using the Cavalieri volume estimation method (magnification: 20×; grid spacing: 40 μm; Cavalieri estimator module in Stereo Investigator, MBF Bioscience).
Retrovirus production and stereotaxic surgery
Retroviral labeling of new neurons.
New neurons were labeled using a pCAG-GFP replication-deficient retroviral vector (based on the Moloney murine leukemia virus) (Tashiro et al., 2006b). Retroviral vectors were harvested from a stable producing cell line, and concentrated by ultraspeed centrifugation (average of 3.5 × 109 IU/ml), as previously described (Stone et al., 2011).
Stereotaxic surgery.
Mice were treated with atropine sulfate (0.1 mg/kg, i.p.) and anesthetized with chloral hydrate (400 mg/kg, i.p.) before being placed in a stereotactic frame. The scalp was incised and retracted to drill holes through the skull above the hippocampi. Viral vectors were injected bilaterally into the DG (anteroposterior: −2.2 mm; mediolateral: 1.8 mm; and dorsoventral −2.2 mm) via a glass micropipette (outer diameter, ∼50 μm) connected to a Hamilton syringe. The injection volume (0.5 μl) and rate (0.1 μl/min) were controlled by an infusion pump (SP100i; WPI). Micropipettes were left in place for an additional 10 min to ensure complete diffusion of the virus.
Morphological analysis
Eight weeks after the viral injections, mice from all groups were transcardially perfused with PBS (0.1 m) and 4% PFA. The brains were postfixed in 4% PFA at −4°C until processed. Vibratome sections (50 μm) of the hippocampus were collected and stored in PBS/glycerol/NaAzide solution until further processed.
Dendritic morphology.
All images were acquired using a confocal (LSM 710 Zeiss) microscope. To characterize neuronal arborization confocal 2 μm Z-stack images (40 objective) were obtained using ZEN software (Zeiss). Dendrites from GFP+ granule cells (α-CaMKIIf/f, n = 118 dendrite segments from 5 mice; α-CaMKII-iKO, n = 81 dendrite segments from 8 mice) were traced with Neurolucida software (MBF Bioscience) by an experimenter blind to the experimental groups. Measures of dendritic complexity (number of branches and bifurcations, proportion of neurons with single vs multiple primary dendrites) were batch-analyzed with the LMeasure software (Scorcioni et al., 2008).
Spine and synapse analysis.
High-resolution (1024 × 1024 pixels), high-magnification (63× objective) Z-stack images (0.45 μm step) were acquired. Every stack was subjected to iterative deconvolution with inverse filtering (ImagePro Plus, MediaCybernetics) to deblur the VGlut1 (red channel) and GFP (green channel) signals. For spine and synapse analysis, a random sample of dendritic segments (length = 20 μm) of GFP+ cells (α-CaMKIIf/f, n = 41 dendrite segments from 4 mice; α-CaMKII-iKO, n = 47 dendrite segments from 5 mice) was extracted. Dendritic spines were defined as protrusions (length <2 μm) stemming from the dendritic shaft (spines analyzed: α-CaMKIIf/f, n = 1360; α-CaMKII-iKO, n = 1489). Spine density was expressed as the number of protrusions per 10 μm. Putative synapses were determined by the juxtaposition of VGlut1 puncta on the terminal tip of the GFP-positive dendritic spines. The proportion of spines directly apposed to VGlut1+ puncta (as a proxy for functional synaptic terminals) was used to estimate the proportion of putative synapses.
General behavioral apparatus and procedures
In the fear conditioning experiments, context A (the training context) consisted of a stainless steel conditioning chamber (31 cm × 24 cm × 21 cm; Med Associates) with a stainless steel shock-grid floor. The grid floor was positioned over a stainless-steel drop-pan, which was lightly cleaned with 70% ethyl alcohol to provide a background odor. Shock grid bars (diameter 3.2 mm) were spaced 7.9 mm apart. The front, top, and back of the chamber were made of clear acrylic and the two sides made of modular aluminum. Mouse freezing behavior was monitored via overhead cameras. Freezing was assessed using an automated scoring system (Actimetrics), which digitized the video signal at 4 Hz and compared movement frame by frame to determine the amount of freezing.
During training, mice were placed in context A. In the weak training experiment, mice were placed in the context and after 120 s presented with a 2 s footshock (0.5 mA). Mice remained in the context for a further 60 s before being returned to their home cage. In the strong training experiment, mice were placed in the context and presented with 2 s footshocks (0.5 mA) at 120, 180, and 240 s. After the final footshock, mice remained in the context for an additional 60 s. Shock responsivity was estimated by comparing mouse velocity in 2 s windows immediately preceding versus during shock presentation using the following formula: (velocityshock − velocitypre-shock)/(velocityshock + velocitypre-shock). Twenty-four hours after training, mice were placed back in the context and freezing assessed in a 5 min test.
Specific experimental protocols
Characterization of α-CaMKII expression in dentate granule cells (DGCs).
Four- to 5-week-old WT C57BL/6 mice were perfused, and brain tissue was processed for immunohistochemistry for α-CaMKII and different markers (n = 5 or 6/group).
Characterization of efficiency of TAM-induced knock-out of α-CaMKII in recombined cells.
Four- to 5-week-old α-CaMKII-iKO mice (n = 5) were treated with TAM. Eight weeks later, mice were perfused and brain tissue was processed for immunohistochemistry for α-CaMKII and LacZ.
Characterization of TAM-induced knock-out of α-CaMKII: proliferation and survival.
Four- to 5-week-old α-CaMKII+/+ and α-CaMKII-iKO mice were treated with TAM. For the proliferation analysis, α-CaMKII+/+ (n = 5) and α-CaMKII-iKO (n = 6) mice received a single injection of BrdU (100 mg/kg) 24 h after the last TAM injection. They were then perfused 24 h later. For the survival analysis, α-CaMKII+/+ (n = 6) and α-CaMKII-iKO (n = 9) mice received a series of BrdU injections (100 mg/kg, 2 per day for 3 d), starting 1 d after the last TAM injection. Eight weeks after the start of TAM, mice were perfused and tissue was processed for immunohistochemistry.
Characterization of TAM-induced knock-out of α-CaMKII: maturation analysis.
α-CaMKII+/+ (n = 6–8/marker) and α-CaMKII-iKO (n = 6–9/marker) mice were treated with TAM. Immunohistochemistry analysis of expression of LacZ and different markers occurred 8 weeks later.
Characterization of α-CaMKII expression in newly generated neurons.
To characterize the onset of α-CaMKII expression, 6-week-old WT mice (n = 3) were injected with BrdU (200 mg/kg) and perfused 1, 3, or 6 weeks after injection.
Characterization of TAM-induced knock-out of α-CaMKII: IEG expression.
α-CaMKII-iKO mice (n = 4) were trained in context fear conditioning (see below). Ninety minutes after the completion of training, mice were perfused and tissue was processed for LacZ and Zif268 immunohistchemistry. Home cage α-CaMKII-iKO animals (n = 2) were perfused and processed at the same time.
Characterization of TAM-induced knock-out of α-CaMKII: dendritic morphology and synapse analysis.
α-CaMKIIf/fand α-CaMKII-iKO mice were treated with TAM. One week after the start of TAM, mice received bilateral infusions of pCAG-GFP retrovirus into the DG. GFP+ cells were analyzed 8 weeks later. Regardless of genotype, brain sections were stained for GFP and LacZ, and in α-CaMKII-iKO mice only double labeled cells were analyzed.
Open field.
For open field testing, 8 weeks after TAM treatment, α-CaMKIIf/f (n = 10) and α-CaMKII-iKO (n = 7) mice were placed in the center of a square-shaped arena (45 cm × 45 cm × 20 cm height) and allowed to explore for 10 min. The open field apparatus was constructed of Plexiglas and was dimly lit from above. Mouse location was tracked by a camera located above the open field, and total distance traveled as well as time spent in three different zones (outer, middle, inner) was measured (Limelight2, Actimetrics). Total distance traveled was used as a measure of spontaneous motor activity, and distribution of activity in different regions of the arena was used as a measure of anxiety-related behavior (Archer, 1973).
Forced swim test.
To assess depressive-like behaviors, we used the forced swim test (Porsolt, 1979). α-CaMKIIf/f (n = 8) and α-CaMKII-iKO (n = 14) mice were treated with TAM. Eight weeks later, mice were placed in a cylinder (radius, 6.5 cm, height 20 cm) for 6 min. The cylinder was filled to a depth of 12 cm with 25 ± 1°C water. The time spent motionless was recorded for each mouse.
Contextual fear conditioning.
α-CaMKIIf/f and α-CaMKII-iKO mice were treated with TAM. Eight weeks after the start of TAM treatment, α-CaMKIIf/f (n = 10) and α-CaMKII-iKO (n = 9) mice were trained with a single footshock, and freezing was assessed in the training context 24 h later. A separate group of α-CaMKIIf/f (n = 10) and α-CaMKII-iKO (n = 13) mice were trained with 3 footshocks, and tested 24 h later. For the Zif268 quantification, α-CaMKII-iKO (n = 4) mice were trained with 5 footshocks (0.7 mAmp) and perfused 90 min later.
Data analysis
Cell counts and morphological analyses are displayed as mean ± SEM. Unless otherwise specified, data were analyzed using ANOVAs and t tests. Between-group pairwise comparison of morphological features (number of branches and bifurcations) and spine density and synapse number were performed by using the nonparametric Wilcoxon rank-sum test because these datasets were not normally distributed.
Results
α-CaMKII expression pattern in DGCs
We first examined α-CaMKII expression in maturing DGCs in both the juvenile and adult mouse brain by staining for proteins expressed at different stages of cell differentiation (Kempermann et al., 2004). In 4- to 5-week-old WT mice, α-CaMKII was rarely expressed in cells expressing progenitor markers, such as nestin (Fig. 1A) or Ki67 (Fig. 1B). However, α-CaMKII was expressed in ∼20% (DCX) and 40% (calretinin) of cells expressing neuroblast markers (Fig. 1C,D), and in virtually all cells expressing mature neuronal markers, such as NeuN and calbindin (Fig. 1E,F; Table 1). We found an equivalent pattern of expression in 12-week-old mice (data not shown). In a separate experiment, 5- to 6-week-old mice were injected with the proliferation marker BrdU, and α-CaMKII expression was examined 1, 3, or 6 weeks later. In the 1 week group, few BrdU+ cells coexpressed α-CaMKII (Fig. 1G). However, the proportion of BrdU+ cells expressing α-CaMKII increased markedly with the postinjection delay (Fig. 1H,I). Therefore, together these data indicate that α-CaMKII is expressed postmitotically, and the onset of expression coincides with the late neuroblast stage (Nakashiba et al., 2012).
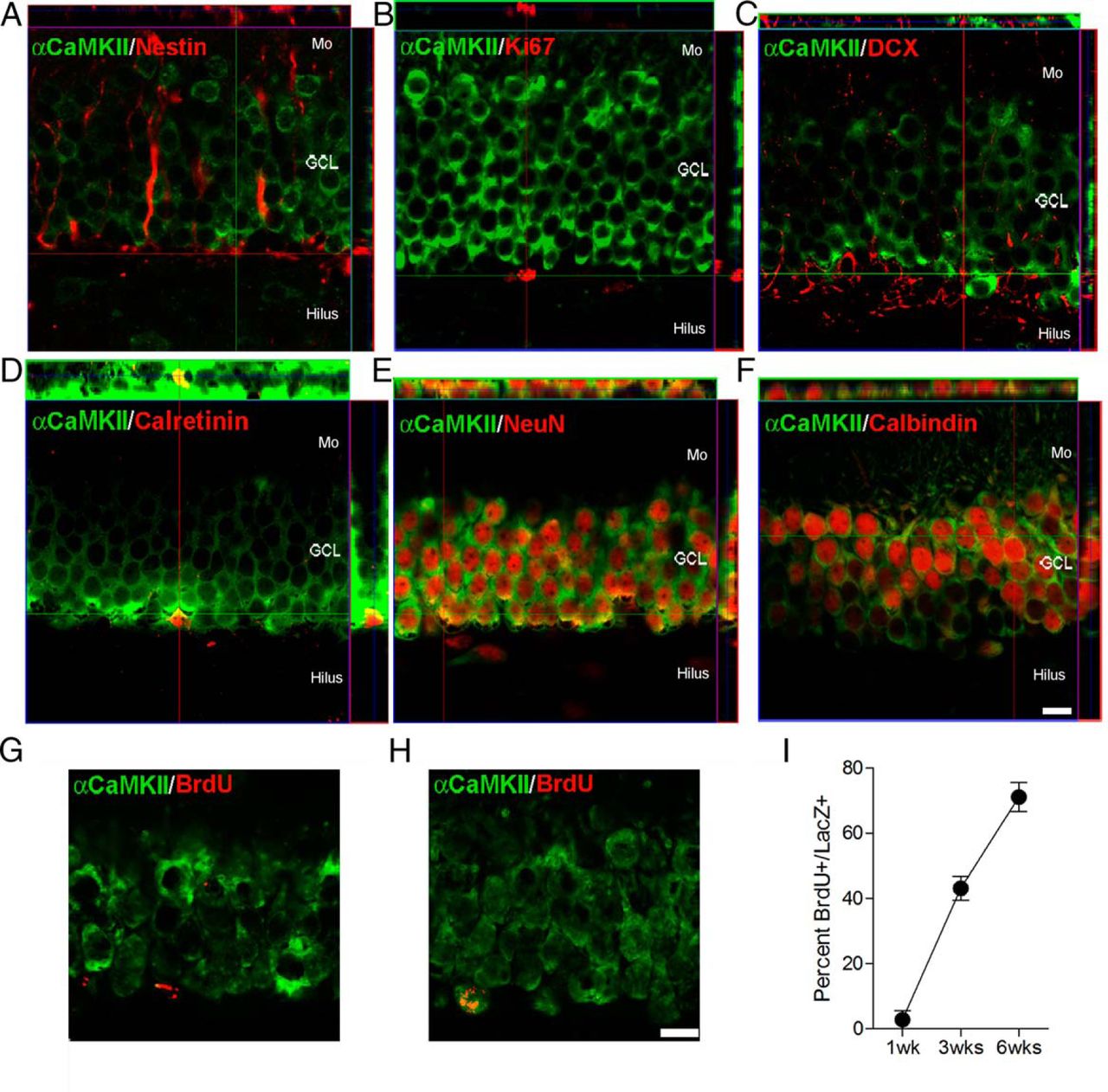
Expression of α-CaMKII in dentate granule cells. Colocalization of α-CaMKII (green) expression with (A) nestin (red), (B) Ki67 (red), (C) DCX (red), (D) calretinin (red), (E) NeuN (red), and (F) calbindin (red) in the dentate gyrus of 4- to 5-week-old mice. G–I, Time course of α-CaMKII expression. WT mice were injected with BrdU and killed 1, 3, or 6 weeks later. Examples of BrdU+ cells at (G) 1 week and (H) 6 weeks. I, Percentage of BrdU+ cells coexpressing α-CaMKII. Scale bar, 10 μm. Mo, Molecular layer; GCL, granule cell layer.
α-CaMKII expression pattern in DGCs
Conditional deletion of α-CaMKII from adult-generated DGCs
To conditionally delete α-CaMKII from NPCs, we crossed mice expressing a TAM-inducible cre-recombinase driven by a progenitor specific promoter (nestin-CreERT2 mice) (Imayoshi et al., 2008; Arruda-Carvalho et al., 2011) with mice in which α-CaMKII is floxed (α-CaMKIIf/f) (Achterberg et al., 2014). In double mutant offspring from this cross (α-CaMKII-iKO mice), TAM treatment induces excision of the flanked sequences and deletion of α-CaMKII in neural progenitor cells and their progeny (Fig. 2A). To visualize recombined cells, α-CaMKII-iKO mice were additionally crossed with a ROSA26-LacZ mouse line, in which a stop codon preventing LacZ expression is flanked by lox P sites.

Conditional deletion of α-CaMKII impairs morphological development of newly generated dentate granule cells. A, Schematic of the strategy for conditional deletion of α-CaMKII from NPCs. In nestin-CreERT2+/α-CaMKIIf/f mice (α-CaMKII-iKO), TAM administration leads to excision of α-CaMKII exon 2 flanked sequences and deletion of α-CaMKII in neural progenitor cells and their progeny. B, Eight weeks after TAM treatment, the vast majority of recombined cells (LacZ+; red) lacked α-CaMKII (green). Scale bars, 10 μm. C, Retrovirally labeled dentate granule cells in control (left; α-CaMKII f/f) and α-CaMKII-iKO (right) mice, 8 weeks after infection. In α-CaMKII-iKO (right) mice, GFP-labeled dentate granule cells dendrites had more (D) bifurcations and (E) branches. Eight weeks after TAM treatment, expression of (F) nestin (green), (G) DCX (green), (H) NeuN (green), and (I) calbindin (green) in recombined (LacZ+; red) cells was similar in control (top; α-CaMKIIf/f) and α-CaMKII-iKO (bottom) mice. J, Left, Eight weeks after TAM treatment, expression of zif268 (green) in recombined (LacZ+; red) in control (α-CaMKIIf/f) and α-CaMKII-iKO mice. Right, Contextual fear conditioning (trained) induced zif268 expression in recombined (LacZ+) cells in α-CaMKII-iKO mice compared with home cage (HC) controls. Scale bar, 10 μm. *p < 0.05.
Eight weeks after TAM treatment, 56.4 ± 6.7% of nestin+ cells in the DG expressed LacZ, indicating that TAM-induced recombination was efficient, as previously observed (Imayoshi et al., 2008; Arruda-Carvalho et al., 2011). Stereological counts revealed that there were 30,912 ± 2832 LacZ+ cells in the DG. These cells were uniformly distributed throughout the anterior–posterior extent of the DG, suggesting no intraregional differences in TAM-induced recombination efficiency. The vast majority of the recombined (i.e., LacZ+) cell population (92.2 ± 0.4%) lacked α-CaMKII (Fig. 2B), confirming the efficiency of the cre-recombinase dependent deletion.
Abnormal morphological development of α-CaMKII-deficient, adult-generated DGCs
In WT mice, our immunohistochemical analyses indicated that the onset of α-CaMKII expression coincides with the late neuroblast stage, when the cells are ∼3 weeks of age. This corresponds to a period when adult-generated granule cells are becoming synaptically integrated into the entorhinal-DG-CA3 circuit (Zhao et al., 2006; Ge et al., 2007, 2012). Thereafter, dendritogenesis and spinogenesis/synaptogenesis continue, with structural changes stabilizing after ∼8 weeks (Zhao et al., 2006). We therefore asked whether α-CaMKII deletion impacts dendritic development of adult-generated DGCs. To do this, α-CaMKII+/+ and α-CaMKII-iKO mice were treated with TAM, and then 1 week later microinfused with a retrovirus expressing GFP into the DG (Stone et al., 2011). Retroviruses infect dividing neural progenitors and their progeny (Tashiro et al., 2006b). As GFP is expressed throughout the cell body and processes of progeny, this strategy can be used to conduct detailed morphological analysis of adult-generated granule cells. Accordingly, 8 weeks after TAM treatment, dendritic morphology of infected granule cells was examined in α-CaMKIIf/f and α-CaMKII-iKO mice (Fig. 2C). Infected cells in α-CaMKII-iKO mice had more complex dendritic morphology with a higher number of bifurcations (W = 3068, p < 0.0001) and branches (W = 3585, p < 0.005) (Fig. 2C–E). The proportion of infected cells with multiple versus single primary dendrites did not differ between groups (α-CaMKIIf/f = 18%; α-CaMKII-iKO = 26.5%) (χ2 = 0.73; p = 0.39). These phenotypes in the 8-week-old knock-out cells are consistent with previous studies showing that α-CaMKII inhibition is associated with more complex dendritic arborization (Wu and Cline, 1998; Zou and Cline, 1999) and, overall, is consistent with a role for α-CaMKII in activity-dependent structural stabilization.
In contrast to the morphological analyses, we found no differences in expression of proteins expressed at different stages of differentiation in α-CaMKII+/+ and α-CaMKII-iKO mice (Fig. 2F–I: [nestin: α-CaMKII+/+ = 11.10 ± 1.26, α-CaMKII-iKO = 13.76 ± 1.86, t(14) = 1.27, p = 0.11; DCX: α-CaMKII+/+ = 11.31 ± 1.39, α-CaMKII-iKO = 14.63 ± 1.64, t(14) = 1.54, p = 0.07; NeuN: α-CaMKII+/+ = 77.18 ± 2.02, α-CaMKII-iKO = 79.04 ± 4.61, t(15) = 0.38, p = 0.35; calbindin: α-CaMKII+/+ = 82.72 ± 1.72, α-CaMKII-iKO = 80.43 ± 3.17, t(10) = 0.63, p = 0.27]). Because the onset of α-CaMKII expression coincides with the late neuroblast stage, the absence of differences at earlier stages of differentiation is expected. Furthermore, equivalent expression of mature neuronal markers, such as NeuN and calbindin, indicates that α-CaMKII deficiency does not alter neuronal fate. Finally, recombined (LacZ+) cells in α-CaMKII-iKO mice expressed the activity-regulated gene Zif268 after behavioral testing (Fig. 2J) (t(4) = 2.23, p < 0.05). This suggests that, although dendritic and synaptic structure was altered, α-CaMKII-deficient cells respond to external stimulation.
We next examined whether deletion of α-CaMKII impacted synaptic integration of newly generated granule cells into adult hippocampal circuits. To do this, in infected granule cells, we quantified the number of spines and the proportion of spines directly apposed to vesicular glutamate transporter 1 (VGlut1+) puncta (as a proxy for functional synaptic terminals) in α-CaMKIIf/f and α-CaMKII-iKO mice. Eight weeks following TAM treatment, spine density was not affected by α-CaMKII deletion (W = 1135, p = 0.15) (Fig. 3A). However, in α-CaMKII-iKO mice, there was a reduction in the proportion of spines in direct apposition to VGlut1+ puncta (W = 1242, p = 0.02) (Fig. 3B). This suggests that there is a reduction in the proportion of functional synaptic terminals onto α-CaMKII-deficient granule cells. Consistent with previous data in the developing Xenopus (Wu et al., 1996), these data indicate that α-CaMKII regulates synaptogenesis and synaptic maturation in the adult brain.

Conditional deletion of α-CaMKII impairs integration and survival of newly generated dentate granule cells. A, Retrovirally labeled granule cell dendritic segments (green) in control (left; α-CaMKIIf/f) and α-CaMKII-iKO (right) mice, 8 weeks after infection. Spine density was similar in α-CaMKII-iKO (closed bars) compared with α-CaMKIIf/f (open bars) mice. Scale bar, 1 μm. B, Retrovirally labeled granule cell spine terminals (green) and VGlut1+ puncta (red). Examples of nonjuxtaposed (top) and juxtaposed (bottom) spine terminals. In α-CaMKII-iKO mice, there was a reduction in the proportion of spines in direct apposition to VGlut1+ puncta. Scale bar, 0.5 μm. C, α-CaMKII+/+ and α-CaMKII-iKO mice were injected with TAM and subsequently injected with BrdU to label newly generated neurons. BrdU was injected twice a day for 3 d, 24 h after the last TAM injection. D, Eight weeks later, the number of BrdU+ cells (red) was reduced in α-CaMKII-iKO (open bars) compared with α-CaMKII+/+ (closed bars) mice. Examples of BrdU+ cells at higher magnification are shown from the insets (right). Scale bars: left, 100 μm; right, 10 μm. *p < 0.05.
Finally, we found that altered dendritic and synaptic maturation in α-CaMKII-deficient adult-generated granule cells was associated with an overall reduction in survival. Control (α-CaMKII+/+) and α-CaMKII-iKO mice were treated with TAM and subsequently with BrdU to label dividing cells (Fig. 3C). Eight weeks later, there was a reduction in number of BrdU+ cells in α-CaMKII-iKO brains (Fig. 3D) (t(13) = 2.41, p < 0.05). This was not reflected in a difference in total granule cell layer volume (t(12) = 1.009, p = 0.335; granule cell layer volume for α-CaMKII+/+ = 537.086 ± 21.228 mm3 × 10−3 and α-CaMKII-iKO = 574.628 ± 30.834 mm3 × 10−3). Because proliferation was unaltered (data not shown), these data suggest that newly generated, α-CaMKII-deficient granule cells are less likely to survive, possibly because of abnormal dendritic and synaptic maturation.
Impaired hippocampus-dependent learning in α-CaMKII-iKO mice
Deletion of α-CaMKII from progenitor cells led to altered granule cell morphology and a reduction in the number of putative functional synaptic terminals. We therefore next asked whether these mice exhibited impairments in hippocampus-dependent learning. To do this, we trained α-CaMKIIf/f and α-CaMKII-iKO mice in a contextual fear conditioning task, in which mice learn an association between a context and an aversive event (i.e., the delivery of a mild footshock). When returned to the same context, contextual fear memory is inferred from an increase in freezing behavior (Kim and Fanselow, 1992) (Fig. 4A). Importantly, several previous studies have shown that disruption of adult neurogenesis impairs learning in this task (Winocur et al., 2006; Warner-Schmidt et al., 2008; Hernández-Rabaza et al., 2009; Ko et al., 2009; Drew et al., 2010; Denny et al., 2012; but see Zhang et al., 2008; Deng et al., 2009). During training, both α-CaMKIIf/f and α-CaMKII-iKO mice exhibited similar reactivity to the footshock (t(17) = 0.75, p = 0.46) (Fig. 4B) and similar levels of freezing before and after shock delivery (Genotype × Training phase ANOVA, Training phase effect, F(1,18) = 14.65, p < 0.01; no Genotype × Training phase interaction, F(1,18) = 0.39, p = 0.50). Planned comparisons indicated that there were no differences in freezing levels in the postshock phase of training (t(18) = 0.80, p = 0.43) (Fig. 4C). These data indicate that the absence of α-CaMKII did not alter nociception or interfere with the ability to freeze. However, when placed back into the same context 24 h later, α-CaMKII-iKO froze less than their α-CaMKIIf/f littermates (Fig. 4D,E) (t(17) = 1.79, p < 0.05). These results indicate that α-CaMKII-iKO mice are impaired in a form of one-trial fear learning.

Conditional deletion of α-CaMKII impairs contextual fear learning. A, Experimental design. Mice underwent contextual fear conditioning (1 shock) 8 weeks after TAM administration and were tested 1 d later. During training, (B) shock reactivity and (C) postshock freezing were similar in control (α-CaMKII f/f; closed bars) and α-CaMKII-iKO (open bars) mice. During testing, α-CaMKII-iKO showed reduced freezing levels (D, minute-by-minute freezing; E, total freezing in the 5 min test). F, Experimental design. Mice underwent contextual fear conditioning (3 shocks) 8 weeks after TAM administration and were tested 1 d later. During training, (G) shock reactivity and (H) postshock freezing were similar in control (α-CaMKIIf/f; closed bars) and α-CaMKII-iKO (open bars) mice. During testing, α-CaMKII f/f and α-CaMKII-iKO mice exhibited equivalent freezing levels (I, minute-by-minute freezing; J, total freezing in the 5 min test). In the open field, α-CaMKII f/f (closed bars) and α-CaMKII-iKO (open bars) mice (K) covered equivalent total distance and (L) spent equivalent time in different regions of the arena. In the forced swim test, (M) latency to become immobile and (N) total time spent immobile were equivalent in α-CaMKIIf/f (closed bars) and α-CaMKII-iKO (open bars) mice. *p < 0.05.
A previous study found that suppression of adult neurogenesis similarly impaired acquisition of contextual fear conditioning under these weak, but not strong, training conditions (Drew et al., 2010). To evaluate whether contextual fear conditioning was impacted after strong training, we next trained α-CaMKIIf/f and α-CaMKII-iKO with three shocks (Fig. 4F). As before, during training shock reactivity and freezing levels were similar in α-CaMKIIf/f and α-CaMKII-iKO mice (Genotype × Training phase ANOVA, Training phase effect, F(3,22) = 63.55, p < 0.0001; no Genotype × Training phase interaction, F(3,22) = 2.26, p = 0.09). Planned comparisons indicated that there were no differences in shock reactivity (t(22) = 0.48, p = 0.64) or freezing levels in the postshock phase of training (t(18) = 0.80, p = 0.43) (Fig. 4G,H). However, in contrast to the weak training conditioning, following strong training α-CaMKIIf/f and α-CaMKII-iKO mice exhibited equivalent levels of conditioned fear when tested 24 h later (t(21) = 0.56, p = 0.58) (Fig. 4I,J). These results indicate that stronger training may overcome learning deficits observed in α-CaMKII-iKO mice, and confirm that conditional deletion of α-CaMKII from NPCs does not interfere with the propensity to freeze.
Adult neurogenesis is implicated in regulation of mood/anxiety (Sahay and Hen, 2007). To evaluate whether emotionality was altered, we tested separate groups of α-CaMKIIf/f and α-CaMKII-iKO mice in the open field and forced swim tests. In the open field, total locomotor activity was similar in α-CaMKIIf/f and α-CaMKII-iKO mice, suggesting that exploratory behavior is unaffected (t(15) = 0.31, p = 0.76) (Fig. 4K). Furthermore, α-CaMKIIf/f and α-CaMKII-iKO mice spent equivalently little time in the center of the open field, suggesting that anxiety is unaltered (two-way ANOVA; effect of zone only F(2,30) = 64.60, p < 0.0001) (Fig. 4L). In the forced swim test, latency to become immobile (t(22) = 0.94, p = 0.36) and total time immobile (t(22) = 0.71, p = 0.48) was equivalent in α-CaMKIIf/f and α-CaMKII-iKO mice (Fig. 4M,N), suggesting no differences in basal levels of depressive-like behaviors. These data further suggest that that α-CaMKII deficiency in newly generated granule cells does not lead to a general alteration in behavior.
Discussion
Here we developed a transgenic strategy to conditionally delete α-CaMKII from neural progenitor cells and their progeny, and studied the cell-autonomous role of α-CaMKII in morphological and synaptic development in the adult hippocampus. There were three primary findings. First, selective deletion of α-CaMKII from newly generated dentate granule cells led to an increase in dendritic complexity. Second, α-CaMKII deletion led to a reduction in number of mature synapses and cell survival. Third, consistent with altered dendritic and synaptic development, acquisition of one-trial contextual fear conditioning was impaired in α-CaMKII-iKO mice. Previous work in C. elegans (Rongo and Kaplan, 1999), Drosophila (Wang et al., 1994), Xenopus (Wu et al., 1996; Zou and Cline, 1999; Wu and Cline, 1998), and mouse (Pattinson et al., 2006) identified α-CaMKII as playing a key role in the stabilization of dendritic and synaptic structure in the developing nervous system. The current study indicates α-CaMKII plays a similar, cell-autonomous role in the adult hippocampus, and, in addition, reveals that the loss of α-CaMKII from adult-generated granule cells is associated with impaired hippocampus-dependent learning.
The vast majority of cells generated in the subgranular zone of the hippocampus differentiate into dentate granule cells, extending apical dendrites into the molecular layer of the hippocampus and mossy fiber axons toward the CA3 region. However, it is not until ∼2.5 weeks that these cells form input and output synaptic connections, a time point that coincides with the onset of α-CaMKII expression. Thereafter continues a period of continued growth and eventually stabilization of dendritic and synaptic structures, where connections are likely refined in an activity-dependent manner (Tronel et al., 2010). In the developing nervous system, α-CaMKII has been shown to play a critical role in this refinement process, acting, for example, to attenuate dendritogenesis and stabilize dendritic structure. Our data here suggest that this role is conserved as newly generated granule cells integrate into adult hippocampal circuits. We found that α-CaMKII-deficient, newly generated granule cells exhibited increased dendritic complexity (more dendritic bifurcations and branches), consistent with the idea that α-CaMKII acts as a brake on continued dendritogenesis (Cline, 2001). Additionally, there was a reduction in number of mature synapses on α-CaMKII deficient granule cells, consistent with a role for α-CaMKII in promoting synapse maturation. In our study, α-CaMKII deletion was limited to NPCs and their progeny and did not impact postmitotic neuronal populations. Therefore, our data additionally indicate that α-CaMKII regulates these processes in a cell-autonomous manner.
The refinement of neural circuits during development is regulated in an activity-dependent manner (Katz and Shatz, 1996), with synaptic strengthening leading to the stabilization of dendritic structure (Cline, 2001). Because α-CaMKII plays an essential role in synaptic strengthening, it is thought to provide an essential link between local activity levels and the stabilization of dendritic structure, and it is likely that α-CaMKII plays a similar role in the integration of adult-generated granule cells into adult hippocampal circuits. Coordinated presynaptic and postsynaptic activity can lead to long-lasting strengthening of synaptic connections through LTP. During LTP, opening of NMDARs leads to an increase in intracellular calcium levels that activates α-CaMKII (Lisman et al., 2012; Hell, 2014). Once activated, α-CaMKII translocates to the synapse (Shen and Meyer, 1999), binds to NMDARs (Barria and Malinow, 2005), and strengthens synaptic transmission through a signaling cascade that increases AMPA receptor number (Shi et al., 1999; Hayashi et al., 2000) and conductance (Derkach et al., 1999; Poncer et al., 2002; Lüthi et al., 2004). NMDAR- α-CaMKII complexes are necessary for LTP (Barria and Malinow, 2005) and can be persistent once formed (Bayer et al., 2006). Consistent with the idea that NMDA-α-CaMKII signaling plays an essential role in stabilization of dendritic arborization and synapse maturation, the conditional deletion of the NMDAR subunit NR1 from NPCs during adulthood leads to an increase in cell death during a critical period marked by high synaptogenesis and the onset of α-CaMKII expression (Tashiro et al., 2006a). The fact that deletion of α-CaMKII in a similar cell population also leads to a decrease in survival supports the hypothesis that activity, potentially through a NMDAR-α-CaMKII pathway, is a crucial survival signal to shape circuits in the continuously evolving DG. Similarly, conditional deletion of the NMDAR subunit NR2B from NPCs during adulthood also disturbed dendritic morphology in the DG and was associated with hippocampal learning deficits (Kheirbek et al., 2012).
Our results suggest that one consequence of perturbing normal integration of newly generated granule cells into hippocampal circuits is impaired hippocampus-dependent learning. The transgenic approach we used allowed for efficient deletion of α-CaMKII from NPCs and their progeny. Nonetheless, 8 weeks after TAM treatment, we estimated that the population of α-CaMKII-deficient newly generated granule cells to be ∼30,000. As the mouse adult DG contains ∼500,000 granule cells (Ihunwo and Schliebs, 2010; Arruda-Carvalho et al., 2011), this subpopulation of α-CaMKII-deficient granule cells represents only ∼6% of the total granule cell population. Therefore, α-CaMKII deficiency in a relatively small proportion of cells is sufficient to produce deficits in hippocampus-dependent learning. Although this may be surprising, similar results have been observed following NPC-specific deletion of other genes using either transgenic (Zhang et al., 2008; Guo et al., 2011; Kheirbek et al., 2012; Pan et al., 2012; Cancino et al., 2013) or even viral (Zhou et al., 2013) strategies, which would impact far fewer adult-generated granule cells. Because the integration of newborn cells into the hippocampus is a fine-tuned process, it is possible that even small perturbations of this process can impact hippocampal function. It is likely that α-CaMKII deficiency not only compromises anatomical integration but also impairs plasticity of those cells once integrated. Therefore, an alternative (but not necessarily mutually exclusive) possibility is that learning deficits are related to the reduced potential for strengthening of synaptic inputs onto α-CaMKII-deficient cells during conditioning.
Footnotes
This work was supported by Canadian Institutes of Health Research Grant MOP86762 to P.W.F. and Grant MOP74650 to S.A.J. and Brain Canada to P.W.F., M.A.-C. was supported by fellowships from the Ontario Graduate Scholarship program and The Hospital for Sick Children. We thank R. Kageyama and I. Imayoshi for the nestin-CreERT2 mice; and B. Richards, G. Vetere, A. de Cristofaro, R. Braybon, M. Yamamoto, and M. Sakaguchi for discussions and technical assistance.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Paul W. Frankland, Program in Neurosciences and Mental Health, The Hospital for Sick Children, 555 University Avenue, Toronto, Ontario M5G 1X8, Canada. paul.frankland{at}sickkids.ca





{kind=link}
{kind=link}
{kind=link}
{kind=link}