

Abstract
Stroke is a major public health concern. The lack of effective therapies heightens the need for new therapeutic targets. Mammalian brain has the ability to rewire itself to restore lost functionalities. Promoting regenerative repair, including neurogenesis and dendritic remodeling, may offer a new therapeutic strategy for the treatment of stroke. Here, we report that interaction of neuronal nitric oxide synthase (nNOS) with the protein postsynaptic density-95 (PSD-95) negatively controls regenerative repair after stroke in rats. Dissociating nNOS–PSD-95 coupling in neurons promotes neuronal differentiation of neural stem cells (NSCs), facilitates the migration of newborn cells into the injured area, and enhances neurite growth of newborn neurons and dendritic spine formation of mature neurons in the ischemic brain of rats. More importantly, blocking nNOS–PSD-95 binding during the recovery stage improves stroke outcome via the promotion of regenerative repair in rats. Histone deacetylase 2 in NSCs may mediate the role of nNOS–PSD-95 association. Thus, nNOS–PSD-95 can serve as a target for regenerative repair after stroke.
Introduction
Stroke is a leading cause of death and adult acquired disability. Many of the sensory, motor, and cognitive impairments caused by stroke eventually improve, suggesting an ability of brain to rewire itself to restore lost functionalities (Carmichael, 2008). The modification of neuronal connections may result from axonal sprouting, neurogenesis, or dendritic arbor remodeling (Arvidsson et al., 2002; Nakatomi et al., 2002; Carmichael, 2008; Clelland et al., 2009; Sun et al., 2009; Mostany et al., 2010; Marín-Burgin et al., 2012; Ueno et al., 2012). Thus, promoting regenerative repair, including neurogenesis, synaptogenesis, and neural regeneration, may offer a new therapeutic strategy for the treatment of stroke.
The protein postsynaptic density-95 (PSD-95) is a scaffolding protein that binds both NMDA receptors (NMDARs) and neuronal nitric oxide synthase (nNOS) at excitatory synapses, and assembles them into a macromolecular signaling complex (Aarts et al., 2002). Activation of nNOS depends on its association with PSD-95 and on NMDAR-mediated calcium influx (Sattler et al., 1999). Recently, we demonstrated that cerebral ischemia or NMDA stimulation induces the interaction of nNOS with PSD-95, thereby leading to nitric oxide (NO) overproduction and neuronal injury, and disrupting this interaction prevents ischemic damage after stroke (Zhou et al., 2010). Here we show that nNOS-PSD-95 interaction impairs regenerative repair after stroke in rats. Dissociating nNOS-PSD-95 coupling in neurons promotes neuronal differentiation of neural stem cells (NSCs), facilitates migration of newborn cells into the injured area, and enhances neurite growth of newborn neurons and dendritic spine formation of mature neurons in the ischemic brain via regulation of histone deacetylase 2 (HDAC2). More importantly, blocking nNOS-PSD-95 binding during the recovery stage improves stroke outcome by promoting regenerative repair.
Materials and Methods
Drugs
ZL006, a small molecular inhibitor of nNOS–PSD-95 interaction, was designed and synthesized in our laboratory (Zhou et al., 2010). DETA/NONOate (3,3-bis(aminoethyl)-1-hydroxy-2-oxo-1-triazene), GSNO (S-nitrosoglutathione), C-PTIO (carboxy-PTIO potassium salt), and TSA (trichostatin A) were purchased from Sigma-Aldrich.
Animals
Homozygous nNOS-deficient mice (B6;129S4-Nos1tm1Plh, nNOS−/−; The Jackson Laboratory) and wild-type (WT) mice with similar genetic background (B6129SF1, nNOS+/+) were maintained in Model Animal Research Center of Nanjing University. Green fluorescent protein (GFP) transgenic Sprague Dawley (SD) rats (a gift from Dr. Cheng He, Second Military Medical University, Shanghai, People's Republic of China) and GFP transgenic C57BL/6 mice (a gift from Dr. Zhengang Yang, Fudan University, Shanghai, People's Republic of China) were maintained in Laboratory Animal Center of Nanjing Medical University. The nNOS−/−, nNOS+/+, and GFP-expressing embryos were used for cell cultures. Adult and embryonic SD rats from Laboratory Animal Center of Nanjing Medical University were also used in this study, for ischemic animal models (male rats) or cell cultures. All experimental protocols using animals were approved by the Institutional Animal Care and Use Committee of Nanjing Medical University.
Focal cerebral ischemia
Focal cerebral ischemia was induced by intraluminal middle cerebral artery occlusion (MCAO), as we described previously (Zhou et al., 2010). In brief, under chloral hydrate anesthesia (350 mg/kg, i.p.), a 4/0 surgical nylon monofilament with rounded tip was introduced into the right internal carotid artery through the external carotid stump, advanced 20–21 mm past the carotid bifurcation until a slight resistance was felt, left in place for 120 min, and then withdrawn to allow reperfusion. Regional cerebral blood flow (rCBF) was measured through a laser Doppler perfusion monitor (Moor Instruments) to ensure that the rCBF decreased by 85–95%.
Cell cultures
Embryonic NSCs were isolated from embryonic day 14 (E14) mouse or E16 rat cortex as we previously described (Luo et al., 2010). Cells were floating cultured in proliferation medium, DMEM/F12 medium (1:1; Invitrogen) containing 20 ng/ml basic fibroblast growth factor (Sigma-Aldrich), 20 ng/ml epidermal growth factor (Sigma-Aldrich), and 2% B27 supplement (Invitrogen), and passaged every 4–6 d when neurospheres ∼100 μm were formed. These embryonic NSCs still possessed proliferation and self-renewal capacity, and were able to generate differentiated progeny until 10th passage (Luo et al., 2010). Embryonic NSCs of the second to the fifth passage were used in this study.
Primary neurons were isolated from E16 mouse or E18 rat cortex and cultured on dishes coated with polyornithine (10 μg/ml; Sigma-Aldrich) in Neurobasal medium (Invitrogen) containing 2% B27 supplement, as we have reported (Luo et al., 2010). The planting density was 1 × 104 cells/cm2 for morphological analysis and 1 × 105 cells/cm2 for biochemical detection. The proportion of β-III-tubulin+ cells at day 10 in vitro was ∼93%.
All cultures were maintained in an incubator (HERAcell 150, Thermo Fisher Scientific) with a humidified atmosphere of 95% air and 5% CO2 at 37°C, except for oxygen-glucose deprivation (OGD) experiments.
Differentiation of cocultured or solo-cultured NSCs
In the coculture of NSCs with neurons, neurons or NSCs were prepared from transgenic animals expressing GFP for distinguishing between neurons and NSCs. Rat NSCs were cocultured with neurons from the transgenic rats expressing GFP in most experiments, except the coculture of NSCs from the transgenic mice expressing GFP with neurons from nNOS−/− mice. Primary neurons were cultured in vitro for 10 d, and neurospheres were triturated to single NSCs and planted on them at 1 × 104 cells/cm2 (morphological analysis) or 1 × 105 cells/cm2 (biochemical detection). For the differentiation of only cultured NSCs, passaged NSCs were planted on polyornithine-coated dishes. Neurobasal medium containing 2% B27 and 0.5% fetal bovine serum was used to induce NSC differentiation for solo cultures or cocultures. After 24 h of differentiation, cultures underwent OGD and/or drug treatment, and then the differentiation was maintained for desired time periods.
Oxygen and glucose deprivation
After 24 h of differentiation, cocultured or only cultured NSCs were exposed to OGD. Cells were rinsed twice with serum- and glucose-free medium and incubated with serum- and glucose-free medium in a hypoxia chamber (MACS MICS jar gassing system, Don Whitley Scientific). The chamber was flushed with a mixture of 85% N2, 10% H2 and 5% CO2 through two anaerobic cycles, and then sealed and maintained at 37°C for 2 h. After OGD, the serum- and glucose-free medium was replaced with normal medium, and the cultures were returned to a normoxic incubator. During 2 h of OGD and 24 h after OGD, 10 μm ZL006 or 50 nm Tat-nNOS-N1–133 was added to the cultures to disrupt the interaction of nNOS–PSD-95.
Recombinant virus production and infection
LV-nNOS-N1–133-GFP.
The recombinant lentivirus, LV-nNOS-N1–133-GFP or its control LV-GFP, was generated as we previously reported (Zhou et al., 2010). LV-nNOS-N1–133-GFP selectively expresses N-terminal amino acid residues 1–133 of nNOS (nNOS-N1–133), a region crucial for nNOS–PSD-95 interaction. Cultured neurons were infected with LV-nNOS-N1–133-GFP or LV-GFP containing 1.0 × 109 transduction units/ml at day 4 in vitro [multiplicity of infection (MOI) = 2.5]. The medium was half changed 8 h later and fully changed 24 h later. The lentivirus-infected neurons were cocultured with NSCs at day 6 after infection. For lentivirus infection in vivo, stereotaxic microinjection of 2 μl of virus suspension to right striatum (posterior, 0.30 mm; lateral, 3.40 mm; depth, 6.00 mm) was carried out. Seven days after lentivirus infusion, the rats were subjected to MCAO.
AD-HDAC2-Flag.
The recombinant adenovirus AD-HDAC2-Flag is produced by GeneChem. The coding sequence of mouse HDAC2 was amplified by RT-PCR. The primer sequences were as follows: forward, 5′-CGG GTA CCG GTC GCC ACC ATG GCG TAC AGT CAA GGA G-3′; reverse, 5′-CGG AAT TCT CAC TTG TCA TCG TCA TCC TTG TAG TCA GGG TTG CTG AGT TGT TCT G-3′. The PCR fragments and the pDC315 plasmid were digested with AgeI and EcoRI, and ligated with T4 DNA ligase to produce pDC315-HDAC2-Flag, which overexpresses mouse HDAC2 protein with a Flag tag in the C terminal. The plasmid was used to transform competent DH5α Escherichia coli bacterial strains for identification. Using 10 μl of Lipofectamine 2000 mixed with 50 μl of DMEM, HEK293 cells were cotransfected with 5 μg of the pDC315-HDAC2-Flag and 5 μg of the pBHG lox ▵E1, 3 Cre plasmid as a helper plasmid to generate the recombinant adenovirus AD-HDAC2-Flag. Fifty days later, the supernatant was harvested from HEK293 cells. After 2× virus amplification, the supernatant was filtered at 0.45 μm and purified using the Adeno-X virus purification kit (BD Bioscience Clontech). After resuspension, serially diluted adenovirus was used to transduce HEK293 cells. Seven days later, labeled HEK293 cells were counted to calculate the viral titer (1.5 × 109 plaque-forming units/ml).
AD-HDAC2-Flag or AD-Flag (control) was added to single-cell suspensions of NSCs when passage (MOI = 3). The medium containing adenovirus was not changed until neurospheres were formed 4 d later. These neurospheres were triturated to single-cell suspension and cocultured with primary neurons.
LV-HDAC2 shRNA.
HDAC2 shRNA (m) lentiviral particles (LV-HDAC2 shRNA, Santa Cruz Biotechnology) are recommended for the inhibition of HDAC2 expression in mouse cells. It is a pool of concentrated, transduction-ready viral particles containing three target-specific constructs that encode 19–25 nt (plus hairpin) shRNA designed to knock down gene expression. Each 200 μl frozen stock contained 1.0 × 106 infectious units of virus. LV-HDAC2 shRNA or LV-Control shRNA (Santa Cruz Biotechnology) was added into cultured NSCs at 10 μl/well (24-well plate) when passaged. Sixteen hours later, the medium was fully changed with fresh proliferation medium. Four days after infection with the lentivirus, NSCs were passaged for experiments.
Tat-nNOS-N1–133 preparation
Gene fragment encoding nNOS1–133 was assembled by RT-PCR, and Tat was added to its 5′ end by add-PCR. The gene fragment was inserted into a T7 RNA polymerase-based expression system pET28a vector (Novagen). We named it pET28a-Tat-nNOS1–133. The pET28a-Tat-nNOS1–133 was transformed into E. coli strain BL21 cultures, which were grown in LB medium containing kanamycin overnight at 37°C, until the OD600 of 0.8 was reached, and then they were cultured for 6 h by adding isopropyl β-d-thiogalactoside (0.7 mm).
The recombinant fusion protein of Tat-nNOS1–133 was expressed into inclusion bodies. Bacteria were resuspended with lysis buffer (50 mm Tris-HCl, 10 mm EDTA, 100 mm NaCl, pH 8.0) and subsequently lysed with ultrasonication. Most of the other bacterial proteins were eliminated during the early steps of washing the resulting pellet with wash buffer (50 mm Tris-HCl, 0.5% Triton X-100, pH 8.0) and 2 m urea. Inclusion bodies were solubilized by 8 m urea, and Tat-nNOS1–133 was refolded by dialysis with a renaturation buffer (50 mm Tris-HCl, 0.5 mm EDTA, 50 mm NaCl, 8% glycerol, pH 8.0) at 4°C. After centrifugation at 12,000 × g for 10 min, supernatant containing refolded protein was loaded on a DEAE-52 column, and Tat-nNOS1–133 was eluted by gradient elution (50 mm Tris-HCl, 0.5 mm EDTA, 0∼200 mm NaCl, pH 8.0). The protein fractions corresponding to Tat-nNOS1–133 were collected and analyzed using SDS-PAGE. Tat-nNOS1–133 was placed into PBS and concentrated with Amicon Ultra-15 Centrifugal Filter Units.
Immunofluorescence
The details of immunofluorescence for brain sections and cultured cells have been described (Luo et al., 2007, 2010). The primary antibodies used were as follows: mouse anti-β-III-tubulin (1:300; Millipore Bioscience Research Reagents), mouse anti-GFAP (1:1000; Millipore Bioscience Research Reagents), mouse anti-GalC (1:1000; Millipore Bioscience Research Reagents), rat anti-BrdU (1:200; Accurate Chemical & Scientific Corporation), rabbit anti-DCX (1:1000; Abcam), rabbit anti-GFP (1:1000, Abcam), and mouse anti-NeuN (1:500; Millipore Bioscience Research Reagents). Secondary antibodies used were goat anti-rat Cy3 (1:200; Millipore Bioscience Research Reagents), goat anti-mouse Cy3 (1:200; Jackson ImmunoResearch), goat anti-rabbit Dylight 488 (1:400; Jackson ImmunoResearch), and goat anti-rabbit Dylight 405 (1:400; Jackson ImmunoResearch). Finally, cultures were counterstained with Hoechst 33258 (Sigma-Aldrich) to label the nuclei.
Morphological analysis
Images were captured with a fluorescence microscope (Axio Imager, Zeiss) or a confocal laser-scanning microscope (LSM700, Zeiss) and analyzed with Imaris 7.3.0 software (BITPLANE). For brain sections, the analysis was conducted on every 10th section in a series of 40 μm coronal sections throughout dentate gyrus (DG) or subventricular zone (SVZ; intravenous injection experiments), or every 4th section in a series of 30 μm coronal sections around the injection site (stereotaxic injection experiments). For cultures, the percentages of cells were calculated systematically in 10 fields across the dish, and the morphological data of neuritis/dendrites were obtained from six to eight cells randomly for each sample. The average was regarded as the final value of one sample. More than five samples (for animals) or three samples (for cultures) from each group were included in the statistics.
Western blot analysis
Western bolt analysis was performed as described previously (Luo et al., 2010; Zhou et al., 2010). The primary antibodies were as follows: mouse anti-HDAC2 (1:3000; Abcam), rabbit anti-biotin (1:2000; Cell Signaling Technology), mouse anti-Flag (1:1000; Sigma-Aldrich), rabbit anti-histone H3 (1:800; Abcam), rabbit anti-acetylated histone H3 (acetyl K9; 1:800; Abcam), rabbit anti-histone H4 (1:500; Abcam), rabbit anti-acetylated histone H4 (acetyl K5; 1:1000; Abcam), rabbit anti-NeuroD (1:2000; Abcam), rabbit anti-pCREB (Ser133; 1:1000; Cell Signaling Technology), or rabbit anti-CREB (1:1000; Cell Signaling Technology). Internal control was performed using mouse anti-GAPDH (1:4000; KangChen Bio-tech) or mouse anti-β-actin (1:1000; Sigma-Aldrich), Appropriate horseradish peroxidase-linked secondary antibodies were used for detection by enhanced chemiluminescence (Pierce).
Biotin-switch assay
This assay was performed in the dark as previously described (Jaffrey and Snyder, 2001). Briefly, cells were lysed in HEN buffer (250 mm HEPES, 1 mm EDTA, and 100 mm neocuproine) and adjusted to contain 0.4% CHAPS. Samples were homogenized, and free cysteines were blocked for 1 h at 50°C in three volumes of blocking buffer [HEN buffer plus 2.5% SDS (HENS)] containing methyl methanethiosulfonate (200 mm; Sigma-Aldrich). Proteins were precipitated with acetone at −20°C and resuspended in 300 μl of HENS solution. After adding fresh ascorbic acid (20 mm; Sigma-Aldrich) and biotin-HPDP [N-[6-(biotinamido)hexyl]-3-pyridyldithio)-propionamide; 1 mm; Sigma-Aldrich], proteins were incubated at room temperature for 1 h. After separation using an SDS-PAGE gel in nonreducing loading buffer, biotinylated proteins were detected by immunoblotting using rabbit anti-biotin. Alternatively, biotinylated proteins were resuspended in 250 μl of HENS buffer plus 500 μl of neutralization buffer (20 mm HEPES, 100 mm NaCl, 1 mm EDTA, 0.5% Triton X-100) and precipitated with 50 μl of prewashed avidin-affinity resin beads (Sigma-Aldrich) at room temperature for 1 h. The beads were washed five times at 4°C using neutralization buffer containing 600 nm NaCl. Biotinylated proteins were eluted using 30 μl of elution buffer (20 mm HEPES, 100 mm NaCl, 1 mm EDTA, 100 mm β-mercaptoethanol) and heated at 100°C for 5 min in reducing SDS-PAGE loading buffer.
HDAC2 activity assay
HDAC activity was assessed using a HDAC fluorometric assay kit (EMD Millipore). For HDAC2-specific activity, immunoprecipitation (IP) with the specific antibody was performed before the assay as described previously (Nott et al., 2008). In brief, cellular lysates prepared in immunoprecipitation buffer (50 mm Tris-HCl, 150 mm NaCl, 5 mm EDTA, 0.5% NP40, pH 8.0, supplemented with 1 mm PMSF) were incubated with 0.5 μg of mouse anti-HDAC2 antibody (Abcam) and 15 μl of protein G-agarose (Sigma-Aldrich) overnight on a tube rotator at 4°C. Then beads were centrifuged at 2000 × g and washed five times in immunoprecipitation buffer. HDAC assay substrate was added to the beads and incubated at 30°C for 45 min. Finally, activator solution containing Trichostatin A (HDAC inhibitor) was used to stop the reaction, and fluorescence was measured according to the instructions of the HDAC fluorometric assay kit.
Coimmunoprecipitation
Lysis and coimmunoprecipitation of cultures and tissues were performed as we described previously (Zhou et al., 2010). Cultured neurons, hippocampal tissues, or striatum penumbra tissues were lysed in 50 mm Tris-HCl, pH 7.4, buffer containing 150 mm NaCl, 1 mm EDTA-Na, 1% NP-40, 0.02% sodium azide, 0.1% SDS, 0.5% sodium deoxycholate, 1% PMSF, 1‰ aprotinin, 1‰ leupeptin, and 0.5‰ pepstatin A. The lysates were centrifuged at 12,000 × g for 15 min at 4°C. The supernatant (200 μl) was preincubated for 1 h at 4°C with 25 μl of protein G-Sepharose beads (Sigma-Aldrich), and then centrifuged to remove proteins that adhered nonspecifically to the beads and to obtain the target supernatant for the following IP experiment. Protein G Sepharose beads were incubated with rabbit anti-nNOS (1:100; Affinity BioReagents) or rabbit anti-PSD-95 (1:100; Cell Signaling Technology) for 3–4 h. The antibody-conjugated protein G-Sepharose beads and the target supernatant were added for incubation overnight at 4°C. Immune complexes were isolated by centrifugation, washed five times with 0.05 m HEPES buffer, pH 7.1, containing 0.15% Triton X-100, 0.15 m NaCl, and 0.1 × 10−3 m sodium orthovanadate; and bound proteins were eluted by heating at 100°C in loading buffer. Proteins were analyzed by immunoblotting using rabbit anti-PSD-95 (1:500; Cell Signaling Technology) or rabbit anti-nNOS (1:800; Invitrogen).
Behavioral assessment
Morris water maze.
The spatial cognitive performance of rats was evaluated by Morris water maze or Barnes circular maze. The protocol of Morris water maze test for rats was similar to that for mice, which has been described in detail in our previous reports (Zhou et al., 2010). Some modifications have been made in this study. In brief, the rat was placed in the opaque water of the circular swimming pool (180 cm in diameter) and trained to locate the hidden platform 1.2 cm under the surface of the water. In the training to find the hidden platform, rats were allowed to swim for a maximum of 60 s in the pool for each trail. One block of four trials per day was given for 5 consecutive days. On the next day, the rats were given one 60 s retention probe test in which the platform was removed from the pool. During retention, the number of crossings of the platform location and the time spent in the target quadrant were measured.
Grid walking.
The grid-walking apparatus was manufactured as described previously (Hernandez and Schallert, 1988; Clarkson et al., 2010), using a 3-cm-square wire mesh with a grid area of 60 × 60 × 60 cm (length × width × height). A mirror was placed beneath the apparatus to allow video recording to assess the animals' stepping errors (foot faults). Each rat was placed individually on top of the elevated wire grid and allowed to freely walk for a period of 5 min. Video footage was analyzed off-line by raters who were blind to the treatment groups. The total number of foot faults for each limb, along with the total number of non-foot-fault steps, was counted, and a ratio between foot faults and total steps taken was calculated. The percentage of foot faults was calculated as follows: number of foot faults/(foot faults + number of non-foot-fault steps) × 100. A ratio between foot faults and the total number of steps taken was used to take into account differences in the degree of locomotion between animals and trials. A step was considered a foot fault if it was not providing support and the foot went through the grid hole. Furthermore, if an animal was resting with the grid at the level of the wrist, this was also considered a fault. If the grid was anywhere forward of the wrist area, then this was considered to be a normal step.
Spontaneous forelimb task (cylinder task).
The protocol of the spontaneous forelimb task for rats was similar to that for mice, which has been described in detail previously (Clarkson et al., 2010). Some modifications have been made in this study. In brief, the spontaneous forelimb task encourages the use of forelimbs for vertical wall exploration/press in a cylinder. When placed in a cylinder, the animal rears to a standing position, while supporting its weight with either one or both of its forelimbs on the side of the cylinder wall. Rats were placed inside a Plexiglas cylinder (30 cm in height with a diameter of 20 cm) and videotaped for 5 min. Videotaped footage of animals in the cylinder was evaluated quantitatively to determine forelimb preference during vertical exploratory movements. While the video footage was played in slow motion (one-fifth real-time speed), the time (in seconds) during each rear that each animal spent on the right forelimb, the left forelimb, or both forelimbs was calculated. Only rears in which both forelimbs could be clearly seen were timed. The percentage of time spent on each limb was calculated, and these data were used to derive an asymmetry index, as follows: (percentage of ipsilateral use) − (percentage of contralateral use).
Golgi-Cox staining
Fresh brains without perfusion and fixation were used for Golgi-Cox staining to show subtle morphological alterations in neuronal dendrites and dendritic spines. Golgi-Cox staining was performed with FD Rapid GolgiStain Kit (FD NeuroTechnologies) according to the user manual. Briefly, the brains were first placed in impregnation solution for 2 weeks followed by 2 d in a 30% sucrose solution. Then they were cut into 100 μm coronal sections using a vibratome (World Precision Instruments) and stained. For morphological analysis, 10 random neurons from each sample were measured, and the average was regarded as the final value of one sample.
Statistical analysis
Comparisons among multiple groups were made with one-way ANOVA (one factor) or two-way ANOVA (two factors) followed by Scheffé's post hoc test. Comparisons between two groups were made with a two-tailed Student's t test. Data were presented as the mean ± SEM, and p < 0.05 was considered statistically significant. Investigators were blind to the group allocation when assessing the outcome.
Results
Ischemia-induced nNOS–PSD-95 association inhibits neurogenesis
Neuron-derived nNOS, a source of extracellular NO for NSCs, exerts a negative control on neurogenesis (Luo et al., 2010). Activation of nNOS in neurons depends on its association with PSD-95 and on NMDAR-mediated Ca2+ influx (Sattler et al., 1999). We thus hypothesized that nNOS–PSD-95 interaction negatively regulates the fate of newborn cells in ischemic brain (Fig. 1A). To address this notion, we tested the effect of ZL006, a drug that selectively blocks nNOS–PSD-95 binding in the ischemic cortex after stroke (Zhou et al., 2010), on the nNOS–PSD-95 interaction in the ischemic hippocampus and striatum, two regions implicated in ischemia-induced neurogenesis (Gould, 2007). As expected, ZL006 significantly decreased nNOS–PSD-95 complex in the hippocampus and striatum of rats subjected to MCAO for 2 h and reperfusion (Fig. 1B). To determine whether the ischemia-induced nNOS–PSD-95 association affects the fate of newborn cells, we treated rats with BrdU to label proliferating cells at day 5 and day 4 before MCAO administration. ZL006 (1.5 mg/kg/d, i.v.) was administered for 7 d beginning immediately after reperfusion. These rats were killed at 48 h after the last ZL006 injection to estimate the number of BrdU+ and BrdU+/DCX+ (a marker for immature neurons) cells (Fig. 1C). Treatment with ZL006 significantly increased the number of BrdU+ and BrdU+/DCX+ cells in the hippocampus (Fig. 1D–F), and increased the number of BrdU+ cells in the granular cell layer (GCL) of DG (Fig. 1G). Additionally, ZL006 increased the number of BrdU+/NeuN+ cells at day 28 after BrdU labeling in the ischemic DG (Fig. 1H).

Dissociation of nNOS–PSD-95 coupling by ZL006 promotes neurogenesis after stroke. A, Illustration showing that ischemia-induced nNOS–PSD-95 interaction negatively regulates neurogenesis in ischemic brain. B, Co-IP showing levels of nNOS–PSD-95 complex in the striatum (left) and hippocampus (right) of rats subjected to MCAO for 2 h (n = 3). C, Schematic representation of experimental design for D–G and I–L. D, Images of the ischemic DG and striatum that were treated with ZL006 or vehicle and stained with BrdU (red) and DCX (green). Scale bars, 20 μm. E–G, Bar graphs showing BrdU+ cells (E) and BrdU+/DCX+ cells (F) in the ipsilateral DG and striatum, and BrdU+ cells in GCL of the ipsilateral DG (G). H, Images of BrdU+/NeuN+ cells in the ipsilateral DG. Scale bar, 50 μm. I, J, Images (I) and bar graph (J) showing DCX+ cells in different areas of the ipsilateral striatum (n = 5 for vehicle, n = 6 for ZL006). K, L, Images (K, left) showing neurites of DCX+ cells and bar graph (K, right) showing total neurite length per DCX+ cell in the ipsilateral striatum, and images (L) showing neurites of DCX+ cells in the ipsilateral DG (n = 5 for vehicle, n = 6 for ZL006). Eight DCX+ cells were measured in each biological sample. Scale bars, 20 μm. Data are the mean ± SEM. *p < 0.05, **p < 0.01; ANOVA for B and E–G, two-tailed t test for J and K.
In the normal striatum, newborn cells were scarce in both vehicle- and ZL006-treated animals (BrdU+ cells: 250.0 ± 17.2 for vehicle and 266.7 ± 17.6 for ZL006; n = 6; p = 0.387). In the ischemic brain, however, many newborn cells migrated from SVZ into striatum. Treatment with ZL006 significantly increased the number of BrdU+ and BrdU+/DCX+ cells (Fig. 1D–F) and the migration distance of DCX+ cells (Fig. 1I, J) in the ischemic striatum. Moreover, this treatment increased the neurite length (Fig. 1K) of DCX+ cells in the ischemic striatum and slightly increased the neurite length of DCX+ cells in the ischemic DG (638.4 ± 42.6 vs 687.1 ± 49.5 μm; Fig. 1L).
Ischemic brain injury stimulates neurogenesis in the mammalian brain (Jin et al., 2010). To test whether dissociating nNOS–PSD-95 interaction by ZL006 treatment upregulates neurogenesis even when lesion volume is decreased, we measured infarct volume at day 9 after MCAO. Indeed, ZL006-treated animals displayed a substantially decreased infarct size compared with vehicle-treated animals [18.9 ± 2.2% (n = 16) vs 31.9 ± 2.8% (n = 11); p = 0.001].
To further ascertain the role of nNOS–PSD-95 association in regulating neurogenesis in the ischemic striatum, we generated a lentiviral vector that selectively expresses nNOS-N1-133, a region crucial for nNOS–PSD-95 interaction (Zhou et al., 2010), and named it LV-nNOS-N1-133-GFP. When microinjected into the striatum, the recombinant virus effectively infected the ischemic striatum (Fig. 2A), produced considerable nNOS-N1-133 peptide (Fig. 2B), and blocked nNOS–PSD-95 interaction in the striatum but not in the hippocampus after stroke (Fig. 2C). Next, we delivered LV-nNOS-N1-133-GFP into the striatum of rats by microinjection, and 2 d later we treated rats with BrdU to label proliferating cells. Rats were subjected to MCAO for 2 h at day 4 after the last BrdU injection, and were killed at day 9 after MCAO to estimate the number of BrdU+ and BrdU+/DCX+ cells (Fig. 2D). LV-nNOS-N1-133-GFP infection significantly increased the number of BrdU+ and BrdU+/DCX+ cells in the striatum but not in the hippocampus (Fig. 2E, F), suggesting a selective enhancement of neurogenesis in the LV-nNOS-N1-133-GFP-infected area. More interestingly, this treatment significantly increased the migration distance (Fig. 2G) and neurite length of DCX+ cells in the ischemic striatum (Fig. 2H, I). We also investigated the neurogenesis in the normal brain after LV-nNOS-N1-133-GFP infection. Like the results from ZL006, almost no newborn cell was found in the normal striatum in either the LV-nNOS-N1-133-GFP or the LV-GFP group. Moreover, neurogenesis in the normal DG was not influenced by striatal LV-nNOS-N1-133-GFP microinfection.

Dissociation of nNOS–PSD-95 coupling by LV-nNOS-N1–133-GFP promotes neurogenesis in the ischemic brain. A, Images of the striatum infected by LV-nNOS-N1–133-GFP (left) and a high-magnification image from a selected area in the leftward image that was stained with GFP and NeuN (red; right). Scale bar, 50 μm. B, Immunoblots showing nNOS-N1-133-GFP peptide level in the striatum infected by LV-nNOS-N1–133-GFP. C, Co-IP showing levels of nNOS–PSD-95 complex in the ischemic striatum infected by LV-nNOS-N1–133-GFP (n = 3). D, Schematic representation of experimental design for E–H. E–G, Bar graphs showing BrdU+ cells (E) and BrdU+/DCX+ cells (F) in the ipsilateral DG and striatum, and DCX+ cells in the different areas of ipsilateral striatum (G). H, Image of the ischemic striatum stained with DCX (blue) and BrdU (red) (left), and bar graph showing neurite length of DCX+ cells (right) in the ischemic striatum (n = 6 for LV-GFP, n = 8 for LV-nNOS-N1–133-GFP). Eight DCX+ cells were measured in each biological sample. Data are the mean ± SEM. *p < 0.05, **p < 0.01, two-tailed t test.
Together, these data suggest that disrupting the nNOS–PSD-95 interaction after stroke benefits the survival and neuronal differentiation of cells that were born before stroke, and promotes the migration of newborn cells into the injured area and neurite growth of newborn neurons in the ischemic brain.
To examine the effect of disassociating nNOS and PSD-95 on ischemia-stimulated newborn cells, we subjected rats to MCAO for 2 h and treated them with ZL006 starting at reperfusion. From day 6 to day 7 after MCAO administration, we treated rats with BrdU to label proliferating cells. The BrdU-treated rats were killed 4 weeks after the last BrdU injection to estimate the number of BrdU+ cells. ZL006 significantly increased the number of BrdU+ cells in the ipsilateral hippocampal DG 4 weeks after BrdU labeling [contralateral: 2557 ± 262.4 cells (vehicle, n = 8) vs 3254 ± 448.1 cells (ZL006, n = 6), p = 0.1441; ipsilateral: 4766 ± 950.3 cells (vehicle, n = 8) vs 7450 ± 640.1 cells (ZL006, n = 6), p = 0.0087], and, moreover, ZL006 increased the survival rate of newborn cells in both contralateral and ipsilateral hippocampus [contralateral, 33.1% (vehicle) vs 53% (ZL006); ipsilateral, 27.9% (vehicle) vs 38.7%], suggesting that disrupting nNOS–PSD-95 coupling also benefits the survival of the cells that are born after a stroke.
Ischemia-induced nNOS–PSD-95 association in neurons negatively controls the fate of NSCs
To answer how the nNOS–PSD-95 interaction affects neurogenesis in the ischemic brain, we established a coculture of NSCs with neurons and treated the coculture with OGD, a cell model used to mimic ischemia in vitro. To distinguish newborn neurons from original neurons in the cocultures, NSCs were cocultured with GFP+ neurons from the transgenic rats expressing GFP. As PSD-95 appeared in neurons, but not in glia cells, and in NSCs, PSD-95 did not appear until 4 d after differentiation (Fig. 3A), we reasoned that the nNOS–PSD-95 interaction must occur in GFP+ neurons only in the cocultures at day 1 after NSC differentiation. We subjected the cocultures differentiated for 24 h to OGD for 2 h and treated them for 26 h with ZL006 or Tat-nNOS-N1–133, a fusion peptide of nNOS-N1–133, and Tat protein. Both ZL006 and Tat-nNOS-N1–133 disrupted OGD-induced nNOS–PSD-95 interaction in neurons (Fig. 3B,C). OGD exposure caused significant decreases in β-III-tubulin+ newborn cells, and newborn neurons with multineurites, both ZL006 and Tat-nNOS-N1–133, reversed the OGD-induced changes (Fig. 3D–H). ZL006 also ameliorated the OGD-induced decrease in neurite length of the newborn neurons with multineurites (Fig. 3I,J).

Interaction of nNOS with PSD-95 in neurons negatively regulates the fate of NSCs. A, Immunoblot showing PSD-95 level in differentiating NSCs at the indicated times, and in neurons and astrocytes (n = 3). B, C, Co-IP showing the amount of nNOS–PSD-95 complex in the cultured neurons subjected to OGD for 2 h and treated with 10 μm ZL006 (B) or 50 nm Tat-nNOS-N1–133 (C) for 3 h beginning at OGD (n = 3). D–J, NSCs cocultured with neurons from the GFP transgenic rats were allowed to differentiate for 24 h, and then subjected to OGD for 2 h. The cocultures were treated with 10 μm ZL006 (D–F, I, and J) or 50 nm Tat-nNOS-N1–133 (G, H) for 26 h beginning at OGD, and stained at days 4∼5 after OGD. D, E, Images of the cocultures (D) and bar graph (E) showing the percentage of newborn neurons, astrocytes, and oligodendrocytes. β-III-tubulin, Neuron marker; GFAP, astrocyte marker; and CalC, oligodendrocyte marker. Scale bar, 50 μm. n = 3. F, Images showing neurites of newborn neurons (left) and bar graph showing the percentage of newborn neurons with multineurites (right) in the cocultures (vs total newborn neurons). n = 3. G, H, Bar graphs showing the percentage of newborn neurons (G) and the percentage of newborn neurons with multineurites (vs total newborn neurons; H) in the cocultures. n = 3. I, J, Images showing newborn neurons with multineurites (I) and bar graph showing the total neurite length per newborn neuron with multineurites (J) in the cocultures. Scale bar, 50 μm. n = 3. Ten newborn neurons with multineurites were measured in each biological sample. Data are the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ANOVA.
To determine whether neurogenic effect of ZL006 in the cocultures under OGD conditions depends on the dissociation of nNOS–PSD-95 coupling in neurons, we subjected the cocultures of the NSCs from the transgenic mice expressing GFP with the neurons from nNOS gene knock-out (nNOS KO) or WT mice to OGD and treated them with ZL006. When cocultured with WT neurons, ZL006 rescued the OGD-induced reductions in β-III-tubulin+ newborn cells and newborn neurons with multineurites, when cocultured with nNOS KO neurons; however, OGD with or without ZL006 did not change the number of β-III-tubulin+ newborn cells and newborn neurons with multineurites under OGD conditions (Fig. 4A–C), suggesting that the nNOS–PSD-95 interaction in neurons is crucial for the effects of OGD and ZL006 on NSC fate in the cocultures.

Blocker of nNOS–PSD-95 association does not affect the fate of NSCs in the solo-cultured NSCs or in the cocultures of NSCs with nNOS KO neurons. A–C, The cocultures of GFP+ NSCs with nNOS−/− (KO) or with WT neurons were allowed to differentiate for 24 h, and then were subjected to OGD for 3 h. The cocultures were treated with 10 μm ZL006 for 27 h beginning at OGD and stained at days 4∼5 after OGD. A, Image of the cocultures of GFP+ NSCs with nNOS−/− neurons. Scale bar, 50 μm. B, C, Bar graph showing the percentage of newborn neurons (B) and the percentage of newborn neurons with multineurites (C; n = 3). D–J, The solo-cultured NSCs were allowed to differentiate for 24 h and then were subjected to OGD for 2 h. The cultures were treated with 10 μm ZL006 (D–H) or 50 nm Tat-nNOS-N1–133 (I, J) for 26 h beginning at OGD and stained at days 4∼5 after OGD. D, Images of solo-cultured NSCs stained with β-III-tubulin, GFAP, and CalC. Scale bar, 50 μm. E, Bar graph showing the percentage of newborn neurons (β-III-tubulin+), astrocytes (GFAP+), and oligodendrocytes (CalC+) in the cultures (n = 4). F–H, Images showing newborn neurons with multineurites (scale bar, 50 μm; F) and bar graph (G) showing the percentage of newborn neurons with multineurites and the total neurite length per newborn neuron with multineurites (H; n = 4). Eight newborn neurons with multineurites were measured in each biological sample. I, J, Bar graph showing the percentage of newborn neurons (β-III-tubulin+) in the cultures (I) and the percentage of newborn neurons with multineurites (J; n = 4). Data are the mean ± SEM. *p < 0.05, **p < 0.01, ANOVA.
However, it is possible that blockers of nNOS–PSD-95 may affect NSC fate by directly targeting NSCs in the cocultures. To exclude this possibility, we subjected only cultured NSCs to OGD and treated them with ZL006 or Tat-nNOS-N1–133. OGD significantly increased neural differentiation, decreased astrocyte differentiation, and caused reductions in the number of newborn neurons with multineurites and the neurite length of newborn neurons; and ZL006 did not affect the OGD-induced alterations (Fig. 4D–H). Similarly, Tat-nNOS-N1–133 also had no effect on the OGD-induced alterations in the only cultured NSCs (Fig. 4I,J). Thus, NSCs are not direct targets of nNOS–PSD-95 blockers.
HDAC2 is implicated in the role of neuronal nNOS–PSD-95 in regulating the fate of NSCs
To explore how the nNOS–PSD-95 association in neurons affects the fate of NSCs, we investigated the role of HDAC2, which is selectively expressed in the neuronal lineage cells (Juliandi et al., 2010; Ma et al., 2010). To regulate HDAC2 specifically, we generated a lentiviral vector containing shRNA of HDAC2 (LV-HDAC2-shRNA) and an adenovirus vector selectively expressing HDAC2 (Ad-HDAC2). LV-HDAC2-shRNA-infected NSCs displayed a significantly increased neuronal differentiation, whereas Ad-HDAC2-infected NSCs exhibited a substantially decreased neuronal differentiation (Fig. 5A,B). Moreover, microinjection of Ad-HDAC2 into the DG of mice caused a significant decrease in BrdU+/DCX+ cells (Fig. 5C). Therefore, HDAC2 is a negative controller of neuronal differentiation.
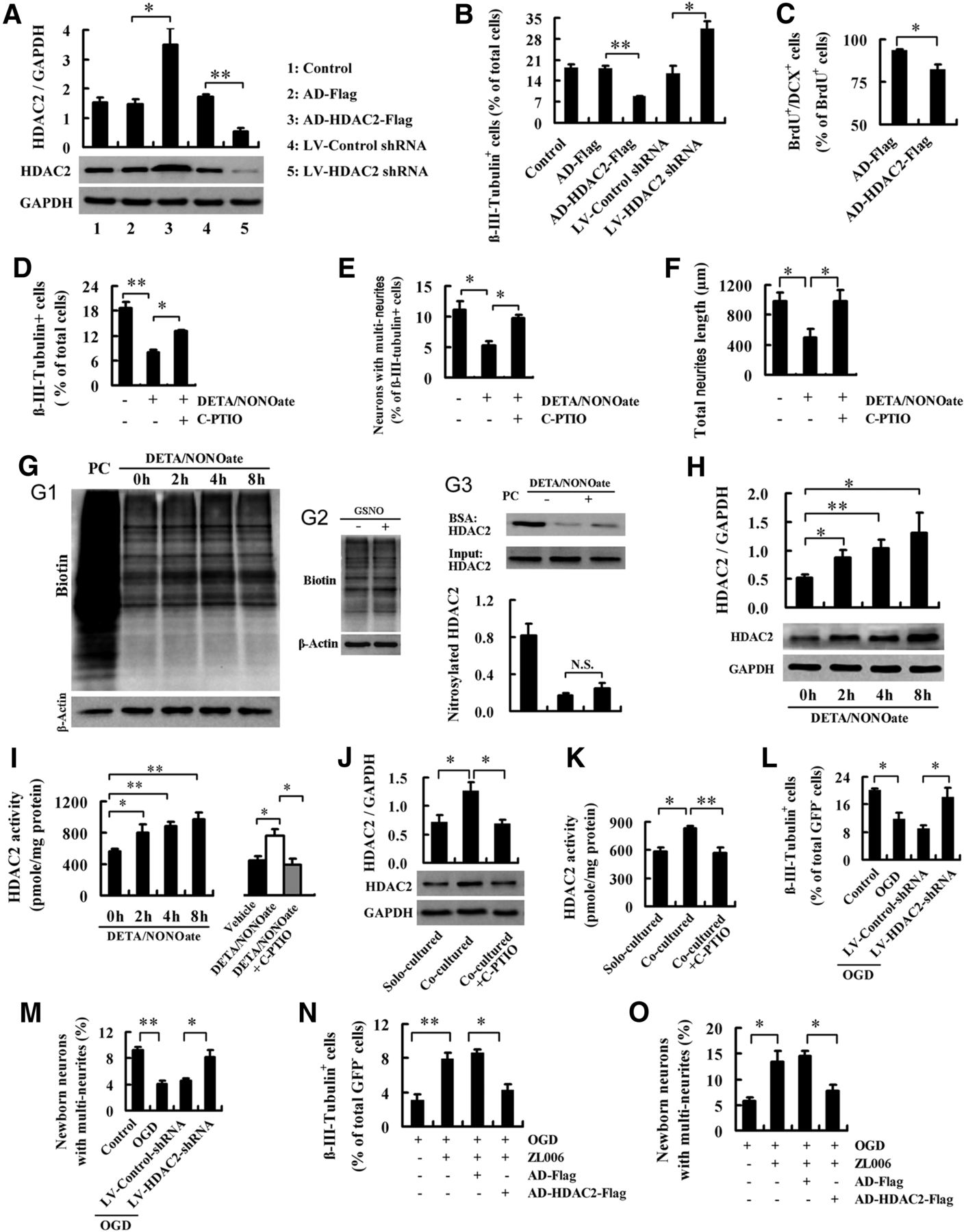
HDAC2 in NSCs is a mediator for the role of neuronal nNOS–PSD-95 association in regulating the fate of NSCs. A, B, Immunoblots showing HDAC2 levels (n = 4; A) and bar graph showing the percentage of newborn neurons (n = 3; B) in the NSCs infected by LV-HDAC2-shRNA, LV-control-shRNA, AD-HDAC2-Flag, or AD-Flag. C, Bar graph showing the percentage of newborn neurons in the DG of rats infected by AD-HDAC2-Flag or AD-Flag (n = 9). These rats were subjected to MCAO at 24 h after virus infection and were killed for BrdU/DCX staining at day 14 after MCAO administration. D–F, Bar graphs showing the percentage of newborn neurons (D), the percentage of neurons with multineurites (E), and total neurite length per newborn neuron with multineurites (F) in the NSCs treated with DETA/NONOate (50 μm) with or without NO scavenger C-PTIO (10 μm) for 24 h at day 2 after differentiation. The cultures were stained 4–5 d after treatment. (n = 3). G, Nitrosylation of total proteins or HDAC2 in NSCs. NSCs were treated with 50 μm DETA/NONOate for the indicated times (G1), with 50 μm GSNO for 4 h (G2), or with 50 μm DETA/NONOate for 4 h (G3) at day 2 after differentiation (n = 4). PC, Positive control, in which the lysate of NSCs was treated with GSNO (200 μm) for 30 min; BSA, biotin-switch assay. H, Immunoblots showing HDAC2 levels in the NSCs treated with 50 μm DETA/NONOate for the indicated times at day 2 after differentiation. (n = 3–5). I, Bar graphs showing HDAC2 activity in the NSCs treated with 50 μm DETA/NONOate for the indicated times (left) or by 50 μm DETA/NONOate with or without 10 μm NO scavenger C-PTIO for 4 h (right) at day 2 after differentiation. n = 3–5. J, K, HDAC2 levels (J; n = 5) and enzymatic activity (K; n = 4) in the solo-cultured or neuron-cocultured NSCs with or without C-PTIO. All cultures were exposed to OGD for 2 h at day 2 after NSC differentiation, and the NSCs were collected 6 h after OGD finish. To measure HDAC2 in NSCs selectively, coculture was performed with neurons in membrane inserts (1 μm pore size) and NSCs in wells. C-PTIO (10 μm) or vehicle was added to the medium of NSCs in wells at the start of OGD exposure. L, M, The percentage of newborn neurons (L) and the percentage of newborn neurons with multineurites (M) in the cocultures of LV-control-shRNA- or LV-HDAC2-shRNA-infected NSCs with GFP+ neurons. The cocultures were subjected to OGD for 2 h at day 2 after differentiation and stained at days 4∼5 after OGD (n = 4). N, O, Bar graphs showing the percentage of newborn neurons (N) and the percentage of newborn neurons with multineurites (O) in the cocultures of AD-HDAC2-flag- or AD-flag-infected NSCs with GFP+ neurons. The cocultures were subjected to OGD for 2 h and treated with 10 μm ZL006 for 26 h beginning at OGD administration, and were stained at days 4∼5 after OGD administration. n = 5. Data are the mean ± SEM. *p < 0.05, **p < 0.01, two-tailed t test for C, ANOVA for others.
The nNOS–PSD-95 interaction in response to ischemia leads to nNOS activation and NO production (Zhou et al., 2010). NO negatively regulated the fate of NSCs in the OGD-treated cocultures of NSCs with neurons (Fig. 5D–F). We thus investigated whether NO production in neurons in response to the nNOS–PSD-95 association regulates HDAC2, thereby affecting the fate of NSCs. We treated NSCs at 24 h after differentiation with NO donor DETA/NONOate or GSNO to mimic neuron-derived NO and found that, although they did not change the nitrosylation of HDAC2 (Fig. 5G), DETA/NONOate dramatically increased HDAC2 expression and enzymatic activity in NSCs (Fig. 5H,I). Meanwhile, the effect of DETA/NONOate on HDAC2 in NSCs was abolished by NO scavenger C-PTIO (Fig. 5I), suggesting an upregulation of HDAC2 in NSCs by NO. To determine whether OGD affects HDAC2 in NSCs in the cocultures, we exposed the NSCs cocultured with neurons to OGD and examined the expression and activity of HDAC2 in the NSCs. As expected, OGD significantly increased HDAC2 expression (1.78 ± 0.18 vs 0.95 ± 0.13; p = 0.0195; n = 3) and enzymatic activity in NSCs (974.1 ± 58.0 vs 741.3 ± 44.5 pmol/mg protein; p = 0.0190; n = 4) compared with no OGD control. To further test whether neuron-derived NO is responsible for the OGD-induced HDAC2 upregulation in NSCs, we exposed the NSCs cocultured with neurons or the solo-cultured NSCs to OGD, and examined the expression and activity of HDAC2 in the NSCs. The HDAC2 protein level and enzymatic activity in the NSCs cocultured with neurons were significantly higher than those in the solo-cultured NSCs, suggesting an upregulation of HDAC2 in NSCs by the OGD-injured neurons. More importantly, scavenging extracellular NO in the neuron-cocultured NSCs by C-PTIO rescued the neuron-mediated upregulation of HDAC2 in NSCs under OGD conditions (Fig. 5J,K). Together, the NO from OGD-injured neurons upregulates HDAC2 in NSCs.
To determine whether HDAC2 in NSCs is essential for the regulation of NSC fate by neuron-derived NO under OGD, we exposed the coculture of neurons with the LV-HDAC2-shRNA-infected or LV-control-shRNA-infected NSCs to OGD. Knockdown of HDAC2 in the NSCs rescued the OGD-induced decreases in β-III-tubulin+ newborn cells and newborn neurons with multineurites (Fig. 5L,M). Subsequently, we cocultured neurons with the Ad-HDAC2-infected NSCs and examined the effect of ZL006. We exposed the cocultures to OGD and treated them with ZL006 to diminish NO production in neurons. OGD exposure caused a significant decrease in β-III-tubulin+ newborn cells and neurite length of newborn neurons with multineurites; ZL006 reversed the OGD-induced neuronal differentiation reduction and neurite growth impairment; and overexpressing HDAC2 abolished these effects of ZL006 (Fig. 5N,O), suggesting that HDAC2 is a mediator for the negative regulation of NSC fate by nNOS–PSD-95 interaction in neurons under ischemic conditions.
Histone acetylation and NeuroD, a neuron-specific transcription factor, is critical for the regulation of neurogenesis (Gao et al., 2009; Jobe et al., 2012). Accordingly, we investigated whether NO-mediated HDAC2 upregulation alters the cellular level of acetylated histones and NeuroD expression. We allowed NSCs to differentiate for 24 h and then treated them with DETA/NONOate for 8 h, and found that it significantly decreased acetylated histone H4 at lysine 5 (acety-H4) and NeuroD levels, and that C-PTIO reversed the effects of DETA/NONOate (Fig. 6A,B). Moreover, HDAC inhibitor TSA and the knockdown of HDAC2 by LV-HDAC2-shRNA abolished the effect of DETA/NONOate on the acetylation of histone H4 and NeuroD expression (Fig. 6C–F). To further confirm the findings from NO donor-treated NSCs, we examined whether OGD exposure affects the acetylation of histone H4 in the NSCs cocultured with neurons. We cocultured the LV-HDAC2-shRNA- or LV-control-shRNA-infected NSCs with neurons, allowed them to differentiate for 24 h and to undergo 2 h of OGD exposure, and examined the levels of histone H4 and acety-H4 in the NSCs at 6 h after the end of OGD exposure. As expected, in the cocultures OGD caused a significant decrease in the acetylation of histone H4 in NSCs, and the knockdown of HDAC2 in the NSCs reversed the OGD-induced decrease in histone acety-H4 (Fig. 6G,H). Collectively, these data suggest that ischemia-induced nNOS–PSD-95 interaction in neurons and consequent NO production may negatively regulate the fate of NSCs via HDAC2-mediated histone deacetylation and NeuroD downregulation.

Association of nNOS–PSD-95 in neurons and consequent NO production may negatively regulate NSC fate via HDAC2-mediated histone deacetylation and NeuroD downregulation. A, B, Immunoblots showing acetyl-H4 (A) and NeuroD (B) levels in the NSCs treated by 50 μm DETA/NONOate with or without 10 μm C-PTIO for 8 h at day 2 after differentiation (n = 4). C, D, Immunoblots showing acetyl-H4 (C) and NeuroD (D) levels in the NSCs treated by 50 μm DETA/NONOate with or without 2 nm TSA (an HDAC inhibitor that was added 30 min before DETA/NONOate) for 8 h at day 2 after differentiation (n = 4). E, F, Immunoblots showing acetyl-H4 (E) and NeuroD (F) levels in the NSCs treated with 50 μm DETA/NONOate with or without LV-HDAC2-shRNA infection for 8 h at day 2 after differentiation (n = 4). DETA/NONOate was added at day 6 after LV-HDAC2-shRNA or LV-control-shRNA infection. G, H, Immunoblots (G) and immunofluorescence (H) showing acetylation of histone H4 in the NSCs cocultured with neurons after OGD. Coculture was performed with neurons in membrane inserts (1 μm pore size) and NSCs in wells (n = 4). Data are the mean ± SEM. *p < 0.05, **p < 0.01, ANOVA.
Blocking nNOS–PSD-95 binding during recovery stage improves stroke outcome
Ischemia-induced nNOS–PSD-95 association lasted for at least 7 d after stroke (Fig. 7A). A delayed treatment with nNOS–PSD-95 blocker during the recovery stage may improve stroke outcome via enhancing regenerative repair without its protection against acute damage. To address this notion, we subjected rats to MCAO for 2 h and treated them with ZL006 (1.5 mg/kg/d, i.v.) for 7 d, starting at day 4 after MCAO. ZL006-treated rats displayed an infarct volume similar to that of vehicle-treated rats (34.5 ± 6.0% vs 35.8 ± 7.1%), suggesting no acute neuroprotection. Under delayed treatment, rats were given BrdU (50 mg/kg, i.p., 4× at 12 h intervals) to label proliferating cells during days 4–5 after MCAO administration. Sensorimotor functions were assessed at days 11, 18, 32, and 46 after MCAO by the grid-walking task and the cylinder task. Spatial cognitive performance in Morris water maze was measured for 5 consecutive days from day 32 to day 36 after MCAO administration. Rats were then killed to measure BrdU+/NeuN+ cells and dendrite spine density (Fig. 7B). Ischemia significantly increased escape latency and decreased the time spent in the target quadrant and target crossings in the Morris water maze (Fig. 7C,D), and increased the number of foot faults on the grid-walking task (Fig. 7E) and forelimb asymmetry scores on the cylinder task (Fig. 7F), suggesting impaired spatial memory and sensorimotor functions. Treatment with ZL006 to block the nNOS–PSD-95 association significantly ameliorated the ischemia-induced impairments of spatial memory and sensorimotor functions (Fig. 7C–F). Ischemia also caused substantial decreases in the number of BrdU+/NeuN+ cells in the ipsilateral hippocampus, and in the dendrite spine density of neurons in the striatum and the motor cortex. ZL006 substantially increased neurogenesis in the DG, and ameliorated the ischemia-induced the loss of dendrite spine in the striatum and the motor cortex (Fig. 7G,H). ZL006 has no effect on cerebral blood flow, platelet aggregation, bleeding time, body weight, arterial blood pressure, partial pressure of arterial oxygen, partial pressure of arterial carbon dioxide, and pH (Zhou et al., 2010).

Dissociating nNOS from PSD-95 improves stroke outcome. A, Co-IP experiment showing the amount of nNOS–PSD-95 complex in the penumbra at indicated times after MCAO. B, Schematic representation of experimental design for C–J. C, D, Escape latency measured during days 32–35 (C), time spent in target quadrant (D, left) and target crossings (D, right) measured at day 36 after MCAO in Morris water maze task (n = 10 for sham; n = 10 for vehicle; n = 11 for ZL006). E, F, Analysis of forelimb foot faults on grid-walking task (E) and assessment of forelimb asymmetry using the cylinder task (F) at indicated times after MCAO (n = 10 for sham; n = 10 for vehicle; n = 11 for ZL006). G, H, Images (G) and bar graph (H) showing BrdU+/NeuN+ cells in the ipsilateral DG at day 46 after MCAO administration (41 d after the last BrdU injection; n = 6). Scale bar, 50 μm. I, J, Images (I) and bar graph (J) showing dendrite spine density in the striatum and motor cortex in the rats subjected to MCAO and treated with ZL006 (n = 3). Eight neurons were measured in each biological sample. Data are the mean ± SEM. C, D, H, and J: *p < 0.05, **p < 0.01, ***p < 0.001, between groups as indicated; E and F: ***p < 0.001, compared with sham controls; #p < 0.05, ##p < 0.01, ###p < 0.001, compared with vehicle, ANOVA.
To further ascertain whether nNOS–PSD-95 coupling is implicated in ischemia-induced structural alterations of neurons, we exposed the cultured neurons to OGD for 2 h and treated them with ZL006. OGD exposure significantly decreased the total process length and dendrite spine density of survival neurons, and ZL006 ameliorated the destructive effects of OGD on process and dendrite spine formation (Fig. 8A,B). Moreover, we delivered LV-nNOS-N1-133-GFP into the striatum of rats to disrupt nNOS–PSD-95 coupling in local tissue and subjected these rats to MCAO, and found that LV-nNOS-N1-133-GFP abolished the ischemia-induced spine density decrease (Fig. 8C). Together, the nNOS–PSD-95 interaction during the recovery stage may affect stroke outcome via impairing regenerative repair, including neurogenesis and neuroregeneration.

Interaction of nNOS with PSD-95 causes reductions in dendritic growth and spine density in neurons following hypoxia or ischemia. A, Images showing representative neurons (left) and bar graph showing total process length per neuron (right). Scale bar, 50 μm. n = 5. Six neurons were measured in each biological sample. B, Images (left) and bar graph (right) showing dendrite spine density of representative neurons. Scale bar, 10 μm. n = 5. Six neurons were measured in each biological sample. A, B, Cultured neurons were subjected to OGD for 2 h and treated with 10 μm ZL006 for 26 h beginning at OGD, and imaging was performed at days 4∼5 after OGD. C, Images (left) and bar graph (right) showing dendrite spine density of neurons in the ischemic striatum of rats infected by LV-nNOS-N1–133-GFP or LV-GFP and subjected to MCAO. LV-nNOS-N1–133-GFP or LV-GFP was microinjected into the right striatum of rats, and 7 d later these rats were subjected to MCAO for 2 h. Dendrite spine density of neurons was measured at day 10 after MCAO. Scale bar, 10 μm. n = 6. Five neurons were measured in each biological sample. Data are the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001, ANOVA.
Discussion
Neurogenesis and neuroregeneration contribute to brain self-repair, thereby improving stroke outcome (Dijkhuizen et al., 2001; Arvidsson et al., 2002; Nakatomi et al., 2002; Carmichael, 2008; Clelland et al., 2009; Sun et al., 2009; Mostany et al., 2010; Marín-Burgin et al., 2012; Ueno et al., 2012). The lack of effective therapies heightens the need for new therapeutic targets, especially to promote regenerative repair. Recently, we demonstrated that the nNOS–PSD-95 association causes neuronal injury and can serve as a potential target for the acute treatment of stroke (Zhou et al., 2010). Here, we show that the nNOS–PSD-95 interaction negatively controls NSC fate and impairs regenerative repair after stroke. Disrupting the nNOS–PSD-95 interaction benefits both neurogenesis and neuroregeneration, and thereby improves stroke outcome.
The activation of NMDARs is crucial for neurogenesis, dendritic plasticity, and synaptogenesis (Arvidsson et al., 2001; Deisseroth et al., 2004; Tashiro et al., 2006; Platel et al., 2010; Chen et al., 2011; Gambrill and Barria, 2011; Kheirbek et al., 2012; Takahashi et al., 2012). NMDAR-mediated Ca2+ influx is responsible for these contributions of NMDAR, because Ca2+ influx plays a crucial role in neurogenesis and neuroregeneration (Deisseroth et al., 2004; Matsuzaki et al., 2004; Spitzer, 2008). Although NMDAR activation triggers the interaction of nNOS with PSD-95, disrupting the nNOS–PSD-95 interaction may have different effects on neurogenesis and neuroregeneration from directly blocking NMDAR, since it does not affect NMDAR-mediated EPSCs (Zhou et al., 2010). Indeed, the current study showed that dissociating nNOS and PSD-95 enhances the survival and neuronal differentiation of NSCs, promotes the migration of newborn cells into the injured area, and facilitates neurite growth in newborn neurons and dendritic spine formation in mature neurons in the ischemic brain. Thus, the interruption of NMDAR-mediated bypassing without affecting NMDAR-mediated Ca2+ influx may be a desirable target for regenerative repair after stroke.
Functional NMDA receptors require the assembly of two GluN1 subunits together with either two GluN2 subunits, of which there are four subtypes, GluN2A-D, with GluN2A and GluN2B predominant in the forebrain (Traynelis et al., 2010). GluN2 subunits have large, evolutionarily divergent cytoplasmic C-terminal domains (CTDs). Among them, the CTD of GluN2B (CTD2B) exhibits stronger physical/functional coupling to the PSD-95-nNOS signaling cassette, which suppresses prosurvival CREB-mediated gene expression, rendering neurons vulnerable to excitotoxic cell death (Martel et al., 2012). Tat-NR2B9c, a PSD-95 inhibitor perturbing the interaction of PSD-95 with CTD2B, improves functional outcome after stroke in gyrencephalic nonhuman primates (Cook et al., 2012). Thus, directly interfering with CTD2B; dissociating CTD2B from PSD-95, such as with Tat-NR2B9c; or disrupting the interaction of PSD-95 with nNOS may have similar effects on neurogenesis and neuroregeneration.
A main question arising from this study is how the nNOS–PSD-95 interaction affects neurogenesis. Recently, we demonstrated that NSC-derived nNOS is essential for neurogenesis, whereas neuron-derived nNOS, a paracrine NO for NSCs, exerts a negative control on neurogenesis (Luo et al., 2010). In the present study, we found that the nNOS–PSD-95 interaction occurred in neurons only. OGD, a cell model used to mimic ischemia in vitro, augmented the NOS–PSD-95 interaction in neurons and led to significantly increased NO production, thereby inhibiting neuronal differentiation of NSCs, and dendritic growth and branching of newborn and mature neurons. In the cocultures of NSCs with neurons, selectively disrupting nNOS–PSD-95 coupling in neurons reversed these deleterious effects of OGD. Thus, the interaction of nNOS with PSD-95 in neurons in response to ischemia leads to NO overproduction, and in turn, NO negatively controls the fate of NSCs through intercellular communication.
HDACs have been implicated in the epigenetic regulation of neurogenesis (Jobe et al., 2012; Ma et al., 2010). Among HDACs, HDAC2 is selectively expressed in the neuronal lineage cells (Juliandi et al., 2010), and functions in modulating the dendritic growth and branching of neurons and long-lasting changes of neural circuits (Nott et al., 2008; Guan et al., 2009; Gräff et al., 2012). Here we showed that HDAC2 exerts a negative control on the neuronal differentiation of NSCs. We used a NO donor to mimic neuron-derived NO, and found that it caused histone deacetylation and NeuroD expression in NSCs via upregulation of HDAC2. Endogenous NO mediates nitrosylation of HDAC2 in neurons, and this nitrosylation promotes dendritic growth and the branching of neurons (Nott et al., 2008), which raise a possibility that a NO donor may regulate the fate of NSCs via the nitrosylation of HDAC2. In NSCs, however, nitrosylation of HDAC2 by the NO donor was slight, and NO-mediated histone deacetylation and NeuroD expression were predominantly due to HDAC2 expression upregulation and activity enhancement. Further investigation will be required to test whether other HDACs implicated in neural stem cells, in addition to HDAC2, contribute to the NO-mediated regulation of the fate of NSCs.
Footnotes
This work was supported by the National Natural Science Foundation of China (Major Research Plan 91232304, Excellent Young Scientist Fund 81222016, Key Program 81030023, General Program 21172108), the National Basic Research Program of China (973 Program; Grant 2011CB504404), the Natural Science Foundation of Jiangsu Province (Special Program Grant BK2011029 and Distinguished Young Scientists Fund BK20130040), and the Collaborative Innovation Center For Cardiovascular Disease Translational Medicine.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Dong-Ya Zhu, School of Pharmacy, Nanjing Medical University, Nanjing 210029, People's Republic of China. dyzhu{at}njmu.edu.cn





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}