

Abstract
The RE1 Silencing Transcription Factor (REST) acts as a governor of the mature neuronal phenotype by repressing a large consortium of neuronal genes in non-neuronal cells. In the developing nervous system, REST is present in progenitors and downregulated at terminal differentiation to promote acquisition of mature neuronal phenotypes. Paradoxically, REST is still detected in some regions of the adult nervous system, but how REST levels are regulated, and whether REST can still repress neuronal genes, is not known. Here, we report that homeostatic levels of REST are maintained in mature peripheral neurons by a constitutive post-transcriptional mechanism. Specifically, using a three-hybrid genetic screen, we identify the RNA binding protein, ZFP36L2, associated previously only with female fertility and hematopoiesis, and show that it regulates REST mRNA stability. Dorsal root ganglia in Zfp36l2 knock-out mice, or wild-type ganglia expressing ZFP36L2 shRNA, show higher steady-state levels of Rest mRNA and protein, and extend thin and disintegrating axons. This phenotype is due, at least in part, to abnormally elevated REST levels in the ganglia because the axonal phenotype is attenuated by acute knockdown of REST in Zfp36l2 KO DRG explants. The higher REST levels result in lower levels of target genes, indicating that REST can still fine-tune gene expression through repression. Thus, REST levels are titrated in mature peripheral neurons, in part through a ZFP36L2-mediated post-transcriptional mechanism, with consequences for axonal integrity.
- axonal integrity
- peripheral nervous system
- post-transcriptional regulation
- REST
- RNA binding protein
- ZFP36L2
Introduction
The specialized gene expression pattern in neurons is established, at least in part, by a global developmental mechanism that represses these genes in non-neuronal cells. The core of this mechanism is the RE1 Silencing Transcription Factor (REST) (Chong et al., 1995; Schoenherr and Anderson, 1995), which functions as a repressor and recruits a corepressor complex, featuring histone deacetylases, to neuronal genes in non-neuronal cells (Tapia-Ramírez et al., 1997; Huang et al., 1999; Naruse et al., 1999; Grimes et al., 2000; Roopra et al., 2000; Ballas et al., 2001). Among the genes repressed by REST are those encoding ion channels, axonal pathfinding proteins, neurosecretory proteins, and neuronal growth factors and their receptors (Schoenherr et al., 1996; Otto et al., 2007; Johnson et al., 2008; Buckley et al., 2009). Predictably, REST is downregulated during neurogenesis, at both the mRNA and protein levels, to allow these genes to be expressed (Ballas et al., 2005; Guardavaccaro et al., 2008; Westbrook et al., 2008; Gao et al., 2011). Forced expression of REST during neurogenesis results in defects in cortical migration and axonal pathfinding (Paquette et al., 2000; Mandel et al., 2011). Consistent with these observations, Rest promoter activity is itself repressed in some postmitotic cortical neurons (Ballas et al., 2005). In other contexts, however, Rest promoter activity is reported as similar between neuronal and non-neuronal cell types (Kojima et al., 2001). Thus, whether post-transcriptional regulation of Rest occurs and whether it is important for mature neuronal functions are open questions.
Mechanisms of post-transcriptional regulation include mRNA splicing, mRNA transport between cellular compartments, and mRNA stabilization. RNA binding proteins (RBPs) are essential components of this regulation, and their activities affect fundamental neuronal properties, such as synaptic plasticity (Goldie and Cairns, 2012). Their importance is further underscored by the findings that mutations in several RBPs underlie neurological diseases (Kapeli and Yeo, 2012). Most studies have focused on the role of RBPs in the CNS, but the Hu family of RBPs has also been implicated in peripheral nerve regeneration (Perrone-Bizzozero and Bird, 2013). Despite the fundamental importance of RNA processing in the nervous system, comprehensive mRNA targets of specific RBPs have not yet been identified, although techniques developed recently for analyzing RBP coimmunoprecipitated RNAs have greatly advanced the field (Darnell, 2010).
Here we identify, using a yeast three-hybrid screen (Putz et al., 1996), an RNA binding factor, ZFP36L2, which recognizes an AU-rich element (ARE) in the 3′ untranslated region (UTR) of Rest mRNA and destabilizes Rest mRNA constitutively in peripheral neurons. We determine that this pathway is important physiologically by examining axonal morphology in peripheral neurons from mice lacking ZFP36L2 (Stumpo et al., 2009) and by testing for rescue of aberrant pathology by reduction of REST levels.
Materials and Methods
Plasmids, libraries: Myc-tagged REST constructs.
Mouse REST cDNA lacking the 5′ and 3′ UTRs (CDS), or containing 3.4 kb of 3′ UTR and/or 258 bp of 5′ UTR, were cloned into pcDNA3.1(−) (Invitrogen). Luciferase reporter constructs. Mouse Rest 3′ UTR (3.4 kb) was cloned downstream from luciferase in plasmid pGL3b (Promega). Cytosine mutations were introduced into the Rest 3′ UTR by overlap extension PCR in the core (UAUUUAU) sequence at position 1915 and 1919 (Rest 3′ UTRCC). Three-hybrid screening constructs. pRevRX, containing the viral protein RevM10 fused to GAL4DBD, and RRE (RNA Recognition Element for RevM10) was a generous gift from Dr. Kuhl (University of Hamburg, Hamburg, Germany). The Rest 3′ UTR was cloned into pRevRX using MluI-NotI restriction sites. To make the mammalian expression vector CMV.EGFP.ZFP36L2, the mouse ZFP36L2 protein coding sequence from the second codon (serine) to the stop codon was PCR amplified from a cDNA library prepared by reverse transcription of total RNA from cultured mouse fibroblasts (line 67+/+) (Lai et al., 2006). The primers used were based on GenBank accession number NM_001001806.2, and were as follows: forward, 5′-ggtacc TCG ACC ACA CTT CTG TCA CCC TTC-3′; reverse, 5′-tctaga TCA GTC GTC GGA GAT GGA GAG GCG-3′, where the uppercase letters correspond to Zfp36l2 sequence and the lowercase letters are the sequences for restriction enzymes Asp718 (F primer) and XbaI (R primer) recognition sites. The start codon ATG was changed to ACC in the 5′ primer to create an Asp718 site, and the 3′ primer contained an XbaI site after the Zfp36l2 stop codon (sequence italicized). This PCR product was digested with Asp718 and XbaI and ligated into the enzyme-digested vector CMV/EGFP/BGH3′/BS+ (described below) so that the ZFP36l2 sequence was C-terminal to the EGFP sequence and in frame. The vector CMV/EGFP/BGH3′/BS+ was made as follows. A DNA fragment encoding the EGFP protein (without the stop codon) was PCR amplified from pEGFP-N1 (Clontech), with a 5′ primer containing a HindIII restriction site and a 3′ primer containing an Asp718 site located immediately after the last amino acid of EGFP. This fragment was digested with HindIII and Asp718 and ligated into the same enzyme-digested vector CMV.BGH3′/BS+ (Lai et al., 1999). In plasmid C176S.CMV.EGFP.ZFP36L2, a zinc-coordinating cysteine in the first finger of the ZFP36L2 TZF domain (residue 176 in GenBank accession number NP_001001806.1) was mutated to a serine to create a non-RNA binding mutant, using the primer-overlapping PCR mutagenesis technique (Lai et al., 1998). The primer representing the upper strand sequence used in the mutation of cysteine 176 to a serine (C176S) was 5′-G TAC GGC GAG AAG TCC CAG TTC GCG CAC-3′, in which the bases encoding serine are italicized.
shRNAs, lentiviral constructs, and virus preparation.
shZFP36L2-1 targeting Zfp36l2 coding sequence CDS (5′-GCAAGTACGGCGAGAAGTG-3′) was designed after validation of siGENOME SMARTpool, Rat LOC298765 (Dharmacon) and cloned into the GFP expressing lentiviral plasmid FUGW downstream of a U6 promoter. shREST-1(5′-GTTAGCACTTTACAGTTTC-3′) was cloned into the lentiviral plasmid FURW downstream of the U6 promoter, containing mCherry. shRNAs sets against mouse REST and ZFP36L2 in pLKO.1 vectors (shREST-2 and shZFP36L2-2, respectively) were obtained from GE Healthcare (RMM4534-EG19712 and RMM3981-EG12193, correspondingly). The most effective shRNA against REST, clone TRCN0000071343, targeting sequence 5′-TTACACTTCTTAGAAGCCGCG-3′, was selected by Western blot analysis of REST in infected NIH3T3 cells. We used qRT-PCR to measure Zfp36l2 transcript levels in lentivirus-transduced murine erythroleukemia cells. The lentivirus carrying shRNA against ZFP36L2, clone TRCN0000175839, with targeting sequence 5′-TTTCATATTGAGTTGTGGTGC-3′ showed the highest knockdown of ZFP36L2. The effectiveness of viral transduction was monitored by qPCR against lentiviral DNA relative to mouse genomic DNA obtained from the infected DRG, using primers in viral 3′-LTR LV forward 5′-TAAAGCTTGCCTTGAGTGCT-3′, LV-R 5′-GTCTGAGGGATCTCTAGTTACCAG-3′, as described previously (Zhang et al., 2012). A scrambled shRNA in pLKO.1 vector (shControl-2) was obtained from Addgene (plasmid #1864) with targeting sequence of 5′-CCTAAGGTTAAGTCGCCCTCGCTC-3′. Viral titers were estimated using qRT-PCR Lentivirus titration kit (ABM) according to manufacturer's instructions and equal titers were used to infect DRG.
Cell culture.
Rat2 fibroblasts were grown in DMEM supplemented with 10% bovine calf serum. PC12 cells (rat adrenal medulla pheochromocytoma cells) were provided by Simon Halegoua (Stony Brook University, Stony Brook, New York) and maintained in DMEM supplemented with 10% horse serum and 5% FBS. Superior cervical ganglia (SCG) from newborn rats or mice (P0-P1) of either sex were dissociated and plated in C2 medium (DMEM/F12 1:1, BSA 0.5 mg/ml, l-glutamate 1.4 mm, selenium 30 nm, transferrin 10 μg/ml, insulin 10 μg/ml) supplemented with 100 U/ml penicillin G, 100 μg streptomycin sulfate, 50 ng/ml nerve growth factor (NGF) (Enzo Life Sciences), and 3% FBS. The neurons were plated on 24-well plates precoated with 100 μg/ml poly-l-lysine (Sigma) and 10 μg/ml collagen Type IV (BD Bioscience). Non-neuronal cells were eliminated by treating the cultures with the anti-mitotic agent, cytosine arabinoside (1 μm; Sigma) for 2 d. Neurons were grown under sterile conditions in a humidified 5% CO2 incubator at 37°C. DRG were dissected from P7 mice and plated onto cover glasses precoated with 100 μg/ml poly-l-lysine (Sigma) and 5 μg/ml laminin (Calbiochem). The organotypic cultures were maintained in Neurobasal A media (Invitrogen) containing B27, 50 ng/ml NGF, 1 μm 5 fluoro-2′-deoxyuridine (Sigma).
Transfection of cultured cells and shRNA lentiviral transduction of DRG.
Rat2 and PC12 cell lines were transfected at 70% confluency in 100 mm dishes, using the Lipofectamine 2000 transfecting reagent (Invitrogen). In all transfections, the amount of plasmid was adjusted on a molar basis, and the total quantity of DNA in the transfection mixture was kept constant by the addition of pBluescript (Agilent). Cells were treated with 50 ng/ml NGF (Enzo Life Sciences) for an additional 72 h where indicated. Infection of organotypic cultures was performed on DIV3 by reducing volume to 250 μl, adding 5–10 μl virus (106 viral particles/μl) for 1 h at 37°C then adding 250 μl of complete media. Cells were fed every 3 d by 50% media replacement and collected for images at DIV7.
Western blotting.
A total of 30 μg of total cell extracts were separated on 5–8% SDS-PAGE gel and transferred to nitrocellulose membranes. The membranes were blocked in 10% nonfat dry milk, 20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.1% Tween 20, followed by incubation with primary antibodies for 2 h at room temperature and then secondary antibody, conjugated with HRP, for 1 h. Proteins were detected using chemiluminescent Super Signal West Pico kit (Thermo Scientific). Antibodies used were as follows: anti-α-tubulin, 1:3000 (AA4.3, Developmental Studies Hybridoma Bank, University of Iowa, Department of Biology, Iowa City, IA), anti-actin, 1:2000 (JLA20, Developmental Studies Hybridoma Bank), anti-hZFP36L2 (1:2000, Cell Signaling Technology, 2119), anti-Myc (1:1000, Millipore, 2276), anti-REST-C (1:1000) (Ballas et al., 2005), anti-neurofilament-H (NF-H, 1:5000, Millipore, 1989), and anti-neurofilament-M (1:10000, Millipore, 1987).
Chromatin immunoprecipitation.
Chromatin immunoprecipitation was carried as described previously (Ballas et al., 2001). Briefly, crosslinked chromatin was sonicated to generate fragments with an average length of 300–400 bp. The chromatin was subjected to immunoprecipitation using 2 μg of anti-REST-C antibodies or anti-rabbit IgG. For quantification, the amount of immunoprecipitated DNA in a sample was expressed as percentage input DNA, using an ABI PRISM 7900HT Real-Time PCR System and SYBR Green PCR mastermix (Applied Biosystems) to perform qRT-PCR. All quantitative ChIP was verified by PCR amplification of the same ChIP DNA and analyzed on agarose gels.
RNA isolation, RT-PCR, and quantitative real-time RT-PCR analysis.
Total RNA was prepared using RNeasy (QIAGEN) or Trizol (Invitrogen) and treated with RNase-free, DNase (Ambion). For reverse transcription, First Strand Superscript III (Invitrogen) was used and quantitative real-time PCR was performed using SYBR Green PCR master mix (Applied Biosystems). Relative abundance of the specific mRNAs purified from cell lines was normalized to 18S mRNA; relative abundance of the specific mRNAs purified from sympathetic primary cultures was normalized to PGP9.5 and or 18S mRNA. Primers used are as follows: rat Rest forward (5′-ACCTCAAGTCAAGCTAACCAGAAGGTCA-3′) and rat Rest reverse (5′-AGGCACTAAGCCAACCTCAATAAGAACC-3′); mouse Rest forward (5′-GGCTGCTCTCAAGGAGTCTG-3′) and mouse Rest reverse (5′-TTCTGCTCAGTGTCCACGTC-3′); Myc-Rest forward (5′-GCAGGAGGAGCGGGAGGA-3′) and Myc-Rest reverse (5′-TCGACGGCGCTATTCAGAT-3′); rat Zfp36l2 forward (5′-CCAACAACGCCTTCACCTTCG-3′) and rat Zfp36l2 reverse (5′-TGCTGCTGGCTGCGGTAGTAGG-3′); mouse Zfp36l2 forward (5′-CTGCATCACAGCCTCAGTTTCT-3′) and mouse Zfp36l2 reverse (5′-CGAGGAGGAACAGGACGAG-3′); rat Nav1.2 forward (5′-TTGTATCTCCAACCACACGACCATAGAA-3′) and rat Nav1.2 reverse (5′-CCACATACTTCTCCACACTGCTGCCTAT-3′); mouse Nav1.2 forward (5′-GGCACAATCAGTGCTGGTACC-3′) and mouse Nav1.2 reverse (5′-CAGCAAGGGATTCCCTGGT-3′); mouse Nav1.8 forward (5′-AGGAAGAGAGCCCCAAAGG-3′) and mouse Nav1.8 reverse (5′-CTGCCTGGTGATGTTCACAC-3′); mouse Snap25 forward (5′-ACGGGAGCAGATGGCCA-3′) and mouse Snap25 reverse (5′-CCCGGGCATCGTTTGTT-3′); Luciferase forward (5′-TGCACATATCGAGGTGGACATCACTTAC-3′) and Luciferase reverse (5′-TTCAGCCCATATCGTTTCATAGCTTCTG-3′); Cherry forward (5′-CAAGACCACCTACAAGGCCAAGAAGC-3′) and Cherry reverse (5′-CGCGTTCGTACTGTTCCACGATG-3′); mouse PGP9.5 forward (5′-CCCGAGATGCTGAACAAAGT-3′) and mouse PGP9.5 reverse (5′-AGTCTCCTCCTCCAGCCCTA-3′); rat 18S forward (5′-TCTGCCCTATCAACTTTCGATGGTAGTC-3′) and rat 18S reverse (5′-CTTCCTTGGATGTGGTAGCCGTTTCT-3′); mouse 18S forward (5′-GTTGGTGGAGCGATTTGTCT-3′) and mouse 18S reverse (5′-GAACGCCACTTGTCCCTCTA-3′).
Native transcript analysis.
For nuclear RNA isolation, cells were detached and washed in 1 ml PBS. The pellet was resuspended in 150 μl buffer RLN (50 mm Tris-HCl, pH 8.0, 140 mm NaCl, 1.5 mm MgCl2, 0.5% Igepal, 1000 U/ml RNase inhibitor (QIAGEN), 1 mm DTT, and incubated for 5 min on ice. Debris and nuclei were pelleted (300 × g, 2 min, 4°C), and the cytoplasmic fraction was discarded; the nuclear pellet was washed in 500 μl PBS, again pelleted for 3 min at 300 × g, and then resuspended in 600 μl RLT buffer (QIAGEN). RNA isolation was then done using an RNeasy Mini kit (QIAGEN) according to the manufacturer's instructions, followed by DNase digestion. Pre-mRNA before splicing was quantified by qRT-PCR using primers positioned within an intron and the adjacent exon: ratRestpre-mRNA forward (5′-TGTGGTTATTTCACCTTGATGACTCCTG-3′) and ratRestpre-mRNA reverse (5′-CGGGTCACTTCGTGCTGATTAGAGG-3′).
Yeast three-hybrid screening.
Yeast three-hybrid screening was performed as described previously (Putz et al., 1996) and using Matchmaker Gal4 Two-Hybrid System (Clontech).
RNA electrophoretic mobility shift assay.
The plasmids carrying EGFP.ZFP36L2 and zinc finger mutant C176S.EGFP.ZFP36L2 fusion constructs were expressed in HEK293 cells by transient transfection using the calcium-phosphate precipitation method, and cytosolic extracts were prepared as described previously (Lai et al., 1999). Gel shift assays were performed as described previously (Kedar et al., 2012), except that the probe was biotinylated. The probe sequence was biotin-5′-UGCAGAAUAUUUAUGUUUUA-3′, from the ZFP36L2 binding site of the Rest 3′ UTR. The assay used 5–10 μg of extract protein, and 25 nm RNA probe.
Generation of a rat anti-mouse REST monoclonal antibody (clone 4A9).
Partial mouse REST CDS (encoding C-terminal amino acids 889–1035) was cloned into pGEX-3X vector (GE Healthcare) and introduced into bacterial host strain BL21(DE3) (Agilent). GST-REST fusion peptide was expressed via IPTG induction followed by glutathione-agarose affinity purification. A total of 100 mg of the purified peptide was used to immunize rats twice, and hybridomas were derived using established procedures (Goding, 1980) (core facility of Oregon Vaccine and Gene Therapy Institute, Oregon Health & Science University). Hybridoma screening was done by ELISA to select candidate cell lines for clonal expansion. Antibody was purified from the supernatant of cloned cell lines using mercaptoethylpyridine chromatography (Pall Biosciences) followed by thiophilic chromatography (Pierce Chemical) using protocols recommended by the manufacturers. The specificity of antibody was tested using Rest−/− (Jørgensen et al., 2009) and wild-type (WT) mouse embryonic stem cells by Western blot analyses and immunofluorescence (see Fig. 4F).
Immunofluorescence.
Primary cultures were fixed with 4% PFA in PBS, pH 7.4, for 10 min at room temperature. The monolayers were permeabilized with 0.1% Triton X-100 in PBS, blocked with 10% donkey serum, and stained with primary antibodies overnight. The primary antibodies used were as follows: rat anti-mouse REST (clone 4A9, 1:300). For detection, species-appropriate secondary antibodies conjugated to AlexaFluor dyes (Invitrogen) were used at a dilution of 1:500 and nuclei were counterstained with DAPI. The images were acquired using a Zeiss Axiocam. For immunofluorescent staining of mouse DRG, the ganglia were dissected from level L1-L4 vertebrae from P7 mice of either sex. The ganglia were fixed with 4% PFA in PBS, pH 7.4, overnight at 4°C, equilibrated in 30% sucrose/PBS, embedded in TFM freezing media (Fisher Scientific), and sectioned at 14 μm thickness. Sections were postfixed in cold acetone and stained with rat anti-mouse REST (clone 4A9; 1:300 dilution), TuJ1 (Covance,1:500), and counterstained with DAPI. For immunostaining of the sciatic nerve, the mice were anesthetized and transcardially perfused with PBS, pH 7.4, followed by ice-cold 4% PFA in PBS. A length of sciatic nerve from the exit from the spinal cord to the knee was removed and postfixed overnight in 4% PFA at 4°C. The nerve was cryo-protected in PBS containing 30% sucrose for 24 h at 4°C, embedded in TFM freezing media (VWR) in liquid nitrogen, and sectioned at 6 μm thickness. Sections were postfixed in cold acetone and stained with anti-NF-H (1:10,000) and anti-myelin basic protein (MBP, 1:500).
Quantification of REST-positive cells in ganglia sections.
Images were acquired at 20× magnification on Zeiss Axiovert confocal microscope using the same exposures, 2 areas 200 × 200 micron close to the edge of the DRG were selected. Channels were converted to 8 bit images using ImageJ software and red channel (REST staining) was adjusted to an15–255 pixel threshold for counting REST positive nuclei. DAPI was counted using 8-bit image.
Quantification of axonal integrity in DRG.
Axons emerging from ganglia were viewed using a Zeiss AxioObserver microscope equipped with a 25× multi-immersion phase objective. Images were taken with an Axiocam MRM in grayscale mode that was controlled by Zen 2011 software. The images were acquired at a resolution of 1024 ×1024 pixels, which corresponded to a total field of 69,696 μ2 with each pixel corresponding to 0.0676 μ2. Images were converted to binary and thresholds were applied for bleb determination on the basis of area using the Particle Analyzer plug-in of ImageJ1.47q. The threshold for bleb determination in ImageJ was set between 20 and 100, corresponding to 1.35 μ2 and 6.76 μ2 for the lower and upper cutoffs, respectively. The total area (axons and blebs) was calculated on basis of 1.35 μ2 to infinity. Data analysis was performed blind to genotype or experimental conditions.
Quantification of nerve fiber diameter in sciatic nerve.
Myelinated nerve fibers were identified by MBP immunostaining on sciatic nerve cross-sections. Cross section diameters were measured directly from slides with two independent measurements per nerve. For calculating the fiber diameters, two areas within the first 4 mm from the spinal cord exit were selected for quantification. The diameters were measured at 40× using ImageJ software. Each bin was 1 μm.
Statistics.
Statistical analyses were performed using Graphpad Prism software, and p values <0.05 were considered statistically significant. Unless otherwise indicated, mean and 95% confidence interval (CI) are shown. Pairwise comparisons within two groups were done using unpaired Student's t tests. Comparisons among more than two groups were analyzed using one-way ANOVA followed, when appropriate (p < 0.05), by Tukey post hoc test. Nonparametric Mann–Whitney t test was used to analyze Figures 4G (right), 5B, C, D, F, and 6D (right). Kruskal–Wallis one-way ANOVA followed by Dunn's multiple-comparison test was used to analyze experiments in Figure 6D (left).
Rodent strains.
All animal studies were approved by the Oregon Health and Science University Institutional Animal Care and Use Committee. Timed pregnant adult Sprague Dawley rats were obtained from Charles River. Zfp36l2 knock-out (KO) mice (Stumpo et al., 2009) were maintained on a C57BL/6NTac background. The Rest KO was a Gene Trap provided from the German Gene Trap Consortium (GenBank Accession #DU821609).
Results
REST expression is regulated tightly in postmitotic peripheral neurons
As a starting point for biochemical studies, we examined the level of Rest transcripts in two cell lines: undifferentiated rat pheochromocytoma PC12 cells, which lack REST and express neuronal target genes typical of neuroendocrine cells; and Rat2 fibroblasts, which express REST and repress neuronal genes. Rest mRNA were approximately fivefold higher in the Rat2 fibroblasts. Previous studies indicate that Rest mRNA and protein levels are downregulated dramatically during neuronal differentiation in the CNS, before terminal neuronal differentiation (Chong et al., 1995). We therefore also examined REST levels in cultured SCG neurons isolated at P0 and cultured for 4 d. Similar to CNS neurons, REST levels were low in the SCG and significantly lower than in the fibroblasts (Fig. 1A, left). To determine whether the low Rest mRNA levels held for peripheral neurons in vivo, we analyzed REST expression in DRG isolated acutely at different postnatal ages (Fig. 1A, right). In contrast, expression of REST target genes encoding neuronal proteins Nav1.6 and SNAP25 increased over the interval of constant REST expression, likely due to robust transcriptional regulation by activators. (Fig. 1B).

Post-transcriptional regulation of Rest mRNA stability. A, Real-time RT-PCR analysis for Rest mRNA from indicated cell types (left) and acutely isolated postnatal DRG (right). For cell types, n = 3 independent experiments. For DRG, n = 4 mice, 8 ganglia/mouse. B, Real-time RT-PCR analysis of REST target genes from DRG in A. C, Real-time RT-PCR analysis of mRNA from dissociated SCG treated with CHX (50 μg/ml) or vehicle alone (DMSO). The results are the mean values from two independent experiments. D, Rest mRNA stability in dissociated SCG. Cells were treated with ActD (5 μg/ml), ActD plus CHX (50 μg/ml), or vehicle alone (DMSO). RNA levels were analyzed by RT-PCR. n = 3 independent experiments. E, Real-time RT-PCR analysis for Rest pre-mRNA in SCG cells. Cells were treated with vehicle (DMSO) or CHX (50 μg/ml) for 5 h before analysis. n = 3 independent experiments. *p < 0.05. **p < 0.01. ***p < 0.001. ns, not significant.
In CNS neurons, REST is regulated, at least in part, at the transcriptional level (Ballas et al., 2005); Rest promoter activity in other cellular contexts is not altered significantly between neuronal and non-neuronal cell types (Kojima et al., 2001). The latter led us to consider the possibility that post-transcriptional mechanisms might play an important role in maintaining the low basal levels of Rest mRNA in peripheral neurons, specifically. Although regulation of mRNA level is often orchestrated by micro-RNAs, they are usually critical during developmental transitions, rather than in mature neurons. Therefore, we tested whether RBPs might underlie regulation of Rest mRNA levels in mature peripheral neurons. To this end, we treated SCG neurons in culture with the protein synthesis inhibitor, cyclohexamide (CHX). Interestingly, Rest mRNA levels increased approximately twofold compared with control as early as after 1 h of CHX treatment, gradually increasing up to ∼10-fold compared with control with 8 h of treatment (Fig. 1C). Similar results were obtained when PC12 cells were treated with CHX (data not shown). These data suggest that a labile protein with short half-life of ∼60 min likely regulates the level of Rest mRNA.
A protein synthesized constitutively confers Rest mRNA instability in peripheral nervous system neurons
The CHX effect could reflect block at the level of transcriptional or post-transcriptional machinery. For example, CHX could either be blocking synthesis of a transcriptional repressor protein, which when removed resulted in increased REST transcription, or of a protein that affected mRNA stability. To distinguish between these two possibilities, we exposed SCG neurons in culture to actinomycin D (ActD), which blocks transcriptional elongation, and asked whether CHX treatment in the absence of transcription could still mediate upregulation of Rest mRNA levels. With ActD alone, Rest mRNA levels were reduced to 40% of initial levels after 1 h (Fig. 1D). In the presence of both ActD and CHX, Rest mRNA levels were still 60% of initial levels after 2 h, suggesting that CHX treatment regulated Rest mRNA stability. We noted that the increase in Rest mRNA levels with CHX treatment at 2 h was significantly higher than the increase after treatment with both CHX and ActD. This difference may reflect simply the difference in conditions, with and without continuous RNA synthesis, or, alternatively, could reflect the contribution of an additional stability mechanism. Consistent with involvement of post-transcriptional mechanisms, two different analyses exclude a significant contribution by transcriptional regulation. First, Rest pre-mRNA levels were not statistically different from controls after CHX treatment in both SCG neurons (Fig. 1E) and PC12 cells (data not shown). Second, chromatin immunoprecipitation analysis of Rest 5′ UTR sequences, using an antibody specific for a marker of transcriptional initiation, RNA polymerase II phosphorylated on serine 5 in the C-terminal domain, indicated no difference with CHX treatment (data not shown).
Sequences in the Rest 3′ untranslated region regulate mRNA stability
To investigate which region of the Rest transcript was the target of the CHX effect, we generated myc-tagged versions of REST cDNA containing the full-length coding region, with and without flanking 5′ and 3′ UTR sequences. The cDNAs were transfected into PC12 cells for analysis of the corresponding mRNAs in the presence or absence of CHX (Fig. 2A). The steady-state levels of myc-tagged Rest transcripts containing only coding sequence (CDS), or coding sequence plus 5′ UTR (5′ UTR-CDS), were significantly higher than constructs expressing CDS with both 5′ and 3′ UTR sequences or just 3′ UTR sequences, consistent with a role for Rest 3′ UTR in transcript stability. Importantly, after CHX treatment, only mRNAs encoded by constructs containing the Rest 3′ UTR were stabilized, supporting the idea that this region is a target for mRNA degradation machinery mediated by an RBP.

Rest mRNA is regulated post-transcriptionally through its 3′ UTR. A, Top, Real-time RT-PCR analysis of mRNA from PC12 cells transfected with Myc-tagged REST cDNAs and treated with CHX (50 μg/ml) or vehicle alone (DMSO). n = 3 independent experiments. Bottom, Schematic showing location of the Myc tag on Rest coding sequence (CDS) and primers (red arrows) used in the analysis. B, Yeast three-hybrid screen. Components are color-coded according to the plasmid of origin. DBD, DNA binding domain of GAL4 activator protein; RevM10, RBP; RRE, HIV-1 RNA recognition element; Rest, 3′ UTR 3.4 kb untranslated region; AD, GAL4 activation domain; GAL4 binding site, upstream activator sequence driving reporter gene activation. C, Representative colony isolate from three-hybrid screen showing that antibiotic resistance and reporter gene expression (blue color) depended on both Rest 3′ UTR and HIV-1 RRE. D, Top, WT and mutant ARE sites in Rest 3′ UTR. Middle, Schematic representation of (Luc)-Rest 3′ UTR constructs. Bottom, RT-PCR analysis of PC12 cells transfected transiently with firefly luciferase (Luc)-Rest 3′ UTR plasmids treated with CHX or vehicle (DMSO). n = 3 independent experiments. ARE are boxed and show A to C mutations. E, Gel mobility shift (left) and Western blot analysis (right) showing binding of ZFP36L2 protein to a biotinylated probe for the predicted ZFP36L2 binding site on Rest 3′ UTR and equivalent expression of Zfp36l2 cDNAs transfected into HEK293 cells. Left, The biotinylated oligonucleotides were incubated alone (lane 1), with cell extracts of HEK293 transfected with either empty vector (lane 2), EGFP expressing vector (lane 3), EGFP- ZPF36L2 fusion vector (lane 4), or a ZPF36L2 construct encoding a mutation in its zinc-finger, EGFP-C176S ZPF36L2 (lane 5) that precludes binding to RNA. Arrow indicates a mobility-retarded complex. Right, Transfection with EGFP control (lane 1), WT EGFP-ZFP36L2 (lane 2), and mutant EGFP-ZFP36L2-C176S (lane 3). F, Western blot analysis of PC12 cells transfected with ZFP36L2-MYC or GFP and treated with 50 μg CHX for indicated times. α-Tubulin (αTu), loading control. HEK293 cell lane shows migration position of endogenous human ZFP36L2 recognized by antibody to human ZFP36L2. Bottom, Quantification of three independent experiments. *p < 0.05. **p < 0.01. ***p < 0.001. ns, not significant.
Identification of proteins that bind to the Rest 3′ UTR
We used a yeast three-hybrid screen (Putz et al., 1996) (Fig. 2B) to identify the RBP in an unbiased manner. This strategy requires three components. One component is a plasmid encoding Rest 3′ UTR sequences fused to the binding site for an HIV RBP (RevM10), as well as a fusion protein between an HIV RBP and the yeast GAL4 activator (GAL4) DNA binding domain. The second component is a plasmid bearing the GAL4 binding site (upstream activator sequence) driving reporter gene expression. Reconstitution of an active GAL4 protein and reporter gene expression requires an intermediary REST RNA binding that is provided by the third component, a library of cDNAs, constructed from an NGF-treated PC12 library, fused to cDNA coding for the GAL4 activation domain (Fig. 2B). Using this system, we obtained ∼1 × 104 clones that conferred growth of 121 yeast colonies under His+/Ade+ selection. After α-galactosidase screening, 57 colonies remained, and these were tested further in yeast by cotransformation with plasmids lacking either the Rest 3′ UTR or RevM10 RNA bridge, resulting in a total of four clones (Fig. 2C). Among them, we identified two proteins that had been reported previously as RNA binding factors: ZFP36L2 and CPEB2. We focused on ZFP36L2 because CPEB2 normally activates translation (Hägele et al., 2009). ZFP36L2 is a member of the TIS11 zinc finger protein family that binds to AREs in the 3′ UTRs of target mRNAs and recruits mRNA decay enzymes (Ciais et al., 2013) and therefore was an excellent candidate for regulation of Rest mRNA stability.
The Rest 3′ UTR contains a functional binding site for ZFP36L2
We identified one consensus ARE (UAUUUAU), within the Rest 3′ UTR (Fig. 2D). Previous biochemical studies indicate that the A nucleotides are critical for its function (Hudson et al., 2004; Lai et al., 2006; Horner et al., 2009). To test whether Rest mRNA stability was regulated by ZFP36L2 binding to this site, we cloned both WT Rest 3′ UTR as well as 3′ UTR containing two C nucleotides in place of the A nucleotides, downstream from a luciferase reporter gene, Luc (Fig. 2D). These constructs, or a construct encoding LUC alone, were introduced into PC12 cells in the presence and absence of CHX. Luciferase mRNA expression programmed by the control vector was unaltered by treatment with CHX. Luciferase mRNA expression programmed by the reporter gene containing the WT Rest 3′ UTR was, as predicted, significantly less than control. Importantly, CHX treatment significantly stabilized the luciferase transcript containing the Rest 3′ UTR (Fig. 2D). Mutating the ARE sequence in 3′ UTR of Rest resulted in rescue of Luc mRNA to levels similar to the Luc mRNA control (Fig. 2D).
We also performed a gel shift analysis to confirm that ZFP36L2 binds directly to the Rest 3′ UTR (Fig. 2E, left). Indeed, Rest 3′ UTR probes were shifted prominently when exposed to extracts from HEK cells expressing WT EGFP-ZFP36L2 protein (Fig. 2E, left, lane 4), whereas HEK cell extracts expressing either mutated ZFP36L2-GFP or GFP proteins (lanes 5 and 3, respectively) did not produce a supershift relative to the probe alone (lane 1). Western blot analysis indicated similar levels of ZFP36L2 and mutant ZFP36L2 expressed in the HEK cells (Fig. 2E, right). Finally, we measured ZFP36L2 protein half-life (Fig. 2F) and found that it is ∼60 min, compatible with the half-life (∼60 min) measurements for REST.
Knockdown of ZFP36L2 stabilizes endogenous Rest mRNA and increases levels of functional REST protein
The results from transient reporter analyses predicted that knockdown of ZFP36L2, in PC12 cells, would result in increased stability of endogenous REST. Before testing this prediction, we measured in two ways knockdown efficiency of ZFP36L2 in PC12 cells. First, we measured endogenous Zfp36l2 mRNA levels in undifferentiated PC12 cells transfected with either control or Zfp36l2-specific shRNA. Levels of Zfp36l2 transcript were decreased approximately threefold compared with control (1.05, CI 0.82–1.29 vs 0.37, CI 0.30–0.44; Fig. 3A). Second, we cotransfected PC12 cells with cDNA encoding a ZFP36L2-Myc tagged protein and shRNA plasmids and then monitored knockdown by Western blotting. Transfection with Zfp36l2 shRNA resulted in a twofold decrease in ZFP36L2-Myc protein levels compared with control levels (0.27, CI 0.20–0.33 vs 0.14, CI 0.05–0.23; Fig. 3A).

Knockdown of ZFP36L2 results in aberrantly higher levels of REST protein and repression of endogenous REST target genes. A, Left, Real-time RT-PCR analysis of knockdown of Zfp36l2 mRNA in PC12 cells transiently transfected with indicated constructs. n = 3 independent experiments. Middle, Western blot analysis of ZFP36L2 knockdown in PC12 cells after cotransfection with ZFP36L2-Myc (lanes 2 and 3) and either scrambled Control shRNA (lane 2) or ZFP36L2 shRNA (lane 3). Lane 1 represents position of endogenous human ZFP36L2 in HEK293 cells using antibody to human ZFP36L2. β-Actin, loading control. Right, Quantification of Western blot at left. B, Real-time RT-PCR analysis of Rest mRNA levels in PC12 cells transiently transfected with scrambled Control or ZFP36L2 shRNAs and treated with ActD (5 μg/ml). n = 3 independent experiments. C, Western blot analysis of REST protein (*) in NIH3T3 fibroblasts (lane 1) and PC12 cells transiently transfected with either scrambled Control (lane 2) or ZFP36L2 shRNAs (lane 3). α-Tubulin (αTu), loading control. D, Chromatin immunoprecipitation analysis of REST occupancy (or IgG control) in PC12 cells transiently transfected with indicated shRNAs and then treated with 50 ng/ml NGF (+NGF) for 72 h. qRT-PCR was performed using primers surrounding the REST binding site in the Nav1.2 promoter region or coding sequence (CDS). n = 3 independent experiments. E, Real-time RT-PCR analysis of Nav1.2 mRNA levels in undifferentiated PC12 cells (−NGF) and PC12 cells treated with NGF, as in D, after transient transfection with indicated shRNAs. n = 3 independent experiments. *p < 0.05. **p < 0.01. ***p < 0.001.
We next transfected PC12 cells with control or Zfp36l2 shRNA, followed by treatment with ActD. Rest mRNA levels at time 0 increased approximately twofold in cells transfected with Zfp36l2 shRNA compared with control, consistent with stabilizing activity of ZFP36L2 (Fig. 3B). At 2 h after ActD treatment, Rest mRNA levels in shZFP36L2-transfected cells were reduced by only 40%, whereas control transfected cells were already reduced by 50% after 60 min of treatment (Fig. 3B). Importantly, the increased Rest mRNA stability was also reflected at the level of protein (Fig. 3C). Furthermore, we detected a significant increase in REST occupancy of the RE1 site in the promoter region of the Nav1.2 gene, a known REST target, which was not found in the Nav1.2 coding sequence lacking a REST binding site (Fig. 3D).
PC12 cells can be induced to differentiate into sympathetic-like neurons by treatment with NGF. REST overexpression in the undifferentiated cells inhibits subsequent NGF induction of Nav1. 2 mRNA and protein, responsible for the action potential and, consequently, membrane excitability (Ballas et al., 2001). Correlating with the increased occupancy of REST protein after ZFP36L2 knockdown, we also observed reduced Nav1.2 mRNA induction after NGF treatment compared with control cells (Fig. 3E).
REST levels are elevated in DRG from Zfp36l2 KO mice
To further pursue the regulation of REST stability by ZFP36L2, we sought to examine peripheral ganglia under different conditions, focusing on DRG because we could extract easily multiple DRG/mouse. We determined that Zfp36l2 mRNA levels in WT ganglia were low at day 4, increasing significantly between days 4 and 7, whereas Rest mRNA levels were low and constant over the entire time interval. This result was a bit surprising. If ZFP36L2 regulates REST steady-state levels through destabilization, we would have expected to see a drop in Rest mRNA levels with increasing amounts of ZFP36L2. One possible explanation is that Zfp36l2 steady-state mRNA levels do not reflect ZFP36L2 protein levels and that the amount of protein is actually constant over that period. Alternatively, Rest may be regulated primarily at the transcriptional level at day 4, after which its steady-state mRNA level becomes regulated primarily post-transcriptionally. In accordance with the latter, previous studies in CNS indicate that REST is itself repressed translationally at terminal differentiation (Ballas et al., 2005).
Consistent with the PC12 knockdown studies, Rest mRNA levels were increased more than threefold in SCG and DRG cultures prepared from Zfp36l2 KO mice compared with WT cultures (Fig. 4B). Reciprocally, mRNA levels of well-characterized REST target genes, Nav1.2 and the peripheral nerve-specific sodium channel, Nav1.8, and the vesicular protein SNAP25, were all reduced by at least 50% in Zfp36l2 KO DRG neurons relative to control neurons (Fig. 4B), indicating that REST still serves as a repressor of these genes in the mature neurons.

Rest transcripts and protein are both upregulated in ZFP36L2 KO mice. A, Real-time RT-PCR analysis of Zfp36l2 mRNA in DRG acutely isolated at indicated postnatal days. n = 3 mice for P4; 4 mice for P7 and P19. 8 DRG/mouse. B, Real-time RT-PCR analysis of Rest mRNA in dissociated neuronal cultures. DRG (P7); SCG (P0), analyzed at DIV3. n = 3 mice. DRG, 8 ganglia/mouse; SCG, 2 ganglia/mouse. C, Real-time RT-PCR analysis of REST target gene mRNAs from DRG cultures in B. D, Immunolabeling for REST in dissociated DRG, DIV3, using a polyclonal rabbit REST antibody against the C terminus (Ballas et al., 2001). E, Western blot analysis of acutely isolated DRG using antibody in D. Lanes correspond to individual mice, 8 DRG/mouse (*), REST protein. Neuron-specific Class III β-tubulin, TuJ1; neuronal α-tubulin (α-Tu), loading control. F, Western blot analysis of mouse embryonic stem cells (left) and brain tissues (right) using a monoclonal antibody to mouse REST epitopes. Actin, loading control; DAPI, blue, nuclei. G, Immunolabeling (left) and quantitation (right) of sections of acutely isolated DRG using the anti-REST antibody in (F). TuJ1 (green) neurons; DAPI, nuclei, blue. Cell counts, n = 3 mice/genotype/ L1–L4 DRG, 2 areas/section, 3 sections/mouse. *p < 0.05. **p < 0.01. ns, not significant.
To examine for morphological changes in the Zfp36l2 KO mouse, we examined dissociated DRG cultures and noticed that the axons exhibited a different distribution of TuJ1 staining and were less complex than control cultures (Fig. 4D). Although we cannot say to what extent the morphological changes were due to increased REST levels, because immunostaining is not quantitative, we performed a Western blot analysis on DRG that were removed acutely (Fig. 4E). This analysis indicated that REST levels were indeed higher in the Zfp36l2 KO DRG. We also examined REST expression by immunostaining in the acutely removed DRG. The specificity of the antibody used in this analysis is shown in Figure 4F using control and Rest−/− embryonic stem cells (left) (Jørgensen et al., 2009), as well as control and Rest KO brain tissue (right). We determined a higher percentage of DAPI-positive nuclei that contained REST epitopes in the Zfp36l2 KO compared with controls, consistent with the Western blot analysis.
The integrity of peripheral axons is compromised in Zfp36l2 KO mice
As reported, we observed that 80% of Zfp36l2 KO mice are smaller at birth and die after 2–3 weeks, most likely because of their hematopoietic abnormalities (Stumpo et al., 2009). We noticed, in addition, that the KO mice exhibited an aberrant shaky gait in hind limbs that could reflect abnormalities in the peripheral nervous system. Because loss of ZFP36L2 increases Rest mRNA stability in primary neuronal cultures from peripheral ganglia and DRG in vivo and because REST represses genes that are important for neuronal morphology and functions, such as axonal growth/pathfinding, we tested whether axonal integrity/outgrowth might be affected in peripheral ganglia in the Zfp36l2 KO mice. To this end, we established organotypic DRG cultures from P7 WT and Zfp36l2 KO mice and examined axons extended by the ganglia after 7 d in vitro (DIV7). Because of the inability to visualize very thin axonal processes by immunofluorescence, we used phase-contrast microscopy to assess the morphological status quantitatively (Sasaki et al., 2009). Representative phase-contrast images are shown in Figure 5A (top). Our measurements indicated an approximately twofold decrease in the total area covered by axons and blebs in the ganglia from Zfp36l2 KO mice compared with WT (12.2, CI 1.8–13.6 vs 7.6, CI 6.7–8.5; Fig. 5B). This result is consistent with the immunostaining of dissociated DRG (Fig. 4D), which showed less complex axons. There was also a threefold increase in blebs in the KO, reminiscent of degeneration (KO 0.47, CI 0.43–0.52 vs 0.15, CI 0.13–0.16; Fig. 5C). The Zfp36l2 KO ganglia begin to sprout axons after 3 d in culture. We were only able to keep the KO ganglia for ∼7 d because they began to detach, whereas WT ganglia were viable for at least another week.

Axonal integrity is compromised in Zfp36l2 KO mice. A, Top, Representative phase-contrast images from DRG, DIV7. Insets, Higher magnification of swellings (blebs). Center, Binarized images of axons and blebs. Bottom, Axons removed to just show blebs. B, Total areas. Each symbol represents an individual ganglion. n = 4 mice/genotype. WT = 10 ganglia, 54 fields; KO = 9 ganglia, 74 fields. C, Quantification of bleb areas in B. D, Quantification of blebs in WT organotypic DRG cultures, DIV7, after transduction with indicated viruses at DIV3. Each symbol represents an individual DRG. Knockdown efficiency of ZFP36L2 shown in Figure 3. E, Left, Real-time RT-PCR analysis of ZFP36L2–2 shRNA knockdown efficiency in murine erythroleukemia cells transduced with the indicated viruses, infected at equivalent titers. Right, Genomic DNA PCR analysis of transduced DRG showing equivalent numbers of viral particles for control and ZFP36L2–2 shRNA viruses. n = 3 independent experiments. F, Quantification of blebs in WT organotypic DRG culture after transduction with indicated shRNA viruses. n = 3 mice, 6 ganglia/genotype. Each symbol represents an individual DRG. ***p < 0.001. ns, not significant.
To test whether the effects on DRG axonal integrity were due to primary defects in the neurons or to secondary effects due to the global hematopoietic phenotype, we sought to transfect WT DRG in organotypic culture with Zfp36l2 or scrambled shRNAs. Toward this end, we first tested the ZFP36L2 shRNA (ZFP36L2-1) that we showed previously was effective in PC12 cells (Fig. 3). We found that transduction with this shRNA virus caused a fourfold increase in axonal blebbing compared with controls (WT 0.64, CI 0.05–0.08 vs 0.16, CI 0.12–0.19; Fig. 5D). To ensure that this was not an off target effect, we repeated the experiment using a different ZFP36L2 shRNA (ZFP36L2-2). Transduction of murine erytholeukemia cells with equal titers of lentivirus showed an approximately fourfold knockdown of Zfp36l2 mRNA compared with transduction with control shRNA (control 0.92, CI 0.57–1.3 vs 0.25, CI 0.23–0.27; Fig. 5E). Consistent with Zfp36l2 KO DRG, and the acute knockdown with ZFP36L2-1 shRNA, there was again more blebbing in ganglia expressing the ZFP36L2–2 shRNA than in the ganglia transfected with control shRNA (KO 0.11, CI 0.08–0.13 vs 0.05, CI 0.04–0.06 Fig. 5F).
ZFP36L2 regulation of REST contributes to the reduced integrity of DRG axons in culture
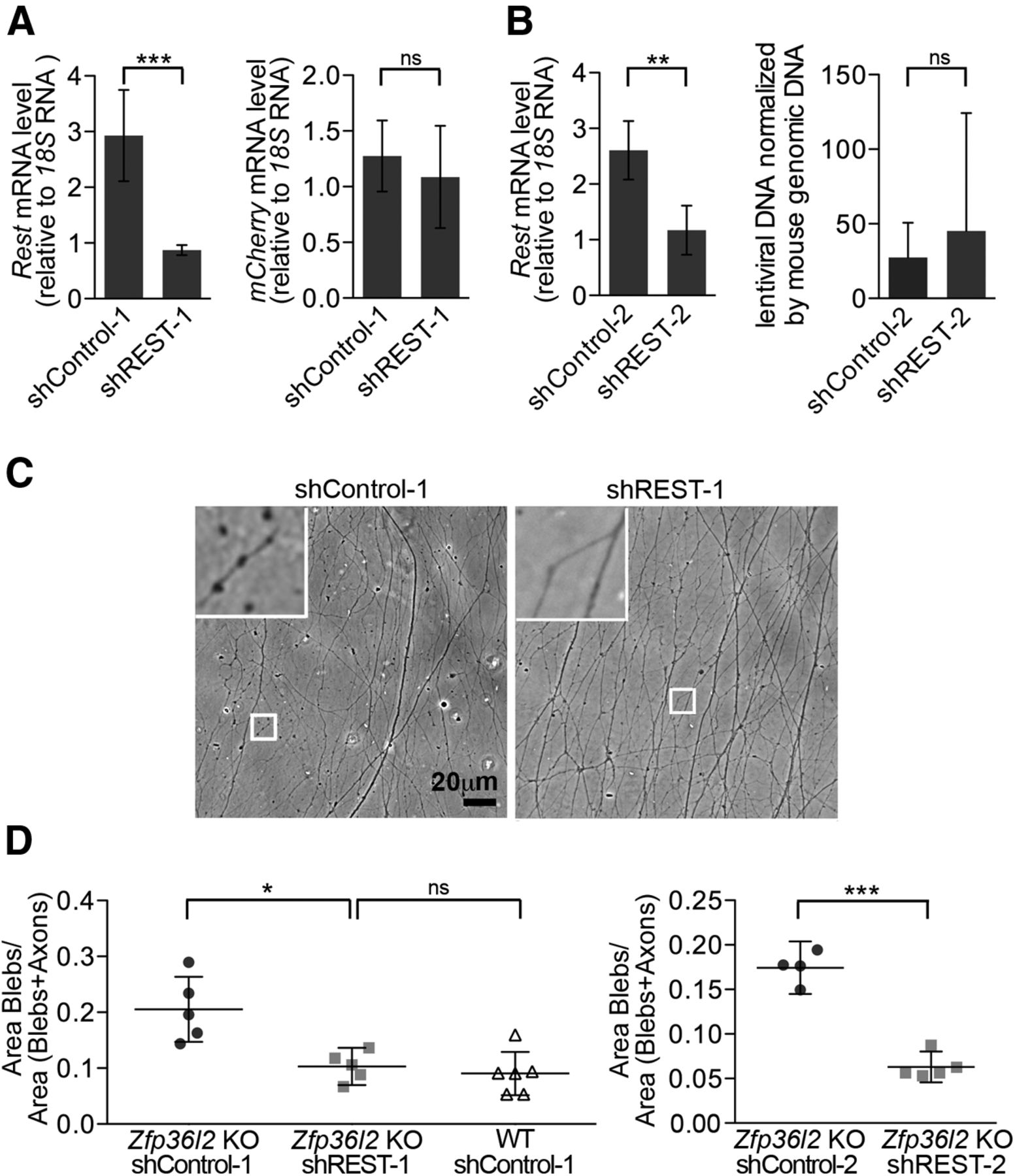
We next tested whether the degenerative axonal phenotype was due, at least in part, to the increased REST levels. To this end, we selected two different REST shRNAs, cloned into lentivirus, which caused significant knockdown of Rest mRNA after transduction into cells (control 2.9, CI 2.1–3.8 vs 0.87, CI 0.778–0.96; Fig. 6A; and control 2.6, CI 2.0–3.1 vs 1.17, CI 0.73–1.61; Fig. 6B). We then transduced DRG organotypic cultures from Zfp36l2 KO mice with lentivirus bearing these REST or scrambled shRNAs. Cultures were maintained for 7 d before analysis. Subsequent quantification of blebs indicated that REST knockdown significantly rescued axonal integrity in the explanted ganglia (control 0.2, CI 0.13–0.27 vs shREST-1, 0.10, CI 0.07–0.13; Fig. 6D, left; and control 0.17, CI 0.14–0.20 vs shREST-2, 0.06, CI 0.04–0.08; Fig. 6D, right). The bleb number in the rescue compared favorably with the number in WT ganglia (0.09, CI 0.05–0.13; Fig. 6D).

Compromised axonal integrity in Zfp36l2 KO DRG is due, at least in part, to upregulation of REST. A, Real-time RT-PCR analyses of (left) REST knockdown efficiency in KO DRG after transduction with Rest shRNA virus. n = 3 mice, 6 ganglia/mouse, and (right) of mCherry mRNA showing that DRG in A were transduced with similar amounts of virus. n = 3 independent experiments. B, Real-time RT-PCR analyses of (left) REST knockdown efficiency in KO DRG after transduction with a different Rest shRNA virus (shREST-2), and (right) PCR analysis of viral genomes showing equivalent transduction efficiencies of control and REST-2 shRNA virus. n = 4 independent experiments. C, Representative phase-contrast images of axons emerging from Zfp36l2 KO DRG after transduction with indicated viruses at DIV7. Insets, Higher magnification of axons. D, Quantification of blebs from Zfp36l2 KO (n = 5 mice) or WT DRG ganglia (n = 6 mice) transduced with two different control and shREST viruses (left and right panels). Analyses were performed as in Figure 5B. Each symbol represents an individual ganglion. *p < 0.05. **p < 0.01. ***p < 0.001. ns, not significant.
Decrease in REST target gene expression correlates with compromised axonal integrity in sciatic nerve of Zfp36l2 KO mice
To investigate whether axonal integrity was affected in Zfp36l2 KO mice in vivo, we examined sciatic nerves in situ, using immunostaining for NF-H, a known axonal REST target, and MBP, a marker of myelin deposition (Fig. 7A). We found that the overall cross-sectional area of the sciatic nerve, calculated from the nerve diameters of the Zfp36l2 KO sciatic nerves (mean diameters WT 321 μm, CI 281–361 vs 254 μm, CI 240–268; Fig. 7B, bottom), was reduced ∼40% compared with controls. The density of fibers associated with MBP staining increased almost to scale (WT 310 μm, CI 280–340 vs 442 μm, CI 408–476; Fig. 7B, top). Similarly, the distribution of the Zfp36l2 KO MBP-associated fibers peaked at lower diameter sizes compared with controls (Fig. 7C). Thus, it appears that the total number of axons, extrapolating from MBP-associated fibers, was not altered between the Zfp36l2 KO and WT sciatic nerves.

The sciatic nerves in Zfp36l2 KO mice have abnormal morphology and decreased levels of REST targets. A, Immunostaining of sciatic nerve cross-sections for Neurofilament-H (NF-H) and myelin basic protein (MBP). B, Top, Density of myelinated fibers. WT, n = 5 mice; Zfp36l2 KO, n = 4 mice; two sections/mouse, two areas/section. Bottom, Quantification of diameters in same specimens as top. C, Myelinated fiber diameters. Each bin = 1 μm. The same areas used in B were used for measuring fiber diameter. D, Immunostaining of sciatic nerve longitudinal sections with NF-H and MBP. Arrows indicate discontinuities (blebs) on fibers. Boxes represent location of higher-magnification images below. E, Top, Western blot analysis of neurofilament proteins in sciatic nerve. Each lane indicates an individual mouse. α-Tu, Loading control. Bottom, Quantification of Western blots from 6 WT and 6 Zfp36l2 KO animals. **p < 0.01. ***p < 0.001.
To determine whether there were signs of aberrant fiber morphology, we stained longitudinal sections with antibody to NF-H and MBP. We detected areas of discontinuities (Fig. 7D), similar to the blebs in DRG, and this was also reflected in lower amounts of neurofilament proteins detected by Western blot (Fig. 7E). The neurofilament genes have consensus REST binding sites within 1 kb of their transcriptional start sites (Ching and Liem, 2009) and would be predicted to be at lower levels in the KO sciatic nerve due to elevated REST levels.
Discussion
The importance of post-transcriptional regulation in the nervous system is only beginning to be appreciated, although many neurological diseases involve either defects in RBPs themselves or gene dosage effects that could be amenable to post-transcriptional intervention. Moreover, and not surprisingly, recent studies examining directly mRNA and protein levels reveal, in some instances, very poor correlation between steady-state mRNA and protein levels (Vogel and Marcotte, 2012). Three fundamental biological processes in which post-transcriptional regulation is thought to be essential in neurons are synaptic plasticity (Goldie and Cairns, 2012; Quadrato and Di Giovanni, 2013), axonal guidance (Loya et al., 2010), and nerve regeneration (Willis et al., 2005; Kapeli and Yeo, 2012; Gao et al., 2013). Because REST repression controls genes directly involved in these processes (Schoenherr et al., 1996; Otto et al., 2007; Johnson et al., 2008), changes in Rest mRNA and protein levels could have profound effects on neuronal biology.
Most prior studies of REST regulation have indicated low or undetectable constitutive levels of Rest transcripts, or none at all, in mature neurons. Interestingly, there are several examples in which aberrantly high levels of REST result in pathology. For example, in adult mouse hippocampus, Rest transcripts are upregulated in response to ischemic insult (Kokaia et al., 1994) and are proposed to contribute to cell death through regulation of glutamate excitotoxicity (Calderone et al., 2003). Rest mRNA levels and RE1 occupancy of REST target genes also increase in peripheral neurons in response to nociceptive stimuli (Michaelevski et al., 2010; Uchida et al., 2010; Ben-Yaakov et al., 2012) and have been associated with epilepsy (McClelland et al., 2011; Pozzi et al., 2013). Mechanisms that regulate Rest mRNA levels in these different functional contexts are unknown, but an early study (Kojima et al., 2001) reported that cell type-specific expression of REST was not strictly promoter-dependent, and our studies herein support the suggestion of post-transcriptional regulation. It is likely that both transcriptional and post-transcriptional mechanisms are used to regulate REST in different cellular contexts. Both micro RNAs and RBPs were good candidates for regulating Rest mRNA levels, but our CHX experiments pointed to a constitutively synthesized protein that destabilized Rest mRNA. Our screen for RBPs, using the REST 3′ UTR, identified ZFP36L2, which could account for this activity. ZFP36L2 is required for normal blood development in vivo (Stumpo et al., 2009). Interestingly, a recent study identified Rest mRNA as a target of ZFP36L2 in erythroid progenitors as well, verifying independently the results of our three-hybrid screen (Zhang et al., 2013). Effects in the nervous system have not been demonstrated previously.
The CHX effects on Rest mRNA half-life were compatible with the ZFP36L2 half-life measurements in PC12 cells (Fig. 2F), as well as with previous half-life measurements of TTP and BRF1 (Tchen et al., 2004), two other members of the TIS11 family. Interestingly, although TTP is also present in PC12 cells, TTP knockdown had no effect on Rest mRNA stability in DRG dissociated cultures (data not shown), pointing to the contribution of sequences other than the consensus ARE for TTP binding or to unique ZFP36L2 binding partners for Rest destabilization. We are not aware of studies comparing these features among the three family members.
No previous studies have examined ZFP36L2 levels in the peripheral nervous system, but our finding of Zfp36l2 expression in undifferentiated PC12 cells, a rat pheochromocytoma, is consistent with previous work showing relatively high levels of ZFP36L2 in adrenal tissue (Stumpo et al., 2009). We were unable to detect a CHX effect on Rest mRNA levels in primary cultures of either P0 hippocampus or cortex (data not shown). Thus, it is possible that a specialized role for the ZFP36L2 mechanism exists in peripheral neurons at this early stage or that ZFP36L2 has region- and temporal-specific effects in brain. Importantly, both DRG culture and sciatic nerve in vivo showed evidence of axonal and fiber degeneration, respectively, in the Zfp36l2 KO mouse, suggesting that the effects were not due to restricted culture outgrowth conditions. Because we did not have markers for sensory or motor neuron populations within sciatic nerve, we cannot relate the blebbing abnormalities to specific types of sensory or motor neurons. Additionally, it is possible that the axonal degenerative phenotype could be a secondary effect of the global KO, which has a robust hematopoietic phenotype and postnatal lethality. The ZFP36L2 shRNA knockdown experiments in early postnatal DRG, and the REST knockdown axonal rescue experiments, however, make this possibility less likely. We suspect, however, that other ZFP36L2 targets, in addition to REST, also contribute to the loss of axonal integrity. Future studies identifying all of the primary ZFP36L2 targets in DRG will help determine the relative contributions of REST and other physiological pathways to peripheral neuron biology.
An interesting candidate to mediate the axonal integrity effects of upregulation of REST in peripheral neurons is the gene encoding NMNAT2, which contains a REST binding site in the first intron downstream of the transcriptional start site occupied by REST in vivo (data not shown). In addition to a role in peripheral nerve development and maintenance, NMNAT2 has also been implicated in Wallerian axonal degeneration (Sasaki et al., 2009; Gilley and Coleman, 2010; Ljungberg et al., 2012; Xiong et al., 2012; Milde et al., 2013). Although we found decreased NMNAT2 mRNA in the Zfp36l2 KO mice (data not shown), it is also likely that more than one target contributes to REST control of axon integrity. Further, abnormal regulation of REST stability in postmitotic peripheral neurons may have additional functional consequences. For example, in conjunction with stabilized REST, we also observed a decrease in expression of a REST target gene encoding the peripheral nerve specific sodium channel Nav1.8 (Fig. 4B), and abnormal sodium channel activity is associated with painful neuropathies in humans (Waxman, 2013). Abnormal regulation of specific sodium channel types could contribute to the secondary peripheral neuropathy that occurs when myeloma patients are treated with proteasomal inhibitors, which also cause increased REST stability. A comprehensive RNA analysis of peripheral ganglia and sciatic nerve overexpressing REST should provide new clues into these physiological pathways.
Footnotes
This work was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences to D.J.S. and P.J.B. and National Institutes of Health Grant NS22518 to G.M., G.M. is an Investigator of the Howard Hughes Medical Institute. We thank Andrea Ansari and Travis Polston for excellent support in genotyping and mouse husbandry; Glen Corson and Daniel Cawley for expertise in generating the monoclonal antibodies to REST; and Drs. Wi S. Lai and Stephanie N. Hicks for performing the gel shift analyses.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Gail Mandel, Howard Hughes Medical Institute, Vollum Institute, Oregon Health & Science University, 3181 S.W. Sam Jackson Park Road, Portland, OR 97239. mandelg{at}ohsu.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}