

Abstract
Recent evidence has resurrected the idea that the amino acid aspartate, a selective NMDA receptor agonist, is a neurotransmitter. Using a mouse that lacks the glutamate-selective vesicular transporter VGLUT1, we find that glutamate alone fully accounts for the activation of NMDA receptors at excitatory synapses in the hippocampus. This excludes a role for aspartate and, by extension, a recently proposed role for the sialic acid transporter sialin in excitatory transmission.
SIGNIFICANCE STATEMENT It has been proposed that the amino acid aspartate serves as a neurotransmitter. Although aspartate is a selective agonist for NMDA receptors, we find that glutamate alone fully accounts for neurotransmission at excitatory synapses in the hippocampus, excluding a role for aspartate.
Introduction
The traditional view of neurotransmission is that one neuron releases just a single type of classical transmitter such as acetylcholine, glutamate, GABA, and the monoamines. The discovery of neuropeptides and their release from the same terminals that release classical transmitters increased the versatility of neurotransmission (Hökfelt et al., 2000). It has also been found that multiple classical transmitters can be stored and released from the same vesicle (Jonas et al., 1998). For example, the vesicular GABA transporter VGAT packages glycine as well as GABA (Wojcik et al., 2006) and vesicular content reflects the cytosolic concentration of amino acid in the terminal. The presynaptic level of amino acid in turn depends on expression of glutamic acid decarboxylase, the biosynthetic enzyme for GABA, and/or the glycine transporter GLYT2 (Nicoll and Malenka, 1998). In the case of other classical transmitters released from the same neuron, such as dopamine and glutamate, serotonin and glutamate, and acetylcholine and glutamate, recent evidence has suggested co-storage in the same vesicles, but it remains unclear whether this is always the case (Gras et al., 2008; Hnasko et al., 2010; Hnasko and Edwards, 2012; Tritsch et al., 2012; Nelson et al., 2014).
One of the oldest debates about co-transmission involves glutamate and the closely related aspartate. Both of these excitatory amino acids satisfy many of the criteria required for the role of neurotransmitter. Present in the brain, they are both taken up into nerve terminals and released upon depolarization (Morland et al., 2013). Although previous work has established a role for glutamate as the transmitter at most excitatory synapses in the brain, the role of aspartate has remained unclear (Fleck et al., 1993; Yuzaki et al., 1996; Gundersen et al., 1998; Dingledine et al., 1999). Two properties of aspartate make it of particular interest. First, in contrast to other vesicular neurotransmitter transporters that recognize multiple substrates, the vesicular glutamate transporters VGLUT1–3 do not transport or even recognize aspartate (Reimer and Edwards, 2004). Therefore, if aspartate serves as a transmitter, then another transporter must be involved. Second, aspartate is a highly selective agonist for NMDAR-type glutamate receptors and does not activate AMPA-type glutamate receptors (Patneau and Mayer, 1990). Synapses releasing only aspartate should therefore generate only NMDAR currents despite a full postsynaptic complement of AMPARs. In this case, variation in the vesicular content of glutamate and aspartate should have a profound effect on the relative contribution of AMPARs and NMDARs to synaptic transmission.
Recent work has suggested that sialin, a sialic acid transporter closely related in sequence to the VGLUTs, can also transport both glutamate and aspartate (Miyaji et al., 2008; Miyaji et al., 2011). Further, it has been proposed that sialin localizes to synaptic vesicles. These observations predict that, in the absence of VGLUTs, excitatory synapses should signal using synaptic vesicles with a higher aspartate/glutamate ratio and thus increase the contribution of NMDARs to synaptic transmission relative to synaptic AMPARs. Previous work has shown that mature hippocampal synapses without VGLUT1 exhibit severely reduced AMPAR responses (Fremeau et al., 2004). However, the strength of NMDAR responses at VGLUT1 lacking synapses has not been examined. We now find that, in the absence of VGLUT1, there is no preferential activation of NMDARs. This result shows that, regardless of mechanism (Miyaji et al., 2008; Morland et al., 2013), any aspartate released from synaptic vesicles is at a concentration too low to be physiologically relevant.
Materials and Methods
Animal care and husbandry.
VGLUT1 knock-out (KO) and wild type littermate mice were housed with 12 h light/dark cycle and unlimited food and water. From postnatal day 15 (P15), all mice received Nutra-Gel Diet (BioServ) food supplement. After weaning, VGLUT1 KO mice were separated from littermates and fed daily with Nutra-Gel to promote their survival. Both male and female mice were used and all experiments were conducted in accordance with University of California San Francisco Institutional Animal Care and Use Committee.
RT-PCR.
Region CA3 was dissected from 6- to 9-week-old wild type or VGLUT1 KO mice. Total RNA was extracted using TRIzol (Invitrogen) and cDNA synthesized from 600 ng of RNA using the Superscript III Fist Strand Synthesis SuperMix for qRT-PCR (Life Technologies) according to the manufacturer's guidelines. Real-time quantitative PCR was performed using SYBR GreenER qPCR SuperMix (Invitrogen). The following oligonucleotide primers were used for both RT-PCR and quantitative PCR: 5′-GTATGCTGGCATCCTCTTGG-3′ and 5′-GATACAGAAGACAGTCTGCC-3′ amplifying nucleotides 1321–1441 in sialin cDNA spanning the junction of exons 10 and 11 (Prolo et al., 2009); and 5′-GTATGCTGGCATCCTCTTGG-3′ and 5′-CCCATTCCCACCATCACACCCTGGT-3′ amplifying β-actin cDNA region spanning nucleotides 121–252 (exons 2 and 3).
Electrophysiology.
Voltage-clamp recordings from CA1 pyramidal neurons and field recordings were taken from acute hippocampal slices from P67–P85 mice. Then, 300 μm transverse hippocampal slices were cut using a Microslicer DTK-Zero1 (Ted Pella) in chilled high sucrose cutting solution containing the following (in mm): 2.5 KCl, 7 MgSO4, 1.25 NaH2PO4, 25 NaHCO3, 7 glucose, 210 sucrose, 1.3 ascorbic acid, and 3 sodium pyruvate. The slices were then incubated for 1 h at 34°C in artificial CSF (aCSF) containing the following (in mm): 119 NaCl, 2.5 KCl, 1 NaH2PO4, 26.2 NaHCO3, 11 glucose, 2.5 mm CaCl2, and 1.3 mm MgSO4. The aCSF was bubbled with 95% O2 and 5% CO2 to maintain pH and the slices were allowed to recover at room temperature for 45 min to 1 h before recording at room temperature. While recording, slices were transferred to a perfusion stage on an Olympus BX50WI upright microscope and perfused at 2.5 ml min−1 with aCSF containing 0.1 mm picrotoxin. Where indicated, aCSF containing 30 μm glycine was applied for several minutes before measuring AMPAR- and NMDAR-mediated currents. Synaptic responses were evoked by stimulating with a monopolar glass electrode filled with aCSF in stratum radiatum of CA1. To ensure stable voltage-clamp recording, membrane holding current, input resistance, and pipette series resistance were monitored throughout recording. Cells were discarded if holding currents were larger than 200 pA, series resistance varied by 25% or more during a recording session, or series resistance exceeded 30 MΩ. Data were gathered through a MultiClamp 700B amplifier (Molecular Devices), filtered at 2 kHz, and digitized at 10 kHz.
Simultaneous whole-cell synaptic and field recordings.
Simultaneous whole-cell voltage-clamp and field recordings were performed by placing a field electrode in stratum radiatum of CA1 between the stimulating electrode and the whole-cell voltage-clamped CA1 pyramidal neuron. The field electrode was filled with aCSF. For whole-cell voltage-clamp recordings, the internal recording solution contained the following (in mm): 135 CsMeSO4, 8 NaCl, 10 HEPES, 0.3 EGTA, 5 QX-314, 4 Mg-ATP, and 0.3 Na-GTP. Osmolarity was adjusted to 290–295 mOsm and pH buffered at 7.3–7.4. AMPAR-mediated responses were isolated by voltage clamping the cell at −70 mV, whereas NMDAR responses were recorded at +40 mV with amplitudes taken 100 ms after stimulation to avoid contamination by AMPAR current.
Statistics.
Mann–Whitney U tests, Mann–Whitney Rank sign tests, and one sample t tests were used to determine statistical significance between groups where indicated. Data analysis was performed in Igor Pro (Wavemetrics), KaleidaGraph (Synergy Software), Excel (Microsoft), and Prism 6 (GraphPad).
Results
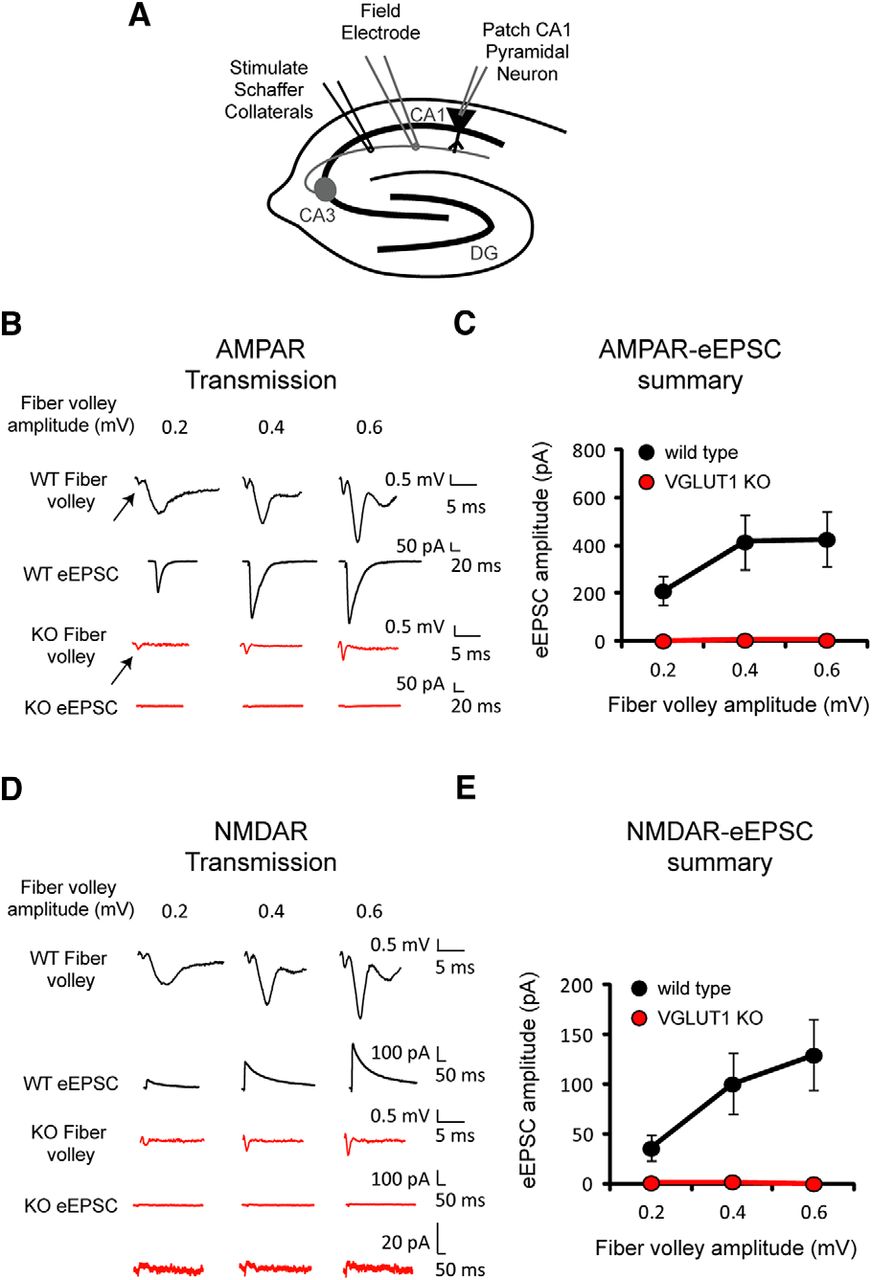
Previous studies have shown that deletion of the glutamate transporter VGLUT1 in CA1 hippocampal pyramidal cells greatly impairs excitatory synaptic transmission (Fremeau et al., 2004). Modest at first due to the transient expression of VGLUT2 early in life, the inhibition becomes essentially complete several weeks after birth. We have therefore used VGLUT1 KO mice at least 2 months old, when AMPAR-mediated field potentials have disappeared. To ensure that the stimulating electrode adequately activated presynaptic axons, we monitored the size of the fiber volley with a field potential electrode placed in stratum radiatum (Fig. 1A,B, arrows). In wild type slices, the fiber volley was associated with large AMPAR-mediated field EPSPs (fEPSPs) and large evoked EPSCs (eEPSCs) simultaneously recorded at −70 mV (Fig. 1B, black traces). In the KO, however, both fEPSPs and eEPSCs were virtually absent (Fig. 1B, red traces). Figure 1C shows the absence of eEPSCs in KO mice at three different fiber volley amplitudes (wild type, n = 7 neurons; VGLUT1 KO, n = 5 neurons), consistent with previous results (Fremeau et al., 2004). The stimulation was then repeated in the same cells while holding the cells at +40 mV to enable detection of NMDAR-mediated eEPSCs (Fig. 1D). In wild type slices, a large NMDAR-eEPSC was recorded (Fig. 1D, black traces). However, no eEPSC was detected in the KO slices (red traces). Figure 1E summarizes these results, showing the absence of NMDAR-eEPSCs in KO slices (wild type, n = 7 neurons; VGLUT1 KO, n = 5 neurons). The data are consistent with the traditional view that glutamate fully accounts for the synaptic response. If aspartate were also released, an NMDAR-eEPSC should have been observed in the KO.

NMDAR–eEPSC amplitude is severely reduced in VGLUT1 KO mice. A, Concurrent recordings of fiber volleys (Field Electrode) and AMPAR- and NMDAR-eEPSCs (Patch CA1 Pyramidal Neuron) in acute hippocampal slices from wild type and VGLUT1 KO mice. B, Representative AMPAR-eEPSC traces from wild type (black) and VGLUT1 KO mice (red) at three fiber volley amplitudes. Arrows point to fiber volleys. C, Average AMPAR-eEPSC amplitudes (mean ± SEM) from wild type (black) and VGLUT1 KO mice (red) at three fiber volley amplitudes (wild type, n = 7 neurons; VGLUT1 KO, n = 5 neurons). D, Sample NMDAR-eEPSC traces from wild type (black) and VGLUT1 KO mice (red) at three fiber volley amplitudes. Vertically expanded eEPSCs are shown for the VGLUT1 KO. E, Average NMDAR-eEPSC amplitudes (mean ± SEM) from wild type (black) and VGLUT1 KO mice (red) at three fiber volley amplitudes (wild type, n = 7 neurons; VGLUT1 KO, n = 5 neurons).
It is possible that the experiments of Figure 1 were not sensitive enough to detect a small contribution of aspartate to the NMDAR-eEPSC. We therefore increased the stimulus strength to record small AMPAR (Fig. 2A) and NMDAR-eEPSCs (Fig. 2B) from VGLUT1 KO neurons. We then calculated the AMPAR/NMDAR-eEPSC amplitude ratio and found no difference between cells from wild type and KO mice (wild type, n = 7 neurons; VGLUT1 KO, n = 6 neurons, Mann–Whitney U test, p = 0.35; Fig. 2C). Because a role for aspartate release might only emerge with strong stimulation, we also examined the response to 10 stimuli given to Schaffer collaterals at 20 Hz. However, the AMPAR/NMDAR ratio of VGLUT1 KO neurons does not differ from wild type neurons under these circumstances (wild type, n = 4 neurons; VGLUT1 KO, n = 4 neurons, Mann–Whitney U test, p = 0.48; Fig. 2D). To ensure that any increase in the NMDA response was detectable, we also measured AMPAR/NMDAR ratios in VGLUT1 KO neurons before and after application of the NMDAR activator glycine for several minutes and found no change in the response (wild type, n = 3 neurons; VGLUT1 KO, n = 3 neurons, Mann–Whitney rank-sign test, p = 0.75; Fig. 2E). Therefore, NMDA receptors in this preparation appear saturated by glycine, indicating the maximum potential to detect an aspartate-mediated change in the AMPAR/NMDAR ratio with the loss of VGLUT1. We also found that sialin is indeed expressed by cells in CA3 (Fig. 3) and that sialin levels do not change with the loss of VGLUT1 (n = 3 animals, one-sample t test, p = 0.803; Fig. 3). The sample analyzed contains glia and inhibitory neurons as well as pyramidal cells forming synapses in CA1, but the abundance of mRNA in pyramidal cells suggests that they had not changed expression of sialin. Although sialin is expressed in CA3, our results thus exclude the possibility that aspartate contributes to the NMDAR-EPSC.

AMPAR/NMDAR–eEPSC amplitude ratios are unchanged in VGLUT1 KO mice. A, Average AMPAR–eEPSC amplitude (mean ± SEM) with maximal Schaffer collateral stimulation in VGLUT1 KO mice (n = 6 neurons). Inset shows sample AMPAR-eEPSC trace. B, Average NMDAR-eEPSC amplitude (mean ± SEM) with maximal Schaffer collateral stimulation in VGLUT1 KO mice (n = 6 neurons). Inset shows sample NMDAR–eEPSC trace. C, Average AMPAR/NMDAR–eEPSC amplitude ratio (mean ± SEM) for wild type (black) and VGLUT1 KO mice (red) (wild type, n = 7 neurons; VGLUT1 KO, n = 6 neurons). Representative AMPAR and NMDAR–eEPSC traces from a wild type (black) and a VGLUT1 KO mouse (red) scaled to peak AMPAR–eEPSC amplitude are shown to the right. D, Average AMPAR/NMDAR–eEPSC amplitude ratio (mean ± SEM) for wild type (black) and VGLUT1 KO mice (red) after stimulation 10 times at 20 Hz (wild type, n = 4 neurons; VGLUT1 KO, n = 4 neurons). Average AMPAR and NMDAR–eEPSC traces from wild type (black) and VGLUT1 KO mice (red) scaled to the peak of the 10th AMPAR–eEPSC are shown to the left. E, Average AMPAR/NMDAR–eEPSC amplitude ratio (mean ± SEM) for VGLUT1 KO mice before (red) and after (blue) treatment with 30 μm glycine (wild type, n = 3 neurons; VGLUT1 KO, n = 3 neurons).

Loss of VGLUT1 does not affect sialin expression in hippocampal region CA3. A, Amplification by RT-PCR shows the expression of sialin (and β-actin control) mRNA in CA3 of both wild type (+/+) and VGLUT1 KO (−/−) animals. B, Quantitative PCR shows no effect of the VGLUT1 KO on sialin expression. Data are presented as sialin amplification curves and as the fold change of sialin expression in VGLUT1 KO relative to wild type (mean ± SEM, n = 3 animals).
Discussion
Our results indicate that aspartate does not contribute to excitatory synaptic transmission in CA1 pyramidal neurons. These results are not consistent with previous work (Miyaji et al., 2008; Miyaji et al., 2011) showing that sialin can transport aspartate as well as glutamate. In fact, the results exclude a mechanism for the accumulation of aspartate into synaptic vesicles dependently or independently of sialin. We do not address the ability of sialin to recognize and transport aspartate and glutamate and it remains possible that synaptic vesicles do contain aspartate. However, the concentration of aspartate released cannot exceed the minimum required to activate NMDARs. Therefore, glutamate transported by VGLUT1 completely accounts for excitatory synaptic transmission in adult CA1 pyramidal cells.
Footnotes
This work was supported by the National Institute of Mental Health–National Institutes of Health (R.A.N., R.H.E., and B.E.H.) and the National Institute on Drug Abuse (R.H.E.). K.S. was supported by a fellowship from the Fondation Fyssen.
The authors declare no competing financial interests.
- Correspondence should be addressed to either Roger A. Nicoll or Robert H. Edwards, Department of Cellular and Molecular Pharmacology, University of California, 600 16th Street, Mission Bay Campus, San Francisco, CA 94158; roger.nicoll{at}ucsf.edu or Robert.Edwards{at}ucsf.edu





{kind=link}
{kind=link}
{kind=link}