

Abstract
After ischemic stroke, various damage-associated molecules are released from the ischemic core and diffuse to the ischemic penumbra, activating microglia and promoting proinflammatory responses that may cause damage to the local tissue. Here we demonstrate using in vivo and in vitro models that, during sublethal ischemia, local neurons rapidly produce interleukin-4 (IL-4), a cytokine with potent anti-inflammatory properties. One such anti-inflammatory property includes its ability to polarize macrophages away from a proinflammatory M1 phenotype to a “healing” M2 phenotype. Using an IL-4 reporter mouse, we demonstrated that IL-4 expression was induced preferentially in neurons in the ischemic penumbra but not in the ischemic core or in brain regions that were spared from ischemia. When added to cultured microglia, IL-4 was able to induce expression of genes typifying the M2 phenotype and peroxisome proliferator activated receptor γ (PPARγ) activation. IL-4 also enhanced expression of the IL-4 receptor on microglia, facilitating a “feedforward” increase in (1) their expression of trophic factors and (2) PPARγ-dependent phagocytosis of apoptotic neurons. Parenteral administration of IL-4 resulted in augmented brain expression of M2- and PPARγ-related genes. Furthermore, IL-4 and PPARγ agonist administration improved functional recovery in a clinically relevant mouse stroke model, even if administered 24 h after the onset of ischemia. We propose that IL-4 is secreted by ischemic neurons as an endogenous defense mechanism, playing a vital role in the regulation of brain cleanup and repair after stroke. Modulation of IL-4 and its associated pathways could represent a potential target for ischemic stroke treatment.
SIGNIFICANCE STATEMENT Depending on the activation signal, microglia/macrophages (MΦ) can behave as “healing” (M2) or “harmful” (M1). In response to ischemia, damaged/necrotic brain cells discharge factors that polarize MΦ to a M1-like phenotype. This polarization emerges early after stroke and persists for days to weeks, driving secondary brain injury via proinflammatory mediators and oxidative damage. Our study demonstrates that, to offset this M1-like polarization process, sublethally ischemic neurons may instead secrete a potent M2 polarizing cytokine, interleukin-4 (IL-4). In the presence of IL-4 (including when IL-4 is administered exogenously), MΦ become more effective in the cleanup of ischemic debris and produce trophic factors that may promote brain repair. We propose that IL-4 could represent a potential target for ischemic stroke treatment/recovery.
Introduction
Under normal conditions, the neurovascular unit (NVU) as a whole generates a signaling circuit whereby a defined set of signaling molecules supporting the functionality and integrity of the unit are expressed. In response to brain injury—for instance, ischemia resulting from cerebral blood vessel occlusion—this homeostatic signaling circuit could be altered or entirely lost. Neurons demonstrate a unique susceptibility to ischemic damage (Baron et al., 2014) and, in response to overwhelming ischemia, may actively (or passively, via lost cell integrity) secrete signaling molecules that signal “danger,” often referred to as damage-associated molecular patterns (DAMPs). By activating various pattern-recognition receptors, DAMPs propagate proinflammatory responses, primarily by activating neighboring microglia (Takeuchi and Akira, 2010). Proinflammatory mediators, proteases, and free radicals are ultimately elaborated, aggravating ischemia-mediated brain damage and even resulting in neuronal loss (Iadecola and Anrather, 2011; An et al., 2014).
Conversely, there is a growing body of literature supporting the notion, as originally proposed by Lo (2008), that sublethally injured tissue (e.g., penumbra) may sustain plasticity and the ability to participate in the repair process. Under these conditions, neurons may act as “first responders,” secreting signaling molecules that support tissue healing and repair rather than potentially deleterious proinflammatory ones (Christopherson et al., 2005; Xing et al., 2014; Neumann et al., 2015). One molecule that may be a part of this endogenous neuronal defense mechanism is interleukin-4 (IL-4). IL-4, a product of select immune cells, is a pleiotropic cytokine involved in the regulation of diverse immune and inflammatory responses (Martinez et al., 2009; Luzina et al., 2012). One of the most intriguing properties of IL-4 is that it polarizes macrophages toward the phenotype that is often termed M2 (or alternatively activated). M2 macrophages aid in the resolution of inflammation via increased trophic input and the augmentation of phagocytosis and proteolysis of dead, diseased cells/proteins, ultimately paving the way for tissue repair (Zhao et al., 2007; Balce et al., 2011; Sica and Mantovani, 2012). Numerous animal model studies representing a vast array of neurological diseases suggest that IL-4 may act as a therapeutic factor (Lyons et al., 2007; Ponomarev et al., 2007; Clarke et al., 2008; Xiong et al., 2011; Gadani et al., 2012). It has also been postulated that activation of the IL-4 receptor (IL-4R) by IL-4 itself leads to activation of the transcription factor peroxisome proliferator activated receptor γ (PPARγ). Augmentation of PPARγ and its pathways—in part by modulating the function of microglia, the resident macrophages of the CNS—has been demonstrated to reduce brain injury after ischemic and hemorrhagic stroke (Luo et al., 2006; Tureyen et al., 2007; Zhao et al., 2007, 2009b).
For the present study, we hypothesized that, in response to sublethal ischemia (e.g., in the area of the ischemic penumbra), neurons would synthesize IL-4. IL-4 would then modulate microglial function by polarizing microglia to an M2 “healing” phenotype. M2 signaling pathways would be activated, resulting in augmentation of phagocytosis-mediated tissue cleanup. We also hypothesized that enhancing such cleanup processes in an animal model of ischemic stroke may result in improved functional recovery.
Materials and Methods
All animal studies followed the guidelines outlined in Guide for the Care and Use of Laboratory Animals from the National Institutes of Health and were approved by the Animal Welfare Committee of the University of Texas Health Science Center at Houston. All studies were performed using a randomization approach, and all analyses were performed by investigators blinded to treatment assignments. Animals were fed a standard mouse diet and housed in standard mouse cages on a 12 h inverted light/dark cycle. Behavioral analyses were performed from the hours of 10:00 A.M. to 4:00 P.M.
Ischemia model in mice.
Focal ischemia was induced by unilateral occlusion of the left middle cerebral artery (MCA) and the left common carotid artery (CCA) (MCA/CCAo), as we have described previously (Aronowski et al., 2000; Zhao et al., 2009b). Briefly, male C57BL/6 mice (10–12 weeks old) or IL-4–green fluorescent protein (GFP) reporter mice (C.129-Il4tm1Lky/J; The Jackson Laboratory) were anesthetized with 0.35 g/kg chloral hydrate intraperitoneally. The rectal temperature was maintained at 36.5 ± 0.5°C during the induction of ischemia and throughout the first hour of reperfusion using a feedforward temperature controller (YSI model 72). An incision was made through the left temporalis muscle perpendicular to a line drawn between the external auditory canal and the lateral canthus of the left eye. Under direct visualization with the surgical microscope, a burr hole was drilled at 1.2 mm rostral to the fusion of the zygomatic arch with the squamosal bone to expose the left MCA rostral to the rhinal fissure. A 0.005-inch-diameter stainless steel wire (Small Parts) was placed underneath the left MCA, proximal to the major bifurcation of the MCA, and distal to the lenticulostriate arteries. The artery was then lifted, and the wire was rotated clockwise. The left CCA was occluded using atraumatic Heifetz aneurysm clips. Reperfusion after 60 min of occlusion was established by first removing the aneurysm clips from the CCA and then rotating the wire counterclockwise and removing it from beneath the MCA. Interruption of flow through the MCA was inspected under the microscope and verified using a laser Doppler flowmeter (model BPM2; Vasamedics). The incision on the skin was closed by a 26 gauge cutting needle with nylon suture (5.0) using simple interrupted pattern. The infarction in this model is limited to the cortical tissue and is used to model MCA stroke in humans.
IL-4 reporter mice to study expression of IL-4.
To determine at a single-cell level the IL-4 expression pattern in the brain after ischemia, we used the “IL-4/enhanced GFP (EGFP)–enhanced transcript” (4Get) allele mice (C.129-Il4tm1Lky/J; The Jackson Laboratory). The 4Get mouse has a bicistronic IRES–EGFP reporter cassette inserted between the translational stop codon and the 3′ UTR of the Il4 gene, thus expressing the endogenous IL-4 gene and EGFP (Mohrs et al., 2001; King and Mohrs, 2009).
To determine the exact cell type expressing IL-4 in the brain after ischemia, we subjected 4Get mice to 30 min of MCA/CCAo, and, 24 h later, we intracardially perfused the mice with saline, followed by 4% Formalin. Dissected brains were cryopreserved in 50% sucrose in PBS before snap freezing and cryosectioning. The sections (10-μm-thick) were treated with sodium citrate buffer (10 mm sodium citrate and 0.05% Tween 20, pH 6.0) for 10 min and then the neurons and EGFP were labeled with anti-NeuN (MAB377; Millipore) and anti-GFP (632592; Clontech) antibodies, respectively. The NeuN- and GFP-immunopositive cells were visualized using goat anti-mouse IgG–Alexa Fluor 546 and goat anti-rabbit IgG–Alexa Fluor 488, and the nuclei were stained with DAPI.
Tissue harvesting.
Animals were anesthetized deeply with chloral hydrate (0.5 g/kg, i.p.) and perfused intracardially with ice-cold PBS. Subsequently, for histology or biochemical analyses, the whole brains or the subdissected tissues representing cortices affected by ischemia were snap frozen by submersion in −80°C 2-methylbutane and then stored in a −80°C freezer before cryosectioning, RNA isolation, or protein analyses.
RNA isolation and RT-PCR.
The ipsilateral cortices around the infarction area were dissected on ice, snap frozen, and processed for mRNA extraction using TRIzol reagents. The primary brain cells in culture were harvested and washed with PBS and then lysed in TRIzol reagent. RT-PCR analyses were done as described previously (Zhao et al., 2006, 2007, 2009a,b). We used glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene as an internal standard. The sequences of primers are listed in Table 1. Measurements of the gene products were normalized to the optical density of GAPDH bands. The results were calculated as percentage change over the control (naive animal, in vivo).
Sequences of primers
Primary cortical neuron cultures.
The cortices of E18 prenatal embryos were dissected and dissociated by trituration as we described previously (Kim et al., 2004; Zhao et al., 2009b), with some modification. The dissociated cells were plated on poly-l-lysine-coated culture plates in Neurobasal medium with B27 at 300–700 cells/mm2. The cells were maintained in a CO2 incubator (5% CO2, 21% O2) at 37.0 ± 0.5°C. Half of the culture medium was changed every 2 d. After a total of 9–12 d in culture, the neuronal cells form extensive axonal and dendritic networks and are ready for the experiments. Using MAP2 immunofluorescence, we confirmed that ≥90% of living cells were MAP2-expressing neurons.
Astrocytes/oligodendrocytes/microglia cultures.
The individual primary cortical glia cell culture was isolated and purified from the rat brain cocultures using P1–P2 pups, as we described previously (Zhao et al., 2007). First, the cells from brain cortices were seeded in 75 cm2 TC flasks and cultured for 14 d. The loosely adherent microglia were harvested from the culture medium after a slight shaking step. After a centrifugation at 400 × g for 5 min, the microglia were collected and replated onto poly-l-lysine-coated TC plates, with or without 12-mm-diameter German glass, at a density of 2–5 × 105 cells/ml. After removing the microglia, the coculture flasks were shaken continuously at a speed of 220 rpm for 30 min to remove the other loosely attached cells, including microglia and some proliferating cells (the cells at the proliferating stage were usually attached loosely to the astrocyte layer and thereby relatively easily shaken off). Then, the coculture flasks were changed into fresh culture medium and agitated at 220 rpm for 16–20 h to harvest the oligodendrocytes. After the above shaking steps, the tightly attached cell layer on the tissue culture flasks primarily consisted of astrocytes.
Using immunolabeling for microglia [cluster of differentiation 68 (CD68), MCA341; Serotec], oligodendrocytes (OSP, Ab7474; Abcam), and astrocytes (GFAP, G3893; Sigma-Aldrich), we confirmed that the microglia cultures at 24 h after replating were ≥96% CD68 positive; the oligodendrocyte-rich cultures were ≥84% pure, and the astrocyte-rich cultures were ≥90% pure.
Oxygen–glucose deprivation injury model.
Nine- to 12-d-old primary cortical neurons in culture were subjected to oxygen–glucose deprivation (OGD) injury, as we reported previously (Zhao et al., 2009b). Briefly, the culture media were replaced with Neurobasal medium (Invitrogen) without glucose, and the cultures were placed in a gas-tight humidified chamber filled with 5% CO2/95% N2 for 15 min, 30 min, 2 h, or 24 h. At the end of OGD, glucose at 4.5 mg/ml was added into the culture dish, and the cells were then returned to their original culture condition and incubated for 6–24 h for reperfusion.
Determination of lactate dehydrogenase activity.
The degree of cell injury was assessed by determining the amount of lactate dehydrogenase (LDH) released into the culture medium using an LDH assay kit (Promega; Zhao et al., 2009b).
Immunofluorescence of MAP2.
MAP2 immunofluorescence was performed using the procedure as we described previously (Zhao et al., 2007). Briefly, the rat cortical neurons grown on German glass were fixed with 95% methanol containing 5% acetic acid for 10 min at −20°C and incubated in mouse anti-MAP2 (M4403; Sigma-Aldrich) overnight at 4°C. Rabbit anti-mouse IgG–Alexa Fluor 488 (Invitrogen) was used to visualize MAP2-labeled neurons. The nuclei were visualized with DAPI.
Phagocytosis assay for dead neurons.
The isolated cortical neuronal cells from E18 prenatal rat pups were seeded on 100 mm culture dishes and cultured in vitro for 5 d. Then, the neurons were subjected to gamma irradiation (20 Gy) to induce apoptosis and then incubated in the CO2 cell culture incubator for 48 h, followed by a short incubation with 1 μg/ml propidium iodide (PI) for nuclear labeling to help visualize apoptotic cells under the epifluorescence microscope, after their engulfment by the microglia. After washing in PBS, the PI-labeled dead neurons were resuspended in Neurobasal medium at 1–5 × 108 cells/ml and used as an indicator/target of phagocytosis.
Three hours after exposing the microglia to the PI-labeled dead neurons, the cells were fixed in 2% paraformaldehyde for 2 min. After a short permeabilization with 0.1% Triton X-100, the cells were labeled with phalloidin–FITC (Invitrogen) to define the shape of individual microglia and Hoechst 33258 to label nuclei. Number of PI-labeled nuclei per microglial cell was determined visually under the fluorescence microscope. We counted 50 microglial cells per each experimental condition.
Microglia were preincubated with IL-4 or rosiglitazone (ROS) for 16 h before adding the PI-labeled dead neurons, the targets of phagocytosis.
Image capture and cell counting.
A Zeiss Axioskop 2 microscope equipped with CCD camera and operated by MetaMorph 7.4 software was used for image acquisition. The fluorescence-labeled cells were visualized and captured using excitation/emission of 490/520 nm for phalloidin–FITC or Alexa Fluor 488, excitation/emission of 550/575 nm for PI-labeled dead neurons or Alexa Fluor 546, and excitation/emission of 365/480 nm for Hoechst 33258 or DAPI. To determine the phagocytosis capacity of the microglia toward the PI-labeled dead neurons, we used a motorized stage and program (MetaMorph software 6.2) that allows image stitching. This computer-controlled image acquisition system permits automatic and unbiased cell counting. Three independent cell culture samples (grown on 12-mm-diameter German glass coverslips) were used for each analysis. Sixteen images from each coverslip were captured under 40× objective for a total area of 1.2 mm2. Fifty microglial cells from the image were observed for each condition, and the number of PI-positive neuronal nuclei in each microglia was counted manually on still images. The average number of phagocytosed dead neurons (PI-labeled nuclei − dead neurons) within each microglia was calculated and used to determine the phagocytosis index.
Western blot.
The protein amount was determined using immunoblot, as we described previously (Zhao et al., 2007). Rabbit anti-IL-4Rα (sc-686; Santa Cruz Biotechnology) or chicken anti-GAPDH (AB2302; Millipore) immunopositive bands were visualized with ECL (Pierce). Semiquantification of luminescence signal intensity on x-ray film was determined by the analyses of optical density, using a computer-assisted Kodak analysis (EDAS) 290 system.
Transfection of IL-4 promoter-driven luciferase reporter plasmids.
To determine the IL-4 gene expressing activity in the primary neuronal cultures, we transfected 1 μg of murine IL-4 promoter luciferase plasmids (plasmid 12195, Addgene; gift from Anjana Rao, La Jolla Institute, San Diego, CA) (Macián et al., 2000) using JetPEI (PolyPlus Transfection) into the 9-d-old primary rat neuron cultures. After 24 h of incubation, the cells were exposed to 15 min of OGD, oxygen deprivation (OD), or glucose deprivation (GD). The cells were rinsed in HBSS after 24 h reperfusion and harvested in 1× lysis reagent for luciferase activity assay using the Luciferase Assay System (E1500; Promega). The luciferase activity (OD) was read using TD-20/20 Luminometer (DLR) and expressed as fold changes over the vehicle control (JetPEI only).
IL-4 ELISA.
The amount of IL-4 in the culture medium was measured using Rat IL-4 Platinum ELISA kit (eBioscience) using the standard at 0–100 pg/ml, according to the instructions of the manufacturer.
Conditioned medium transfer.
The medium from the cultured neurons that were exposed to 30 min OGD (or media without OGD, control) followed by 24 h of incubation under normal conditions (representative of “reperfusion”) was harvested and centrifuged at 400 × g for 5 min. The supernatant was transferred to culture wells containing microglia and incubated for 24 h before harvesting microglia for mRNA analyses.
ROS, GW9662, or IL-4 administration.
For the animal experiments, ROS (Cayman Chemicals) at 1 mg · kg−1 · d−1 was administered intraperitoneally at 24 h, 2 d, 3 d, and 4 d after ischemic stroke injury. Recombinant mouse IL-4 (rIL-4, CYT-282; PROSPEC) at 2 μg · kg−1 · d−1 was administered subcutaneously at 2 h and then 1, 2, and 3 d after ischemic stroke for the “acute” treatment experiment and alternatively at 24 h and then 2, 3, and 4 d for the “delayed” treatment experiment. Saline was used as the vehicle control. For the cell culture experiments, 2 μm GW9662 (GW) was applied to the microglia culture medium at 30 min before adding ROS or IL-4. rIL-4 at 1 ng/ml or 2 μm ROS was applied directly into the culture medium at 16 h before the cells were exposed for phagocytosis assay.
Neurological/functional deficits measurement.
All behavioral tests in mice were conducted in a quiet and low-lit room by an experimenter blinded with respect to the treatment groups. Pretests were done to exclude abnormal animals. Only animals with <20% foot faults and animals that had no preference in forelimb placing were used for the MCA/CCAo injury experiment. An individual test score and a combination test score from a battery of behavioral tests (foot fault, forelimb placing, postural flexing, wire and corner test) was used to measure the neurological/functional deficits (NDs) as we reported previously (Zhao et al., 2007; Zhao et al., 2009b). We also determined the grand NDs score (NDS) by combining all the tests, assuming an equal weight of each of the tests.
Cortical tissue loss (atrophy) measurement.
The infarction volume was measured as we described previously (Waxham et al., 1996; Aronowski et al., 2000). Briefly, the mice were deeply anesthetized with chloral hydrate (0.5 g/kg, i.p.), perfused intracardially with 50 ml of ice-cold PBS, and then decapitated. The brain was removed, rinsed in PBS, and sliced into 2 mm sections. The image of each section was captured using a digital camera, and the tissue loss (cavity size) was determined using a computer-interfaced NIH ImageJ program. The lesion volume represents the integrated infarction size derived from the four rostral sections. The lesion area is a surface area of the lost tissue (at the two sides of each section) from these four sections.
Statistical analyses.
All data were expressed as a mean ± SEM. For the in vitro experiments, we pooled the samples from three culture wells and repeated the experiments three times. We performed statistical analyses using the GraphPad and InStat programs. One-way ANOVA followed by Newman–Keuls post hoc test was used for multiple comparisons. Nonpaired t test was used when two groups were compared. Two-way ANOVA was used to compare the NDs between the three groups at different time points.
Results
Neuronal IL-4 is present in the ischemic penumbra after focal ischemia
During analyses of mouse brain at various time points of reperfusion after 60 min of transient MCA/CCAo, we discovered that ischemia resulted in a rapid and robust induction of the IL-4 gene, with a significant increase noted as early as 1 h after reperfusion (Fig. 1A). The peak of IL-4 mRNA induction occurred between 3 and 24 h, followed by a gradual decline to the baseline level over the next 2 d. Interestingly, the induction of IL-4 occurred much earlier than the appearance of CD68 mRNA, a sensitive biomarker for monocytes/microglia, the cell type that is thought to be the first recruited to stroke-affected brain (Gelderblom et al., 2009).

Induction of IL-4 expression by neurons after ischemia. A, Time course of IL-4 and CD68 mRNA expression in the ischemic mouse brain hemisphere at the indicated time points after 60 min of MCA/CCAo. Sham surgery was used as the control (CON). B, Immunofluorescence for visualization of NeuN (neurons; red) and GFP (IL-4; green) in a 4Get IL-4 reporter mouse brain in the contralateral cerebral cortex (CTX) (top) and ipsilateral cortex (bottom) at 24 h after 30 min of MCA/CCAo. The EGFP-expressing cells are primarily localized at the peri-lesional zone (ischemic penumbra) around the ischemic core of the ipsilateral cortex. There is negligible EGFP signal in the corpus callosum (CC), corpus striatum (CS), contralateral hemisphere, and the ischemic core (IC). C, A magnified highlighted area from B, illustrating EGFP (green), NeuN (red), and EGFP + NeuN (COMB) (yellow) at the peri-ischemic zone in the ipsilateral cortex.
Intrigued by this finding, we set out to identify what brain cell type was contributing to IL-4 expression in the ischemic brain. To answer this question, we used the IL-4–EGFP reporter mice. In these mice, IL-4 gene induction at the single-cell level could be identified by the presence of EGFP. We subjected these mice to 30 min of MCA/CCAo and 24 h of reperfusion, and, by analyzing the presence of EGFP, we noted a robust induction of IL-4 expression only in the cells surrounding the ischemic core (IC) within the ipsilateral cortex. No notable expression of EGFP was present in the contralateral cortex, suggesting that IL-4 expression was tightly linked to ischemia-affected tissue (Fig. 1B). To our surprise, virtually all the EGFP-expressing cells were identified as neurons based on colocalization with the neuronal marker NeuN (Fig. 1B,C).
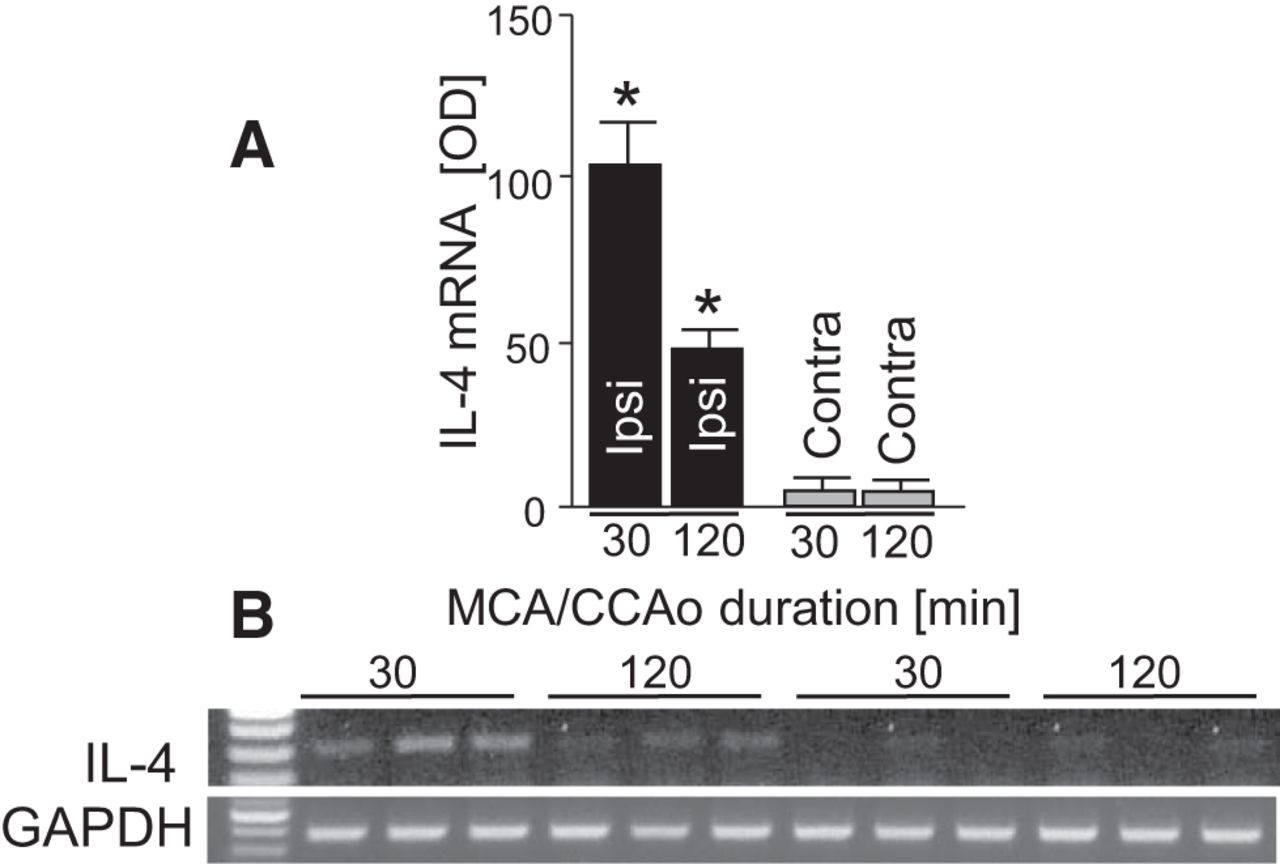
Because short 30 min MCA/CCAo is known to result in considerably mild injury (Waxham et al., 1996), we next wanted to establish whether longer more damaging ischemia is also capable of inducing IL-4 expression. To address this question, we subjected animals to 30 or 120 min of ischemia and measured IL-4 gene expression at 24 h after the onset of ischemia. As anticipated, 120 min of ischemia resulted in a significantly diminished IL-4 induction compared with 30 min of ischemia (Fig. 2A,B), suggesting that a sublethal insult is more potent in inducing IL-4 expression.

Representative PCR gel (B) and a bar graph for IL-4 mRNA expression (A) in the ipsilateral (ipsi; ischemic) and contralateral (Contra) hemisphere at 24 h after 30 or 120 min MCA/CCAo in mouse. *p ≤ 0.05 compared with all other groups. n = 3 per group.
Altogether, our studies strongly suggest that neurons under sublethal ischemic conditions can produce IL-4.
Sublethal level of OGD induces IL-4 generation by neurons
To further understand the relationship between ischemia and IL-4 production, we used an OGD injury model to simulate ischemic events in primary neuronal culture. First, we exposed neurons in culture to various durations of OGD and established that 30 min of transient OGD results in a low to moderate degree of damage (potentially modeling ischemic penumbra) compared with 2 or 24 h of OGD that resulted in more devastating injury (potentially modeling ischemic core; Fig. 3A). Under the assumption that ischemia can induce expression of IL-4, we measured IL-4 gene expression in neurons after exposing them to an increasing duration of OGD. Similar to the MCA/CCAo results, we found that 30 min of OGD potently induced the expression of IL-4 mRNA (Fig. 3B). We also detected a corresponding increase in IL-4 protein (ELISA) in the neuronal culture media (Fig. 3C). These results indicate that neurons subjected to sublethal ischemia can synthesize and secrete IL-4. Finally, using an IL-4 promoter-driven luciferase reporter gene assay, we confirmed the induction of the IL-4 gene by neurons in response to transient/sublethal OGD (Fig. 3D). Interestingly, the secretion of IL-4 by neurons persisted for several hours and led to a gradual accumulation of IL-4 in the culture media. By 24 h after functional “reperfusion,” the IL-4 in the culture medium reached 37.9 pg/ml (Fig. 3E). As anticipated, a lethal dose (2 or 24 h) of OGD-mediated injury failed to induce IL-4 expression/release by the injured neurons (Fig. 3B,C). These findings substantiate the results noted in the ischemic brain model (Fig. 1), essentially that neurons under the proper injury conditions will produce and secrete IL-4. Neurons in the ischemic penumbra can generate IL-4, whereas neurons in severely injured/dead tissue (ischemic core) do not.

IL-4 is produced by neurons in culture. LDH (A), IL-4 mRNA (B; RT-PCR), and IL-4 protein (C; ELISA) produced by rat cortical neurons at 24 h after exposing the cells to OGD for 0.5, 2, and 24 h. In A and C, the data are expressed as mean ± SEM (n = 3). *p ≤ 0.05 compared with the control (0 h) group (no OGD). The MAP2 immunofluorescence below shows the typical maturity stage of rat cortical neurons in culture that were used in these experiments. D, The IL-4 promoter-driven luciferase activity in the rat cortical neuron cultures at 24 h after 15 min of GD, OD, or OGD. The IL-4 promoter-luciferase plasmids were pre-transfected into the neurons using JetPEI reagent 24 h before the OGD injury. The data are expressed as mean ± SEM (n = 5). *p ≤ 0.05 compared with the control (CON, JetPEI only). E, IL-4 protein accumulation in the rat neuronal culture medium at 0, 2, 6, and 24 h after exposing the cells to 30 min of OGD. The data are expressed as mean ± SEM (n = 3). *p ≤ 0.05 compared with the control (0 h) group. F, Cell specificity of IL-4 expression in rat primary astroglia, oligodendroglia, microglia, and neurons cultures. Purity of each of the cell type was established using synaptophysin (SYN1) and neurofilament L (NF-L) for neurons, glial fibrillary acidic protein (GFAP) for astrocytes, myelin basic protein (MBP) for oligodendrocytes, and CD68 for microglia. GAPDH is used as an internal control.
Finally, by assessing the gene induction profile in cultured astroglia, oligodendroglia, microglia, or neurons after exposure to brief OGD, we found that only neurons, not other cell types, expressed the IL-4 gene (Fig. 3F).
rIL-4 and media conditioned by OGD-exposed neurons modulates expression of markers of microglial polarization
It has been established previously that IL-4, acting through the IL-4R on macrophages, can polarize macrophages from the proinflammatory M1 phenotype to the anti-inflammatory healing M2 phenotype. Because microglia are the resident macrophages of the CNS, we hypothesized that IL-4, particularly the IL4 generated by ischemic neurons, would modify the phenotype of adjacent microglia. We first demonstrated that cultured microglia indeed express the IL-4R (Fig. 4A). Next, microglia were exposed to the conditioned media transferred from the sublethal OGD-treated neurons or to rIL-4; these microglia robustly upregulate IL-4R mRNA (Fig. 4A, top) and protein (Fig. 4A, middle). IL-4R induction by IL-4 occurred under fairly selective conditions because neither general proinflammatory lipopolysaccharide (LPS) nor general anti-inflammatory dexamethasone (DEX) affected IL-4R expression (Fig. 4A, bottom).

IL-4 modulates microglia phenotype. A, IL-4R mRNA (RT-PCR; top and bottom) and IL-4R protein (middle; Western blot) production by the rat microglia exposed for 24 h to control culture media (M-CON), culture media conditioned by neurons subjected to 30 min OGD (M-OGD; also refer to the diagram in Fig. 3C), or rIL-4. GAPDH is used as the control. In the bottom, LPS (10 ng/ml) or DEX (0.1 μm) was applied to the culture medium for 24 h and compared with the responses produced by rIL-4 or saline control. B, The RT-PCR product of selected prototypic genes signifying M2 phenotype and PPARγ activation (left) and M1 phenotype (right) expressed by the rat microglia at 24 h after treatment with saline (CON) or rIL-4. C, A diagram of the experiment with the transfer of conditioned medium from the OGD-treated neurons (M-OGD) to the microglia in culture. Right, The expression profile of genes signifying M2 phenotype (gray arrows) and PPARγ activation (black arrow) in the rat microglia that were exposed to the media from control (M-CON) or OGD-treated neurons (M-OGD). *p ≤ 0.05 compared with the M-CON group.
To ascertain whether IL-4 can modulate microglia phenotype, we treated microglia in culture with rIL-4 and measured the induction of several prototypic M2 genes: CD163, CD36, and CD206 (Fig. 4B). We also sought to determine whether conditioned media from the neurons exposed to sublethal OGD (thereby containing IL-4; Fig. 2E) could also modulate microglia phenotype; microglia were thus treated with the media harvested from OGD-exposed neurons (M-OGD) or media harvested from neurons that were not exposed to OGD. M-OGD, similar to rIL-4 (Fig. 4B), induced the expression of CD163, CD204, CD206, and CD36, prototypic M2 genes (Fig. 3C). Under these experimental conditions, IL-4 did not alter the expression of proinflammatory M1 phenotype genes, such as nuclear factor-κB (p50, c-Rel), matrix metalloproteinase 9 (MMP9), or IL-1β, nor the expression of the anti-inflammatory cytokine IL-10 (Fig. 4B).
Interestingly, several of the rIL-4- or M-OGD-induced genes [e.g., CD36, lipoprotein lipase (LPL), CD206, or PPARγ; Fig. 4C] are established target genes of the transcription factor PPARγ, suggesting a possible interplay of IL-4 with PPARγ and PPARγ-regulated genes.
IL-4 enhances microglial phagocytosis
After establishing that IL-4 can alter gene expression in microglia, our next experiment was to determine whether these changes were functionally relevant. One of the central roles of microglia in the setting of ischemia/infarction is to conduct cleanup of injured tissue via phagocytosis of cellular debris and irreparably damaged/dead cells. With our next set of experiments, we examined whether IL-4 could enhance the phagocytosis efficacy of microglia. To simulate post-ischemic cleanup conditions, we used apoptotic neurons as the target of microglial phagocytosis.
After treatment of microglia with rIL-4, we observed a robust augmentation in their ability to engulf apoptotic neurons (Fig. 5A). As demonstrated previously, several of the genes induced by IL-4 are also prototypical PPARγ targets, including CD36 (Fig. 4B), a scavenger receptor likely responsible for promoting phagocytosis (Ren et al., 1995; Zhao et al., 2007; Woo et al., 2012). Treatment of microglia with the PPARγ antagonist GW blocked the pro-phagocytotic effect of rIL-4, as did treatment with the prototypic PPARγ agonist ROS (Fig. 5A). This suggests that there is likely interplay between PPARγ and IL-4 in microglial phagocytosis of apoptotic neurons.

IL-4 increases phagocytic activity and trophin production in microglia. A, Top, A representative image demonstrating a typical phalloidin–FITC-labeled microglial cell (green) that phagocytosed the PI-labeled apoptotic neurons (red). The nuclei are labeled with DAPI (blue). Bottom, Phagocytosis index (number of engulfed PI-labeled cells per microglial cell) for the rat microglia at 3 h after exposure to PI-labeled apoptotic neurons. Cells were pretreated with saline (CON), rIL-4 (1 ng/ml), or rIL-4 plus GW (2 μm), ROS (2 μm), or ROS plus GW for 16 h before assessing for PI. The data are expressed as mean ± SEM (n = 50 microglial cell per condition). *p ≤ 0.05. B, Gene expression profile of rat microglia at 24 h after exposing to OGD (in the presence of neuronal debris) with or without 1 ng/ml rIL-4. *p ≤ 0.05 versus the other two groups.
IL-4 induces expression of trophic factors by microglia
Trophic factors represent a set of molecules that contribute to tissue repair/healing. Microglia can be an important source of these healing factors (Harada et al., 2002; Pollard, 2009). To test whether IL-4 may enhance the pro-trophic capacity of microglia, we exposed microglia in culture to a sublethal level of OGD (again, simulating conditions that result in IL-4 production by neurons) in the presence of neuronal debris (mimicking damaged tissue), subsequently measuring the production of four selected trophic factors demonstrated previously to be relevant in post-stroke repair. Using this protocol, we discovered that OGD selectively induced the expression of BDNF but not IGF-1 or CNTF (Fig. 5B). When we repeated these experiments in the presence of IL-4, there was robust induction of CNTF and IGF-1 (Fig. 5B). IL-4 did not appear to have any effect on the expression of BDNF (Fig. 5B). Neither OGD nor IL-4 treatment affected NGF-1 expression.
IL-4 is protective in an animal model of focal cerebral ischemia
Encouraged by these in vitro results, we sought to explore (as proof-of-concept) whether IL-4 could improve functional neurologic outcome in an animal model of ischemic stroke. First, we subjected mice to MCA/CCAo, administered rIL-4 subcutaneously as described in Materials and Methods (2 h after the arterial occlusion), and then analyzed the affected ipsilateral brain cortex for gene expression changes at 24 and 72 h after reperfusion. In agreement with our in vitro data, we found a significant increase in the expression of several proposed IL-4 target genes. Specifically, we found that IL-4R, CD163, and several gene products regulated by PPARγ (catalase, CD36, and PPARγ itself) were all upregulated after the administration of rIL-4 (Fig. 5A).
Our next set of experiments sought to determine the clinical benefit of IL-4 administration using our IL-4 treatment protocol: 2 μg/kg rIL-4 subcutaneously beginning 2, 24, 48, and 72 h after stroke onset (60 min of MCA/CCAo) compared with saline-treated mice. Behavioral performance 14 d after stroke onset was significantly improved in the rIL-4-treated group across a battery of five tests: corner, forward placing, postural flexing, wire hanging, and foot fault, with the composite score of all the tests referred to as grand NDS (Fig. 6B, top and bottom). Intriguingly, the beneficial effect of rIL-4 was not seen at day 1 and was not statistically significant at day 3 or even day 7, suggesting that the therapeutic mechanism of IL-4 was likely not one of acute cytoprotection but instead one having to do with infarction maturation and brain repair. We also assessed morphological damage after completion of the behavioral assessments (14 d) and found that animals treated with IL-4 had smaller lesion volumes (Fig. 6C).

Peripheral IL-4 affects the gene expression profile and reduces MCA/CCAo-mediated injury. A, Gene expression profile in the ischemic cortices at 24 and 72 h after MCA/CCAo in mice that were treated with saline or rIL-4. *p ≤ 0.05 compared with the saline-treated group. n = 3 per group. B, Neurological dysfunction shown with the grand NDS (top) and five individual tests (bottom) at the indicated time point after 60 min of MCA/CCAo in mice treated with saline of rIL-4. C, Lesion size, presented as a surface area of the missing brain tissue at consecutive rostrocaudal (S1–S7) coronal brain section (a line graph) or as integrated total lesion volume (LV; an inserted bar graph) at 14 d after 60 min of MCA/CCAo in mice treated with saline or rIL-4. In B and C, n = 10, *p ≤ 0.05 compared with the vehicle control group. In A–C, mice received 2 μg/kg rIL-4 subcutaneously at 2 h and then days 1–3.
Because the functional benefits of IL-4 emerged during the later stages of post-stroke recovery (between days 7 and 14), we investigated whether IL-4 administration could also be beneficial with a 24 h treatment “delay.” We also sought to compare functional outcomes in stroke mice treated with IL-4 compared with those treated with the PPARγ agonist ROS, both individually and when administered together (to determine whether there was any type of a synergistic effect). Even delayed treatment with rIL-4 (24 h) was nearly as effective as acute treatment (2 h) in reducing the behavioral deficits 14 d after stroke (Fig. 7A,B). When we compared rIL-4-treated mice with ROS-treated mice, we found a similar profile of neurologic recovery (Fig. 7A,B). Because we have proposed that IL-4 may exert its effects via PPARγ pathways, we hypothesized that combining the two together would not result in a synergistic effect on functional outcome. Indeed, mice treated with IL-4 plus ROS performed similarly to mice treated with IL-4 or ROS alone (Fig. 7A,B).

Delayed treatment with rIL-4 facilitates late stage of recovery after MCA-CCAo. Grand NDS on days 7 and 14 (A), individual behavioral test score (corner, postural flexing, forward placing, hanging, and foot fault) on day 14 (B), infarction volume (C), and rostrocaudal distribution of infarct area on day 14 after 60 min of MCA/CCAo (D). rIL-4, ROS, and the combination of rIL-4 plus ROS (Comb) was administered at 24 h and then on days 2–4. *p ≤ 0.05 compared with the saline control.
Last, after finishing behavioral testing at day 14, we analyzed the mouse brains to determine the degree of tissue loss (e.g., the degree of brain damage; Fig. 7C,D). Again, lesion volume in the treated mice was modestly reduced (by ∼20–30%) in both IL-4- and ROS-treated mice; this is similar to the degree of lesion volume limitation observed in the mice treated “acutely” (e.g., within 2 h).
When analyzing the relationship between the lesion volume and neurological deficit for individual animals included in this study, we found that the correlation coefficient between these two parameters was γ2 = 0.89, indicating considerably strong relationship.
Discussion
IL-4 is an influential cytokine, acting as a pleiotropic regulator of numerous immune/inflammatory pathways (Brown, 2008; Wang and Joyce, 2010). Recognized initially as a product of activated T cells stimulating B-cell proliferation (Howard et al., 1982; Martinez et al., 2009), it has become evident that IL-4 is a central initiator and effector of Th2 immune responses, M2 macrophage polarization, and IgE secretion (Ricardo et al., 2008; Martinez et al., 2009; Luzina et al., 2012). Although classically thought of as being secreted by T cells, mast cells, basophils, or eosinophils, our study is novel in identifying neurons as the source of IL-4 in ischemic brain tissue. We demonstrated experimentally this in (1) primary brain cells in culture in which only neurons expressed the IL-4 gene in response to brief OGD and (2) an animal model of ischemic stroke in which IL-4 expression as early as 1 h after reperfusion occurred primarily in cells expressing the neuronal marker NeuN. At 1 h after stroke, there were no CD3-expressing cells in the brain. Indeed, in other experimental stroke models, T cells did not infiltrate the ischemic brain until after 24 h (Schroeter et al., 1994; Stevens et al., 2002; Arumugam et al., 2005; Hallenbeck et al., 2005; Yilmaz et al., 2006; Gelderblom et al., 2009; Zierath et al., 2010; Iadecola and Anrather, 2011; Zhou et al., 2013). In one of our previous studies, IL-4 mRNA expression was also observed after 15 min of MCA/CCAo, an insult unlikely to induce immune cell infiltration into the brain, lending additional support to the non-immune origin of IL-4 in this setting (Aronowski et al., 1999).
Here, we explored the functional response to IL-4 administration in both microglia cell culture and our animal model of focal brain ischemia. In vitro, IL-4 was able to induce the expression of genes typifying the M2 microglial phenotype and to stimulate PPARγ-dependent phagocytosis of apoptotic neurons. In our animal model, this translated to decreased lesion size and greater neurologic recovery at 14 d after stroke. We believe that our study has identified a unique NVU regulatory signaling circuit whereby ischemic neurons defend against tissue injury via IL-4. We propose that, in areas of sublethal ischemia—representing the ischemic penumbra—neuron-derived IL-4 acts on adjacent microglia, polarizing these microglia toward a healing M2 phenotype with gene expression profiles mirroring those of PPARγ.
In models of cerebral ischemia, the infarcted tissue not only acts as a reservoir of cytotoxic and proinflammatory molecules that may harm adjacent healthy brain, it also forms a physical barrier hampering neural reorganization, repair, and recovery. Microglia, with their phagocytic functions, are therefore primed to be executors of the cleanup/repair processes, beginning with the removal of infarcted debris. We and others have demonstrated that PPARγ signaling upregulates microglial expression of scavenger receptors (Zhao et al., 2007; A-Gonzalez and Hidalgo, 2014), including CD36 (a central PPARγ gene target), thereby augmenting phagocytosis (Ren et al., 1995; Majai et al., 2007; Zhao et al., 2007; Woo et al., 2012). In this study, we were able to demonstrate that microglia treated with IL-4 had a significantly greater phagocytosis efficacy, similar to microglia treated with a PPARγ agonist. This was inhibited by coadministration of a PPARγ blocker, arguing that the pro-phagocytosis effect of IL-4 is likely mediated via PPARγ signaling. In clinically relevant animal models of intracerebral hemorrhage (ICH) (Zhao et al., 2007) and postnatal ischemic stroke (Woo et al., 2012), PPARγ and CD36 have been suggested as key players in cleaning up after acute brain injury. Likewise, studies in patients with ICH have demonstrated that hematoma resorption is impaired in CD36-deficient patients, correlating with poorer short-term neurologic outcomes (Fang et al., 2014).
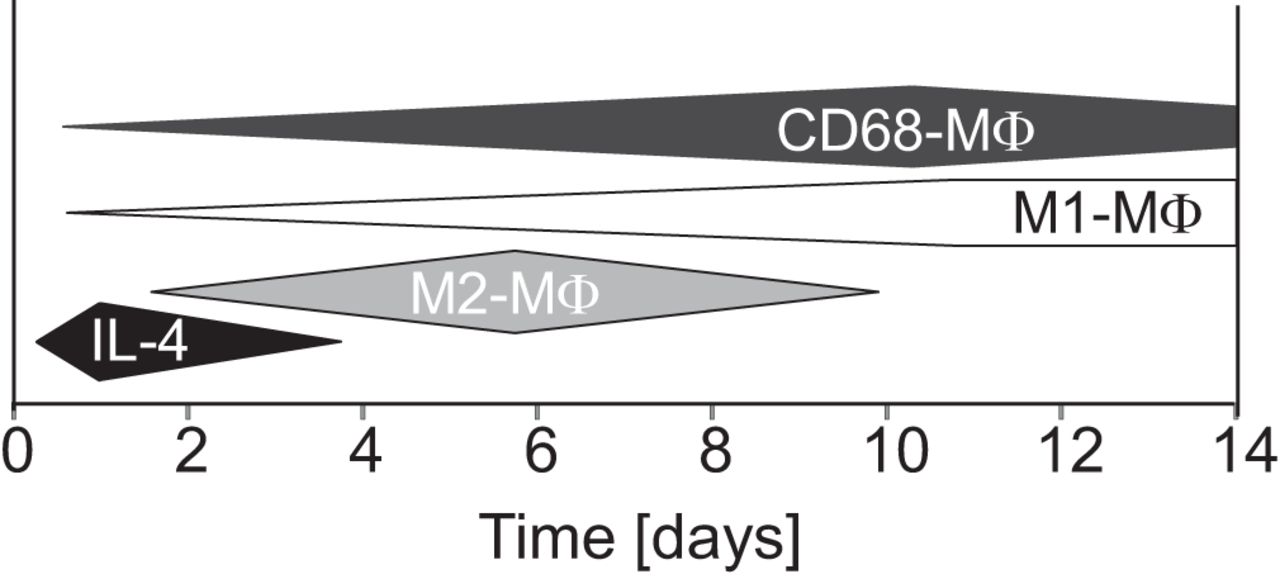
Regarding the time course of the IL-4 signaling circuit, in our in vivo model, CD68-expressing microglia/macrophages (MΦ) were present in the ischemic tissue for weeks. Conversely, the production of IL-4 lasted for only the first 2–3 d (Fig. 8). Interestingly, in a study by Suenaga et al. (2015), the expression of markers indicative of M2 MΦ peaked 7 d after stroke, whereas the expression of M1 markers peaked 14 d after stroke, suggesting a progressive M2-to-M1 phenotype shift. Because neuronal IL-4 production appears to be transient, it may be that, unless IL-4 is replenished by another source, the therapeutic effects of IL-4 polarization to an M2 phenotype are lost within days after stroke onset, allowing the M1 presence to strengthen. In other words, the loss of IL-4 signaling may represent a significant contributor to M2 phenotype loss after stroke.

Time course of endogenous IL-4 expression and microglia phenotype profiles.
In preclinical models of ischemic stroke, the therapeutic time window for acute neuroprotection has proven relatively short. Specifically, no single therapeutic agent of >50 tested in our laboratory during the past two decades resulted in acute infarct volume reduction if intervention was delayed beyond 2 h. Therefore, we were not surprised when rIL-4, ROS, and even the combination of both did not result in significantly improved outcomes at 24 h or 3 d after stroke. The limited utility of IL-4 as an acute neuroprotectant after stroke was also inferred from a study whereby intracerebroventricular administration of rIL-4 to mice 30 min before stroke onset was ineffective in reducing infarct volume/neurologic dysfunction (Xiong et al., 2011). Conversely, IL-4 deficiency in the same stroke model resulted in a greater degree of acute ischemic damage; this damage was reversed by intracerebroventricular rIL-4 pretreatment (Xiong et al., 2011), suggesting that mechanistically, IL-4 is somehow involved in the modulation of ischemic brain damage.
It was rather unexpected when we discovered that IL-4 had a fairly strong delayed therapeutic efficacy in our animals, even when rIL-4 was administered not at 2 h but at 24 h after MCA/CCAo. The mechanisms underlying this delayed recovery are not entirely clear. The polarization of microglia to the healing M2 phenotype may result in the augmentation of subacute to chronic brain repair processes rather than to any acute cytoprotection (Derecki et al., 2011). We have demonstrated in this study increased production of various trophic factors by microglia treated with IL-4. Two of these trophic factors, IGF1 and CNTF, have been associated with “brain rewiring” in other studies (Zhao et al., 2005; Kang et al., 2013). IL-4 itself may also have a direct effect on delayed repair of the NVU, perhaps via modulation of angiogenesis (Fukushi et al., 1998). Recently, we have seen a similar delayed post-stroke recovery in our focal ischemia animals in the setting of cell therapy. In one study of mononuclear cell (MNC) therapy, mice who received a single MNC 24 h after stroke ictus had a modest, albeit reproducible reduction in brain tissue loss as determined 28 d after the stroke (Yang et al., 2013). We should also consider that other neuronally secreted cytokines may be adding to the effect of IL-4. For instance, other studies have demonstrated that ischemic neurons can release lipocalin-2, a “help-me” signal to adjacent neuroglia, resulting in the activation of pathways that ultimately improve the resistance of the neurons to ischemic insults and promote neurovascular recovery (Xing et al., 2014; Wu et al., 2015). Additional studies are needed to define the gamut of ischemic neuronal help-me signals that may be produced alongside IL-4.
IL-4 as a potential therapeutic may have significant clinical relevance. For more than a decade, rIL-4 has been studied as a potential therapy for various types of human cancers (Ghosh et al., 1993). Previous clinical work suggests that rIL-4 is well tolerated at the low end of the dose range; specifically, doses of 0.25–5 μg/kg are likely safe (Prendiville et al., 1993; Leach et al., 1997; Vokes et al., 1998). Therefore, we chose the 2 μg/kg dose, administered subcutaneously, for our MCA/CCAo studies. Previous pharmacokinetic studies have revealed that, after subcutaneous administration, IL-4 is present in the serum for as long as 8–12 h after its administration, whereas it is rapidly cleared (with a half-life of <1 h) after intravenous administration (Prendiville et al., 1993). Needless to say, additional studies clarifying the optimal dose and therapeutic time window of IL-4 in focal brain ischemia models are warranted.
In conclusion, our study results demonstrate uniquely the following: (1) in response to sublethal ischemia, IL-4 is preferentially produced and secreted by neurons; (2) under conditions simulating ischemia, neuronal IL-4 upregulates IL-4R expression on cultured microglia, allowing for a “feedforward” regulation of these cells; (3) IL-4 via PPARγ-dependent mechanisms stimulates microglial phagocytosis, enabling efficient clearance of apoptotic neurons; (4) IL-4 also upregulates microglial expression of trophic factors; and (5) in an animal model of focal ischemia, systemic administration of rIL-4 reduces ischemic lesion volume and is associated with an improved functional neurologic status at 14 d after stroke. We propose that IL-4 secreted by ischemic neurons represents an endogenous defense mechanism, playing a vital role in the regulation of brain cleanup and repair after stroke. Modulation of IL-4 and its associated pathways could represent a potential target for ischemic stroke treatment.
Footnotes
The project was supported by National Institutes of Health/National Institute of Neurological Disorders and Stroke Grant 1R01NS084292.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Jaroslaw Aronowski, University of Texas– Houston, Medical School, Houston, TX 77030. J.Aronowski{at}uth.tmc.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}