

Abstract
Factors providing trophic support to diverse enteric neuron subtypes remain poorly understood. We tested the hypothesis that hepatocyte growth factor (HGF) and the HGF receptor MET might support some types of enteric neurons. HGF and MET are expressed in fetal and adult enteric nervous system. In vitro, HGF increased enteric neuron differentiation and neurite length, but only if vanishingly small amounts (1 pg/ml) of glial cell line-derived neurotrophic factor were included in culture media. HGF effects were blocked by phosphatidylinositol-3 kinase inhibitor and by MET-blocking antibody. Both of these inhibitors and MEK inhibition reduced neurite length. In adult mice, MET was restricted to a subset of calcitonin gene-related peptide-immunoreactive (IR) myenteric plexus neurons thought to be intrinsic primary afferent neurons (IPANs). Conditional MET kinase domain inactivation (Metfl/fl; Wnt1Cre+) caused a dramatic loss of myenteric plexus MET-IR neurites and 1–1′-dioctodecyl-3,3,3′,3′-tetramethylindocarbocyamine perchlorate (DiI) labeling suggested reduced MET-IR neurite length. In vitro, Metfl/fl; Wnt1Cre+ mouse bowel had markedly reduced peristalsis in response to mucosal deformation, but normal response to radial muscle stretch. However, whole-bowel transit, small-bowel transit, and colonic-bead expulsion were normal in Metfl/fl; Wnt1Cre+ mice. Finally, Metfl/fl; Wnt1Cre+ mice had more bowel injury and reduced epithelial cell proliferation compared with WT animals after dextran sodium sulfate treatment. These results suggest that HGF/MET signaling is important for development and function of a subset IPANs and that these cells regulate intestinal motility and epithelial cell proliferation in response to bowel injury.
SIGNIFICANCE STATEMENT The enteric nervous system has many neuronal subtypes that coordinate and control intestinal activity. Trophic factors that support these neuron types and enhance neurite growth after fetal development are not well understood. We show that a subset of adult calcitonin gene-related peptide (CGRP)-expressing myenteric neurons produce MET, the receptor for hepatocyte growth factor, and that loss of MET activity affects peristalsis in response to mucosal stroking, reduces MET-immunoreactive neurites, and increases susceptibility to dextran sodium sulfate-induced bowel injury. These observations may be relevant for understanding and treating intestinal motility disorders and also suggest that enhancing the activity of MET-expressing CGRP neurons might be a useful strategy to reduce bowel inflammation.
- calcitonin gene-related peptide
- dextran sodium sulfate (DSS)
- enteric nervous system
- hepatocyte growth factor
- intrinsic primary afferent neurons
- MET
Introduction
Survival depends on controlled intestinal motility to mix food with digestive enzymes, bring nutrients into contact with gut epithelium, eliminate waste, and facilitate fluid reabsorption. This requires neuronal networks that sense stretch, villus distortion, and luminal content composition, and then alter motility to suit constantly changing conditions. Fortunately, this occurs without conscious thought because the bowel has an intrinsic nervous system called the enteric nervous system (ENS), which controls most aspects of intestinal function (Bornstein et al., 2004; Grundy and Schemann, 2005; Wood, 2008; Furness, 2012; Sasselli et al., 2012; Goldstein et al., 2013). The ENS contains ≥20 neuron subtypes that differ in function, neurotransmitters, axonal projections, and electrophysiology (Furness, 2006b). Some signals governing ENS development and maintenance are known (Sasselli et al., 2012; Lake and Heuckeroth, 2013; Obermayr et al., 2013), but it is unclear how diverse neuronal populations are established or what factors support most enteric neurons after birth. Trophic factors that affect ENS development, maintenance, and function include glial cell line-derived neurotrophic factor (GDNF; Moore et al., 1996; Sánchez et al., 1996; Treanor et al., 1996; Chalazonitis et al., 1998; Hearn et al., 1998; Heuckeroth et al., 1998), neurturin (Heuckeroth et al., 1998, 1999), nerve growth factor (NGF; Mulholland et al., 1994), brain derived neurotrophic factor (Grider et al., 1997b; Boesmans et al., 2008), ciliary neurotrophic factor (Grider et al., 1997a; Chalazonitis et al., 2001; Schäfer et al., 2003), and neurotrophin-3 (Chalazonitis et al., 1994, 1998, 2001). We hypothesized, that hepatocyte growth factor (HGF) and its receptor MET might also be important because HGF supports spinal motor neurons (Ebens et al., 1996), dorsal root ganglion (DRG) subtypes (Maina et al., 1997), retinal ganglion cells (Tönges et al., 2011), and hippocampal neurons (Lim and Walikonis, 2008). Our prior studies also suggested HGF expression in the ENS (Vohra et al., 2006). Finally, we were intrigued by the protective effect of HGF in rodent colitis models (Tahara et al., 2003; Mukoyama et al., 2005; Numata et al., 2005; Oh et al., 2005; Hanawa et al., 2006; Kanbe et al., 2006), and hypothesized that this might be mediated through MET-expressing enteric neurons.
We now demonstrate MET immunoreactivity in most ENS precursors and in a subset of adult calcitonin gene-related peptide (CGRP)-expressing myenteric neurons thought to be intrinsic primary afferent neurons (IPANs; i.e., sensory neurons; Furness et al., 2004a,b). In vitro, HGF/MET signaling influences ENS precursor neurite growth and neuronal differentiation, but conditional Met-null mutations driven by Wnt1Cre [i.e., Metfl/fl; Wnt1Cre+ (Met cKO)] did not cause major ENS developmental defects. Met cKO mice had a normal density of MET-immunoreactive (IR) myenteric neurons, but fewer or shorter MET-IR neurites. Met cKO mice also had a specific defect in the peristaltic response elicited by mechanical deformation of intestinal villi. However, in vivo tests of bowel motility were unaltered. Finally, Met cKO mice had increased susceptibility to dextran sodium sulfate (DSS)-induced mucosal damage, suggesting that CGRP-expressing enteric neurons protect the bowel from injury and that HGF's ability to protect the bowel might depend on signaling within the ENS.
Materials and Methods
Animals.
c-Metfl/WT mice (129SV/C57BL/6 background; Huh et al., 2004) were generously provided by Dr. Snorri S. Thorgeirsson (National Cancer Institute, National Institutes of Health, Bethesda, MD). Wnt1Cre mice [STOCK Tg(Wnt1-Cre)11Rth Tg(Wnt1-GAL4)11Rth/J, Stock #003829, C57BL/6; Swiss albino mixed background] and R26R-EYFP reporter mice (B6.129X1-Gt(ROSA)26Sortm1(EYFP)Cos/J, Stock #006148, C57BL/6J) were from The Jackson Laboratory. RET-EGFP mice were previously described (Jain et al., 2006). Etv5M/M and Etv4 −/−;Etv5lacZ/WT mice were kindly provided by Dr. Kenneth Murphy (Washington University School of Medicine, St. Louis, MO) and Dr. Silvia Arber (University of Basel, Switzerland). CF-1 mice were from Charles River. The morning of vaginal plug was considered embryonic day (E) 0.5. Mice of either sex were studied. The use and care of mice were accredited and approved by the Washington University Animal Care Committee and by The Children's Hospital of Philadelphia Research Institute Institutional Animal Care and Use Committee.
Antibodies and reagents.
Primary antibodies for mouse analysis were as follows: p75NTR antibody (rabbit, 1:1000; #AB1554, EMD Millipore), Choline acetyltransferases (ChAT; goat, 1:10; #AB144P, Millipore), calretinin (rabbit, 1:2500; #AB5054, EMD Millipore), HGF (goat, 1:100; #sc-1357, Santa Cruz Biotechnology), HuC/D (mouse, 1:200; #A21272, Invitrogen), GFP (chicken, 1:1000; #GFP-1020, Aves Labs), S100B (rabbit, 1:800; DAKO), PGP9.5 (guinea pig, 1:100; #GP14104, Neuromics), TuJ1 (rabbit, 1:10,000; #PRB-435P, Covance), TuJ1 (mouse; #MMS-410P, 1:100, Covance), RET (goat, 1:800; #GT15002, 1:800, Neuromics), RET (R787) (Rabbit, 1:100; #18121, Immuno-Biological Laboratories), MET (goat, 1:100; AF527, R&D Systems), CGRP (rabbit, 1:100; #C8198, Sigma-Aldrich), phosphohistone 3 (pH3; rabbit, 1:800; #AB06-570, EMD Millipore), neuronal nitric oxide synthase (rabbit, 1:1000; AB#5380, EMD Millipore), substance P (rabbit, 1:1000; Inestar), vasoactive intestinal polypeptide (VIP; rabbit, 1:1000; Peninsula), NF145 (rabbit, 1:100; #AB1987, EMD Millipore). Primary antibodies for human gut tissue were as follows: PGP9.5 (rabbit, 1:100; #7863-0504, Serotec) and c-MET (goat, 1:100; #AF276, R&D Systems). Secondary antibodies were as follows: donkey anti-goat Alexa 594 (1:400; Invitrogen), donkey anti-rabbit Alexa 488 (1:400; Invitrogen), donkey anti-mouse Alexa 647 (1:400; Invitrogen). Tissue culture reagents included GDNF (Creedon et al., 1997), HGF (mouse; #2207-HG, R&D Systems), Neurobasal media (Life Technologies), B27 (Life Technologies), DMEM, glutamine (Fisher), penicillin, and streptomycin (Fisher). Inhibitors were as follows: PD98059 (MEK1 inhibitor; #EI360-0005, Enzo Life Sciences) and LY294002 (PI3K inhibitor; #ST420-0005, Enzo Life Sciences).
Quantitative ENS analysis.
Whole-mount myenteric plexus analysis was performed using 8–12-week-old mice (n = 3–6) as described previously (Wang et al., 2010). Briefly, gut was opened along the mesenteric border, pinned to Sylgard, fixed [4% paraformaldehyde (PFA), 30 min, 25°C], and then dissected to separate muscle layers from submucosa. After immunohistochemistry or NADPH diaphorase staining, quantitative analysis was performed. For CGRP antibody staining, peeled gut muscle layers were cultured with colchicine (0.1 mg/ml; C9754, Sigma-Aldrich), DMEM, glutamine (2 mm), penicillin (100 IU/ml), and streptomycin (100 μg/ml) for 24 h before fixation. Neuronal density was quantified by counting cells within 20 randomly selected 20× fields per mouse. At least three mice of each genotype were analyzed.
Immunohistochemistry and image processing.
After fixation, cells, organs, or peeled gut muscle layers were kept in TBST (100 mm Tris, 150 mm NaCl, 0.5% Triton X-100) for 30 min at 37°C, blocked with 5% donkey serum/TBST (30 min, 37°C), and then incubated with primary antibody (overnight, 4°C). Images were obtained with an Olympus BX60 microscope, Axiocam and AxioVision software (Zeiss) or with Zeiss Axio Imager.A2, AxioCam MRm Rev.3 Camera, and ZEN software. Image processing included only cropping and uniform adjustments of brightness, contrast, and saturation.
Human gut.
Paraformaldehyde-fixed, paraffin-embedded human colon was obtained from the Washington University Digestive Disease Research Core Center after approval from the Institutional Review Board at Washington University School of Medicine. Five micrometer sections were deparaffinized and rehydrated for immunohistochemistry.
One-1′-dioctodecyl-3,3,3′,3′-tetramethylindocarbocyamine perchlorate labeling combined with immunohistochemistry.
Adult mouse bowel was dissected, fixed, and peeled as for quantitative whole-mount analysis. Muscle layers from distal small intestine were cut into 3-cm-long pieces and pinned out on a Sylgard dish. A dissecting pin dipped in NeuroTrace DiI Tissue-Labeling Paste (#N-22880, Life Technologies) was inserted into the middle of each tissue piece. Pierced samples were kept in 4% PFA at 37°C for 3 weeks. Immunohistochemistry for MET was performed as described above, except that instead of Triton X-100, 1000 μg/ml digitonin (#D141, Sigma-Aldrich) was used to permeabilize tissue while preserving 1-1′-dioctodecyl-3,3,3′,3′-tetramethylindocarbocyamine perchlorate (DiI) staining (Matsubayashi et al., 2008). For cell counting, tissue pieces were evaluated using a 5 × 7 grid of 20× fields centered on the pin insertion site. The grid was additionally subdivided into three zones of varying distances from the pin (see Fig. 5 K) and cells within each 20× field were counted.
Dissociated cell culture.
E12.5 CF-1 ENS precursor cells from dissociated bowel were immunoselected with p75NTR antibody (1:1000) and maintained in culture as previously described (Sato and Heuckeroth, 2008) except that GDNF was not included in media for cell dissociation or immunoselection. Briefly, whole bowel was treated with collagenase (0.5 mg/ml) and dispase (0.5 mg/ml), triturated, and filtered through a 40 μm cell strainer before incubation with p75NTR antibody and goat anti-rabbit-coupled paramagnetic beads (Miltenyi Biotec). After separation of p75NTR-expressing cells using a MACS Separation column (Miltenyi Biotec), immunoselected cells were plated at 6000 cells/well on poly-d-lysine and laminin-coated eight-well chamber slides. Cells were cultured in Neurobasal media supplemented with B27 (2%), glutamine (2 mm), penicillin (100 IU/ml), and streptomycin (100 μg/ml) for 48 h before fixation with 4% PFA and analysis by immunohistochemistry.
Slice culture.
E12.5 CF-1 gut slice cultures were performed as described (Fu et al., 2006) with minor modifications. Briefly, 300–500-μm-long small-bowel slices were cultured on fibronectin-coated plastic chamber slides (Nunc Lab-Tek, Thermo Scientific) in DMEM, B27 (2%), glutamine (2 mm), penicillin (100 IU/ml), and streptomycin (100 μg/ml). Immediately after plating, slices were treated with PBS (vehicle), HGF, or GDNF at the indicated concentration for 24 h before fixation (4% PFA, 15 min, 25°C) and processing for immunohistochemistry. For analysis, the distance from the edge of the explant to the most distant TuJ1+ neurites or RET+ cells was determined in ≥3 and ≤8 regions per explant.
In vitro peristaltic response.
The colon of adult mice was opened along mesenteric attachments to form flat sheets and pinned mucosal side up in a three-chambered organ bath as previously described (Grider and Jin, 1994; Grider et al., 2010). Force-displacement transducers were attached to the circular muscle to record ascending contraction in the orad peripheral compartment and descending relaxation in the caudad compartment. A sensory stimulus that initiates the peristaltic reflex was applied to the bowel in the central chamber. We used a hook-and-pulley system to produce graded (2–8 g) radial stretch of the circular muscle layer to test the stretch-activated sensory pathway. Mechanical deformation of villi was tested using graded mucosal stroking with a fine brush to stimulate the mucosal-activated pathway.
Whole gastrointestinal transit assay.
Adult mice were fed by intragastric gavage with 300 μl of 6% carmine red dye solution (#C1022, Sigma-Aldrich) dissolved in distilled water containing 0.5% methylcellulose (#274429, Sigma-Aldrich). Mice then were placed into individual cages without bedding. A white sheet of paper covered the cage bottom to facilitate detection of carmine in fecal pellets. Following gavage, cage bottoms were checked for dyed fecal pellets at 10 min intervals. Each mouse was tested three times with ≥3 d between tests.
Colon motility assay.
After adult mice were anesthetized with isoflurane, a fire-polished glass rod (3 mm in diameter, custom made by University of Pennsylvania Glass Shop) was used to insert a glass bead (3 mm in diameter; #Z143928, Sigma-Aldrich) into the rectum, 2 cm from the anal verge. The glass rod and beads were lubricated with sterile corn oil (#C8267, Sigma-Aldrich) before insertion. The time required to eject the bead was measured as an estimate of colonic motility. Each mouse was tested three times with ≥1 d between trials.
Small-intestine transit assay.
Adult mice were fasted overnight and then fed by intragastric gavage with 100 μl of 10 mg/ml fluorescein isothiocyanate-dextran (FITC-dextran; average molecular weight, 70,000; #46945 Sigma-Aldrich) dissolved in distilled water containing 2% methylcellulose. Animals were killed 90 min later and the stomach, small intestine, cecum, and colon were collected in 1× PBS. Small intestine was divided into 10 segments, cecum into two segments, and colon into three segments. Each segment was opened along the mesenteric border without losing luminal content and placed into an individual 1 ml Eppendorf tube containing 500 μl 1× PBS. Tubes were vortexed 15 s and then centrifuged (2000 × g, 10 min) to obtain FITC-dextran-containing supernatant. FITC fluorescence was measured in 100 μl aliquots of supernatant in a 96-well plate using a FilterMax F5 (Molecular Devices) plate reader. Small-intestine transit was evaluated by determining the geometric center of the FITC-dextran in the bowel. The geometric center (Miller et al., 1981) was calculated as follows: geometric center = ∑ (fluorescence in each segment × segment number)/total fluorescence recovered.
DSS injury.
Colitis was induced with DSS (2.5% in drinking water) as previously described (Pull et al., 2005). Control littermate mice were placed in separate cages at the time of the experiment and received water without DSS. Anatomic analysis of the colon was done on day 14 after starting DSS or water with the exception of bromodeoxyuridine (BrdU) experiments, which were completed on day 7. For BrdU studies, mice received intraperitoneal injections (10 mg/ml, 100 μg/g body weight) and were analyzed 1 h later. Colons were pinned flat mucosal side up and fixed (4% PFA, 30 min, 25°C) before gross morphologic analysis. Longitudinal 5 μm sections of paraffin-embedded distal and proximal colon were stained with hematoxylin and eosin for additional analysis.
Quantitative reverse transcriptase PCR.
Total RNA isolated using TRI Reagent (Sigma-Aldrich) and purified using RNeasy Mini kit (Qiagen) was reverse-transcribed using SuperScript II Reverse Transcriptase (Invitrogen). Quantitative reverse transcriptase PCR (qRT-PCR) was performed in duplicate using SYBR green PCR Master mix (Applied Biosystems) and an iCycler iQ (Bio-Rad). Primers are in Table 1.
Primers for qRT-PCR
Statistical analysis
SigmaPlot 11 (Systat Software) was used for statistical analyses. All studies included ≥3 biological replicates. Measurements were made by observers blinded to conditions used for studies. Student's t test or one-way ANOVA with post hoc multiple-comparisons tests (Dunn or Holm–Sidak) were used for statistical analysis. Log-rank testing was performed for analysis of the Kaplan–Meier survival curves. Data are plotted as mean ± SEM for all graphs. For all tests, p < 0.05 was considered significant.
Results
MET and HGF are expressed in a subset of adult enteric neurons
Immunohistochemical analysis of adult mouse small bowel demonstrated that MET is present in 34 ± 6% of HuC/D+ myenteric neurons (Fig. 1 A) and that all MET+ cells express the pan-neuronal marker HuC/D+ (Table 2). As expected, MET immunoreactivity was not detected in S100B+ enteric glia (Fig. 1 B). MET-IR myenteric neurons were also detected in cross sections of human colon, where MET was coexpressed with the neuronal marker PGP9.5 (Fig. 1 C,D). To determine which enteric neuron types express MET, remaining studies used adult mouse small-bowel whole-mount preparations to facilitate analysis of many cells at once. Interestingly, RET and MET were detected in mutually exclusive subsets of myenteric neurons as confirmed by immunohistochemistry (Fig. 1 E) and by using a RET-EGFP reporter mouse thought to faithfully reproduce normal Ret expression patterns (Fig. 1 F). One hundred percent of MET+ neurons were CGRP-IR and 49.9 ± 2.0% of CGRP+ neurons were MET+ (Fig. 1 G–I). MET/calretinin staining demonstrated MET immunoreactivity in 16 ± 5% of calretinin+ cells and that 8 ± 2% of MET+ cells are calretinin+ (Fig. 1 J–L). Consistent with this observation, since ChAT is largely expressed in the same cell population, we also detected MET immunoreactivity in 12 ± 0.1% of ChAT-IR neurons and found that 8 ± 0.1% of MET+ neurons were ChAT+ (Fig. 1 M–O). There was no overlap between MET and NADPH diaphorase-stained nitric oxide-producing neurons (Fig. 1 P–R). Thus, MET appears to be primarily expressed in a subset of adult CGRP-expressing cells that are likely to be IPANs. To determine where the MET ligand HGF was expressed in adult mouse bowel muscle layers, HGF/MET double-label immunohistochemistry was performed. Surprisingly, HGF was detected in 43 ± 11% of MET+ neurons and 100% of the HGF+ neurons had MET immunoreactivity (Fig. 1 S–U). Finally, MET and HGF were also detected in cross sections of E14.5 fetal bowel (Fig. 1 V–X). HGF immunoreactivity (Fig. 1 V) was prominent in the mesenchymal cells that surround the developing ENS (seen with TuJ1 in Fig. 1 W) and also was detected at lower levels in developing gut submucosa. MET immunoreactivity was present in the region of the developing ENS, but prominent signal was also detected in gut epithelium and in the mesenchymal cells surrounding the ENS, as well as in developing enteric neurons (Fig. 1 X). This fetal immunoreactivity for HGF and MET suggested that these proteins might have roles during development as well as in the adult ENS.

MET immunoreactivity was detected in a subset of myenteric plexus CGRP+ IPANs. A , MET immunoreactivity was detected in 34 ± 6% of HuC/D+ myenteric neurons of the adult mouse small bowel. All MET+ cells expressed the pan-neuronal marker HuC/D. B , MET staining was absent from S100B+ enteric glia. C , D , MET was detected in human myenteric neurons using colon cross sections. E , F , MET and RET are detected in mutually exclusive sets of neurons as confirmed by immunohistochemistry ( E ) and by using a RET-EGFP reporter mouse ( F ). G–I , 100% of MET+ neurons were CGRP+ and 50% of CGRP+ neurons were MET+. J–L , MET was also found in 16 ± 5% of calretinin+ cells and 8 ± 2% of MET+ cells are calretinin+. Arrows highlight a MET+ calretinin+ neuron. M–O , 12 ± 0.1% of ChAT+ cells were MET+ and 8 ± 0.1% of MET+ neurons were ChAT+. P–R , There was no overlap between MET and NADPH diaphorase-stained nitric oxide-producing neurons. S–U , HGF was detected in 43 ± 11% of MET+ neurons and 100% of the HGF+ neurons were MET+. V , In cross sections of E14.5 fetal bowel, HGF-IR was found in mesenchymal cells surrounding developing ENS stained with TuJ1 ( W ). X , At E14.5, MET+ cells were present in the region of developing ENS as well as in gut epithelium and mesenchymal cells. Scale bars in U applies to A , B , E–U . Scale bar in D applies to C and D . Scale bar in X applies to V–X . N ≥ 3 replicates/staining condition.
Immunohistochemical localization of MET and HGF in the adult mouse small-bowel myenteric plexus
HGF and MET signaling support fetal ENS neurogenesis and neurite growth in vitro
To determine whether HGF/MET signaling could affect fetal ENS development, we cultured ENS precursors from E12.5 bowel after immunoselection with p75NTR antibody. In these dissociated cell cultures, we initially tried adding HGF at a range of concentrations (0–100 ng/ml) to Neurobasal media, with B-27 supplement and l-glutamine, but in the absence of added GDNF, ENS precursors grew poorly or died and there was no evidence that HGF had any effect. When we tried including GDNF at commonly used concentrations (e.g., 50 or 100 ng/ml) in the media, the trophic effects of GDNF were so strong that no additional effect of HGF could be discerned. Recognizing that the concentrations of GDNF used commonly in culture are higher than the ED50 for GDNF-induced ENS precursor proliferation (1.5 ng/ml; Heuckeroth et al., 1998) and dramatically higher than the dissociation constant value (Kd) for GDNF binding to GFRα1 (30 pg/ml; Jing et al., 1996), we decided to titrate the GDNF to low levels that might be more physiologic, yet support ENS precursor survival and permit effects of HGF to be observed. Under these conditions, all TuJ1+ enteric neurons were also MET antibody-IR (Fig. 2 A–C). Remarkably, including GDNF at 1 pg/ml (50,000–100,000-fold less than is typically used in culture) led to robust and dose-dependent effects of HGF (Fig. 2 D,E). ENS precursors were therefore immunoselected with p75NTR and cultured at low density with 1 pg/ml GDNF plus either 0, 1, 20, 50, or 100 ng/ml HGF. In cultures containing only low levels of GDNF, there were very few TuJ1+ cells in culture after 48 h. Increasing HGF doses led to progressively more TuJ1+ cells in culture, with a sixfold increase in TuJ1+ cells in cultures containing 100 ng/ml HGF plus 1 pg/ml GDNF compared with cells maintained in GDNF alone (Fig. 2 M). Including HGF in culture media also dramatically increased average neurite length compared with 1 pg/ml GDNF alone (Fig. 2 N). We confirmed that the HGF effects are MET dependent using MET-blocking antibody and 50 ng/ml HGF (Fig. 2 E,F,M,N). Under these conditions, the blocking antibody almost completely prevented HGF effects on TuJ1+ cell number and neurite length.

HGF promoted neurogenesis and neurite growth in cultured E12.5 ENS precursor cells. A–C , E12.5 ENS precursors immunoselected with p75NTR antibody were maintained in culture for 48 h in the presence of HGF plus 1 pg/ml GDNF before TuJ1 and/or MET immunohistochemistry and DAPI nuclear staining. All TuJ1+ enteric neurons were MET-IR. D–F , M , N , HGF caused a dose-dependent increase in TuJ1-IR neuron number and neurite length. *p < 0.05, ANOVA with Dunn's multiple-comparison test. F , M , N , MET blocking antibody (Aby) reduced TuJ1+ neuron number and neurite length in surviving cells. Control (Ctrl) is 50 ng/ml HGF plus 1 pg/ml GDNF. **p < 0.01, Student's t test. G–L , O , P , When ENS precursors were grown in GDNF alone, the MEK inhibitor PD98059 (PD) had no effect on neuron number or neurite length, but the PI-3K inhibitor LY294002 (LY) reduced neuron number and neurite length. In contrast, in HGF (50 ng/ml) plus GDNF (1 pg/ml)-treated cells, both MEK and PI-3K inhibition reduced neurite length ( P ), whereas only PI-3K inhibition reduced neuron number ( O ). *p < 0.01, ANOVA with Dunn's multiple-comparison test. Scale bar in C applies to A–F . Scale bar in L applies to G–L . (N ≥ 3 biological replicates/group; 12 individual wells/group).
Given the similarity of HGF and GDNF effects on the number of TuJ1+ cells in culture and on neurite length, we hypothesized that GDNF and HGF depend on the same signaling pathways to support neurogenesis and neurite growth. We previously reported, using rat enteric neurons, that GDNF-induced increases in neuron number and neurite length depend on phosphatidylinositol 3-kinase (PI3K), but not MEK signaling (Srinivasan et al., 2005). Using the MEK inhibitor PD98059 and the PI3K inhibitor LY294002, we confirmed these findings in mice using p75NTR-immunoselected E12.5 ENS precursors in culture (Fig. 2 G–I,O,P). Similar to the results obtained using GDNF alone at 50 ng/ml, the number of TuJ1+ cells present after 48 h in culture with HGF (50 ng/ml) plus GDNF (1 pg/ml) was reduced by the PI3K inhibitor, but not by the MEK inhibitor. In contrast, neurite length was reduced by both PI3K and MEK inhibitors in HGF-containing cultures (Fig. 2 J–L), but only by the PI3K inhibitor in the GDNF (50 ng/ml) cultures (Fig. 2 P). This difference in downstream effectors suggests that HGF/MET-induced neurite growth and GDNF/RET-induced neurite growth support ENS precursors via partially overlapping signaling pathways.
HGF/MET signaling enhances ENS precursor differentiation into neurons in vitro
The increase in TuJ1+ cells in dissociated ENS precursor cultures in response to HGF plus 1 pg/ml of GDNF could occur because of increased precursor proliferation, reduced cell death, or enhanced differentiation of RET+/TuJ1− precursors into RET+/TuJ1+ cells. To distinguish between these possibilities, dissociated p75NTR-immunoselected cells from E12.5 mouse bowel were cultured in media with GDNF (1 pg/ml) with and without HGF (50 ng/ml) for 48 h and then stained with TuJ1, RET, and pH3 antibodies (Fig. 3 A–H). RET is expressed in ENS precursors and differentiated neurons and TuJ1 immunoreactivity is a marker of neuronal differentiation, whereas pH3 identifies mitotic cells. While the total number of RET+ cells was not significantly altered with HGF (Fig. 3 I), the number of TuJ1+ cells and the proportion of RET+ cells that are TuJ1-IR was increased by HGF (Fig. 3 J,K). HGF also decreased the number of RET+TuJ1− cells in culture, and reduced the number of dividing (pH3+/RET+) ENS precursors (Fig. 3 L). Collectively these data suggest that HGF increased the number of TuJ1+ enteric neurons in vitro by enhancing ENS precursor differentiation into neurons instead of through increased precursor proliferation or survival.

HGF/MET signaling enhanced ENS precursor differentiation into neurons in vitro. A–H , E12.5 ENS precursors were maintained in culture after p75NTR immunoselection for 2 d in the presence or absence of 50 ng/ml HGF before immunohistochemistry using RET ( A , B ), TuJ1 ( C , D ), and pH3 ( E , F ) antibodies as well as DAPI nuclear staining ( A–H ). G , H , Merged images. I–K , While the total number of RET+ cells was not altered by HGF ( I ), the total number of TuJ1+ neurons ( J ) and the percentage of RET+ cells that were TuJ1+ ( K ) increased with HGF treatment. L , The number of dividing precursor cells (pH3 and RET double positive) decreased with HGF treatment, suggesting that HGF increased neuronal differentiation and decreased proliferation. White arrows, Nonmitotic RET+ pH3− ENS precursors. Yellow arrow, Mitotic RET+PH3+ ENS precursors. White arrowhead, RET+TuJ1+ neurons. Scale bar in G applies to all images (N = 3 biological replicates/group; 12 individual wells/group; *p < 0.01, Student's t test).
HGF/MET signaling did not increase ENS precursor migration in vitro
Since HGF and MET are expressed in fetal bowel when ENS precursors migrate and have well known effects on migration of other neuronal cell types (Giacobini et al., 2007; Garzotto et al., 2008), we hypothesized that HGF/MET signaling might influence ENS precursor migration. To test this hypothesis, E12.5 gut slices were cultured on fibronectin-coated culture dishes and ENS precursors were allowed to migrate from the slice onto the culture dish for 24 h. Addition of GDNF (100 ng/ml) to the media markedly increased the distance that ENS precursors migrated onto the culture dish (Fig. 4). In contrast, HGF (50 or 100 ng/ml) did not increase enteric neural crest-derived cell (ENCDC) migration onto the culture dish compared with no added factor, suggesting that HGF may not be needed for ENCDC migration in vivo.

HGF/MET signaling did not increase ENS precursor migration in culture. A–C , E12.5 gut slices were cultured 24 h on fibronectin-coated dishes with no added factor, HGF, or GDNF before staining for RET and TuJ1. A , ENCDCs migrate from the gut slice onto the culture dish even without any added factors. B , HGF did not increase the distance that ENCDCs migrated from the edge of the gut slice. C , GDNF markedly increased the distance ENCDCs migrate from the edge of the gut slice. D , Quantitative data (no added factor, 15 slices; 50 ng/ml HGF, 14 slices; 100 ng/ml HGF, 19 slices; 100 ng/ml GDNF, 30 slices; N = 3 independent experiments). *p < 0.001 for GDNF versus no added factor, ANOVA with Dunn's multiple-comparison test.
MET inactivation within ENS precursors causes selective defects in MET-expressing enteric neurons
Met −/− mice die in utero between E13.5 and E16.5 (Huh et al., 2004). Therefore, to investigate the role of HGF/MET signaling in the ENS in vivo, we bred mice with LoxP sites surrounding Met exon 16 to Wnt1Cre transgenic animals to generate Met cKO mice (Metfl/fl; Wnt1Cre+). CRE-dependent recombination inactivates MET by removing the intracellular kinase domain, but should permit production of a truncated protein that includes the extracellular and transmembrane domain. The Wnt1Cre transgene is expressed in the developing neural tube and neural crest derivatives, including the ENS, but not in other cells within the bowel. Thus Met cKO mice should have selective loss of MET activity in ENS precursors within the bowel without affecting other intestinal cell lineages. Met cKO mice survive to adulthood and are born at rates that are not statistically different from expected Mendelian ratios (p > 0.99, χ2 test).
To test the hypothesis that Met mutations might slow migration of ENS precursors down the fetal bowel, we evaluated ENS structure in Met cKO and control littermates (Met WT or CRE deficient) using TuJ1 and RET antibodies. Consistent with in vitro migration studies, control and Met cKO mice had ENCDCs in the entire small bowel and half the colon at E12.5 and there were no obvious differences in ENS structure (data not shown). We then examined the adult mouse myenteric plexus to test the hypothesis that HGF/MET signaling influences the development of MET-expressing CGRP+ enteric neurons. Using an antibody to the MET extracellular domain to stain the myenteric plexus, we found that MET+ neuronal cell bodies are easy to identify in Met cKO mice and that these cells were normal in size (Fig. 5 A) and abundance (Fig. 5 B). In contrast, MET-IR interganglionic and intraganglionic neurites are very difficult to see in Met cKO mice compared with control animals (Fig. 5 C,D). To determine whether this staining pattern reflects a difference in neurite length for myenteric plexus MET-expressing neurons, we performed DiI labeling combined with MET immunohistochemistry. DiI paste on a dissecting pin was inserted into fixed small-bowel muscle layers containing the myenteric plexus and tissue was incubated for 3 weeks. DiI taken up by axons undergoes passive retrograde diffusion through lipid membranes to label cell bodies away from the pin site (Fig. 5 E–J). Costaining of DiI-labeled samples with MET antibody revealed that the number of DiI and MET-double-positive cell bodies was dramatically reduced in Met cKO mice compared with controls, especially as the distance from the DiI-labeling site increased (Fig. 5 K–N). In contrast, the average number of MET-positive cell bodies was not different at varying distances away from the pin site (Fig. 5 M,O). Furthermore, the number of MET-negative neuron cell bodies labeled by DiI did not significantly differ in WT and Met cKO mice (data not shown). These data suggest that neurites in MET-IR myenteric neurons of Met cKO mice are shorter than in WT mice.
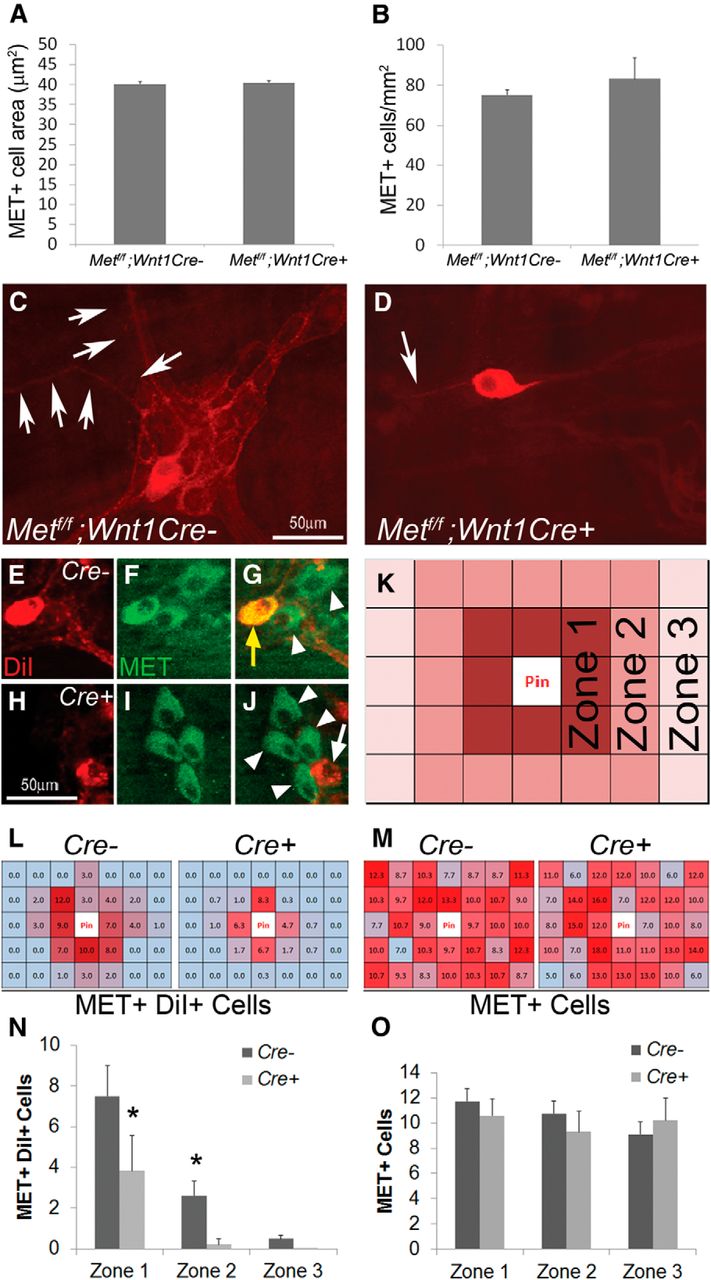
MET deletion within ENS precursors caused selective defects in MET-expressing enteric neurons in adult mice. A , B , The myenteric plexus of mice lacking functional MET receptors in the ENS (Mettf/f ; Wnt1Cre+) contained MET+ neuronal cell bodies of normal size and abundance (N = 3 mice of each genotype/condition). C , D , Mettf/f ; Wnt1Cre+ mice had few interganglionic and intraganglionic MET-IR neurites (N = 6 mice of each genotype/condition). E–J , Double-labeling for myenteric neurons combining DiI labeling and MET immunohistochemistry. Cells shown were two 20× fields away from the DiI application site. Yellow arrow, DiI+ MET+ cell body. White arrow, DiI+ MET− cell body. White arrowheads, DiI− MET+ cell bodies (N = 4 mice of each genotype; N > 8 distal small-bowel DiI-labeled regions/genotype). K , Schematic for analysis of DiI/MET-labeled samples: tissue pieces were divided into a 5 × 7 grid of 20× fields centered on the pin insertion site. The grid was additionally subdivided into three zones of varying distances from the pin. L , M , Heat map representations of the average number of MET+ DiI+ cells ( L ) and total MET+ cells ( M ) in each 20× field of the grid. Dark red > light red > blue for the number of cells in each region. N , The number of DiI+ MET+ cell bodies was dramatically reduced in Met cKO mice compared with controls in Zones 1 and 2. *p < 0.02 (Student's t test). O , The number of total MET+ cell bodies did not differ at varying distances from the pin site.
Functional analysis of gut motility
To test the hypothesis that HGF/MET signaling is important for intestinal motility, the peristaltic response to stretch or to mechanical stimulation of the mucosal lining was measured in WT and Met cKO mice using an oxygenated three-compartment organ bath. These studies showed that the ascending and descending components of the peristaltic reflex were strongly blunted in Metfl/fl; Wnt1Cre+ mice in response to gentle brushing of the mucosa (Fig. 6 A,B,E). In contrast, both components were similar in WT and Met cKO mice when the peristaltic reflex was elicited by muscle stretch (Fig. 6 C,D). These data suggest that Met mutations cause a selective defect in one of the sensory signaling modalities capable of initiating peristalsis (i.e., the reflex elicited by mucosal stimulation). Despite this defined defect in the peristaltic reflex, whole gastrointestinal transit as measured using carmine dye (Fig. 6 F), gastric emptying and small-intestine transit measured by FITC-dextran gavage (Fig. 6 G), and colonic motility measured by expulsion of a glass bead from the rectum (Fig. 6 H) were not altered in Met cKO mice compared with controls. These data suggest that intact sensory response to villus deformation is not required for normal transit of luminal contents through the bowel.

MET deletion within ENS precursors resulted in selective defects in the sensory arm of the peristaltic response. A , B , E , Mice lacking functional MET receptors in the ENS (Metfl/fl; Wnt1 Cre+) had an abnormal peristaltic reflex in response to mechanical stimulation of the villi, as evidenced by a severely blunted ascending contraction ( A ) and descending relaxation ( B ). C , D , In contrast, the peristaltic reflex elicited by circular muscle stretch had normal ascending contraction ( C ) and descending relaxation ( D ) in Metfl/fl;Wnt1 Cre+ mice, suggesting a selective sensory defect (N = 3 mice/genotype; *p < 0.01, Student's t test). F , Whole gastrointestinal transit as measured by the time needed to pass orally gavage-fed carmine dye in stool was not altered in Metfl/fl;Wnt1 Cre+ mice (N = 8 Met cKO and 7 control mice). G , Small-bowel transit as measured by determining the geometric center of FITC-dextran within the bowel 90 min following oral gavage was not altered in Metfl/fl;Wnt1 Cre+ mice (N = 4 Met cKO and 4 control mice). H , Colonic transit, as measured by the time taken to expel a bead placed 2 cm into the distal colon was not altered in Metfl/fl;Wnt1 Cre+ mice (N = 8 Met cKO and 7 control mice).
HGF/MET signaling and bowel injury
HGF/MET signaling potently reduces bowel injury in response to the toxins DSS and 2,4,6-trinitrobenzene sulfonic acid in rodent models and supports epithelial cell proliferation (Tahara et al., 2003; Mukoyama et al., 2005; Numata et al., 2005; Oh et al., 2005; Hanawa et al., 2006; Kanbe et al., 2006; Setoyama et al., 2011). We hypothesized that these effects might be mediated by the newly discovered MET-expressing neurons. To test this hypothesis, we treated Metfl/fl; Wnt1Cre+ mice or control littermate animals with 2.5% DSS in drinking water and analyzed the colon after 2 weeks of DSS treatment. Metfl/fl; Wnt1Cre+ mice had significantly more mucosal damage than control animals after 14 d of DSS treatment (Fig. 7 A–F) and higher death rates (Fig. 7 G). Because increased damage in specific models, including DSS, is linked to diminished epithelial proliferation, we examined intestinal stem and progenitor cell proliferation and found that Met cKO mice had reduced proliferation compared with control animals after 7 d of DSS (Fig. 7 H–J). Collectively these data suggest that HGF/MET signaling protects the intestinal mucosa from DSS-induced injury through the activity of MET-expressing enteric neurons since the only cells in the bowel that express CRE in this model are in the ENS.

MET inactivation in ENS precursors increased mucosal injury in response to DSS treatment. A–F , Metfl/fl; Wnt1Cre+ mice and Metfl/fl; Wnt1Cre control animals were treated with 2.5% DSS in drinking water for 14 d and then examined using a dissecting microscope ( A , B ) or after paraffin sectioning and hematoxylin and eosin staining ( C , D ). E , F , Quantitative analysis of ulcer area in the descending colon and ulcer length in the rectum demonstrated increased ulcers in Metf/f; Wnt1Cre+ mice compared with controls. *p < 0.01, Student's t test. G , Kaplan–Meier analysis demonstrated that DSS-treated Metfl/fl; Wnt1Cre+ mice had higher death rates than controls (N = 8 Met cKO and 11 control mice). p < 0.05, log-rank test. H–J , BrdU labeling after 7 d of DSS treatment showed reduced colonic epithelial cell proliferation within crypts of Metf/f; Wnt1Cre mice compared with control animals. *p < 0.01, Student's t test. Yellow arrows: ulcerated regions (N = 5 Met cKO and 4 control animals/group for ulcer analysis and BrdU labeling).
GDNF/RET signaling increased Met and Etv5 mRNA in cultured ENS precursors, but MET protein levels in vivo do not depend on Etv5/Etv4
The requirement for very small amounts of GDNF to detect any HGF effects on neurite growth and neuron numbers is striking, especially since MET and RET are closely related tyrosine kinase receptors. One possible explanation is that GDNF/RET signaling is needed to induce Met expression in cultured ENS precursors as occurs in the kidney and motor neurons via the Etv4 (Pea3) and Etv5 (Erm) transcription factors (Haase et al., 2002; Livet et al., 2002; Lu et al., 2009; Kuure et al., 2010). To test this hypothesis, we cultured E12.5 immunoselected ENS precursors for 18 h with or without 1 pg/ml GDNF. Although extended culture without GDNF results in death of ENS precursors, there were many healthy-appearing ENS precursors in the GDNF-deprived cultures and in cultures containing 1 pg/ml of GDNF when the mRNA was collected after 18 h in culture. Relative mRNA levels for Etv4, Etv5, and Met were analyzed by real-time qRT-PCR. We found that compared with cells cultured without GDNF, ENS precursors grown with 1 pg/ml GDNF had an eightfold increase in Etv5 mRNA, a 230-fold increase in Met mRNA, but no change in Etv4 mRNA. These data suggest that low levels of GDNF induced Met expression, allowing HGF to affect ENS precursor development (Fig. 8 A,B).

GDNF/RET signaling increased Met and Etv5 mRNA in cultured ENS precursors, but MET protein immunoreactivity appeared normal in mice with Etv4 and Etv5 mutations. A , ENS precursors grown with 1 pg/ml GDNF had an eightfold increase in Etv5 mRNA, but not in Etv4 mRNA when compared with precursors cultured without GDNF. B , ENS precursors grown with 1 pg/ml GDNF also had a 230-fold increase in Met mRNA (N = 3 biological replicates). C–H , The myenteric plexus of adult Etv5M/M mice and P14 Etv4 −/− ; Etv5lacZ/WT compound mutants appeared grossly normal with no differences in MET-IR neuron density (N = 3 mice for each group). *p < 0.01 (Student's t test).
To further explore the role of ETV5 signaling in MET expression in vivo, we examined MET expression in the ENS of Etv5M/M animals, as well as in Etv4 −/− ; Etv5lacZ/WT compound mutants (Lu et al., 2009). Etv5M/M allele is a weak allele that permits survival to adulthood. In contrast, the Etv5lacZ allele used in the Etv4 −/− ; Etv5lacZ/WT mice causes early fetal lethality, prohibiting analysis of the Etv5 lacZ/lacZ ENS. We found that the ENS of Etv5M/M and Etv4 −/− ; Etv5lacZ/WT compound mutants was grossly normal, with MET-IR neuron density comparable to that of WT littermates (Fig. 8 C–H). In contrast, in the developing kidney, where GDNF also induces Met expression, the Etv4 −/− ; Etv5lacZ/WT compound mutants fail to express Met in the ureteric bud, causing serious defects in renal development (Lu et al., 2009). This suggests that ETV4 and ETV5 are dispensable for MET expression in enteric neurons or that at single allele of Etv5 is adequate for MET expression in the ENS.
Discussion
HGF enhanced fetal enteric neuron differentiation and neurite growth in vitro, but did not affect ENS precursor migration from gut slices or bowel colonization by ENS precursors in vivo. In adults, MET immunoreactivity was found in a subset CGRP+ myenteric neurons thought to be IPANs (Qu et al., 2008). MET-IR neuron density was normal in Met cKO mice, but MET-IR neurites were short and sparse. Met cKO mice also had reduced peristalsis after mucosal deformation and increased mucosal injury after DSS exposure. These data suggest HGF and MET support a subset of CGRP-expressing IPANs that regulate intestinal motility and epithelial function.
HGF, MET, GDNF, and RET
MET influences many cellular functions (Trusolino et al., 2010), including neuron survival (spinal motor, sympathetic, sensory, and retinal neurons; Ebens et al., 1996; Maina et al., 1997; Thompson et al., 2004; Lamballe et al., 2011; Tönges et al., 2011), differentiation (sensory and hippocampal neurons; Maina et al., 1997; Lim and Walikonis, 2008), axon outgrowth and guidance (spinal motor, gonadotropin-releasing hormone (GNRH), sensory, and retinal neurons; Ebens et al., 1996; Maina et al., 1997; Giacobini et al., 2007; Tönges et al., 2011), precursor migration (cortical and GNRH neurons; Giacobini et al., 2007; Garzotto et al., 2008), and synaptic plasticity (hippocampal neurons; Akimoto et al., 2004). Our data reveal previously unsuspected roles for MET and HGF in the ENS, but there is more to learn. One intriguing question is whether HGF in some MET-IR neurons acts as a chemoattractant to support formation of the extensive network of IPAN-to-IPAN connections (Furness et al., 2004a). HGF is chemoattractive for spinal motor axons (Ebens et al., 1996), but has autocrine roles in sympathetic neurons (Maina et al., 1998) and may have similar ENS functions. Another interesting finding was that HGF effects required small amounts of GDNF, which increased Met and Etv5 mRNA (Lu et al., 2009; Costantini, 2010). In kidney, Met was absent in Etv4 −/− ; Etv5lacZ/WT mice, but MET was readily detected in myenteric neurons, suggesting single Etv5 alleles may support ENS Met expression. Unfortunately, Etv5lacZ/lacZ mice die early (Lu et al., 2009) and milder Etv5M/M mutations did not affect MET-IR myenteric neuron density, so ETV5 function in the ENS remains uncertain. Finally, MET and RET appear in nonoverlapping adult myenteric neuron populations, highlighting the need to define mechanisms restricting receptor tyrosine kinases (e.g., RET, NTRK3, and MET) to specific enteric neuron subtypes (Chalazonitis et al., 1994, 2001; Schuchardt et al., 1994; Heuckeroth et al., 1999; Uesaka et al., 2007, 2008).
HGF/MET and ENS development
The requirement for 1 pg/ml GDNF to observe HGF effects on ENS precursors is reminiscent of synergistic HGF and NGF effects in DRG. HGF alone did not support DRG neuron survival or axon outgrowth, but HGF enhanced survival, differentiation, and axonogenesis with NGF present (Maina et al., 1997, 1998). In the ENS, low GDNF levels maintain cells as progenitors (Uesaka et al., 2013) and may support survival while enhancing HGF responsiveness. In contrast, high GDNF triggers neuronal differentiation and migration, masking HGF effects. GDNF concentrations in vivo are unknown but are probably below the 50–100 ng/ml commonly used in vitro, since increased and reduced GDNF alters enteric neuron number (Gianino et al., 2003; Wang et al., 2010) and the Kd for GDNF binding to GFRα1 is only 30 pg/ml (Jing et al., 1996). These observations suggest that it may be appropriate to evaluate how other factors affect ENS precursors in the presence of low concentrations of GDNF instead of the levels typically used in culture.
HGF/MET and IPAN subtypes
Functional data suggest IPANs are heterogeneous (Clerc and Furness, 2004; Furness et al., 2004a; Furness, 2006a). For example, stretch opens IPAN gadolinium-insensitive mechanosensitive ion channels (Kunze et al., 1999), whereas mucosal deformation triggers serotonin and ATP release from enteroendocrine cells to activate IPAN 5HT3/5HT4 or P2X receptors (Grider and Jin, 1994; Pan and Gershon, 2000; Raybould et al., 2004; Patel, 2014). These IPANs all express CGRP. However, only 49% of CGRP+ neurons are MET+. MET signaling is not needed for survival of MET-IR neurons, but Met cKO mice have impaired peristalsis in vitro after mucosal stroking consistent with a functional defect in a subset of IPANs. In contrast, bowel stretch-responsive IPANS are either MET-negative or do not require MET for function. It is not clear whether the selective functional defect results from reduced neurite growth in MET+ neurons or from reduced function. In DRG neurons, for example, MET enhances nociceptor peptidergic differentiation (Gascon et al., 2010). Nonetheless, in contrast to in vitro results, whole-bowel transit, gastric emptying, small-bowel transit, and colonic-bead expulsion were normal in vivo in Met cKO mice. These data add to recent studies suggesting that mucosal deformation-induced peristalsis is not required for normal transit through the bowel, at least when stretch response is intact (Li et al., 2011; Heredia et al., 2013).
HGF, the ENS, and intestinal injury
HGF's ability to reduce bowel injury is fascinating (Tahara et al., 2003; Arthur et al., 2004; Mukoyama et al., 2005; Numata et al., 2005; Oh et al., 2005; Ohda et al., 2005; Hanawa et al., 2006; Kanbe et al., 2006; Gong, 2008; Setoyama et al., 2011). Remarkably, Met cKO mice had increased mucosal damage, reduced epithelial stem and progenitor cell proliferation in response to injury, and increased mortality compared with controls after DSS treatment. Although gut epithelial cells express Met (Prat et al., 1991), Metfl/fl Wnt1Cre mice do not express CRE in epithelium (Danielian et al., 1998), suggesting that MET effects on epithelial proliferation after DSS are not cell-autonomous.
Many mechanisms might underlie our observations. One hypothesis is that impaired injury response results from reduced CGRP release from MET+ enteric neurons. CGRP mutations increased colonic damage in DSS-treated mice (Thompson et al., 2008). Furthermore, CGRP is a potent vasodilator (Pawlik et al., 2000) and adequate mucosal blood flow may facilitate injured bowel repair. CGRP also supports bowel epithelial proliferation via mast cells and fibroblasts producing transforming growth factor α (Hoffmann et al., 2010) and by regulating gene expression in macrophages (Baliu-Piqué et al., 2014) that influence the set point of intestinal epithelial proliferation (Baliu-Piqué et al., 2014; Sun et al., 2015). MET+ neurons might also support epithelial proliferation via acetylcholine release from ChAT+MET+ neurons, since acetylcholine enhances epithelial growth (Tutton, 1975; Lundgren et al., 2011; Gross et al., 2012).
Consistent with ENS support for bowel epithelium, enteric neurons express the receptor for glucagon-like peptide 2, a potent epithelial mitogen (Bjerknes and Cheng, 2001; Guan et al., 2006), and neuronal serotonin increases epithelial proliferation (Gross et al., 2012). However, myenteric plexus ablation increases epithelial cell proliferation (Zucoloto et al., 1988; Holle, 1991; Hadzijahic et al., 1993; Holle et al., 2003) and hypomorphic Ret +/− mice (Gianino et al., 2003) had increased epithelial proliferation after small-bowel resection (Hitch et al., 2012). These data suggest distinct enteric neuron subtypes enhance or inhibit intestinal epithelial proliferation, but it is unclear how these processes are integrated.
We also note that CRE-induced MET mutations are not restricted to the ENS in Met cKO mice (Danielian et al., 1998). MET should be disrupted in CGRP-expressing DRG neurons (Gascon et al., 2010) that might normally enhance mucosal repair (Takami et al., 2009; Engel et al., 2011, 2012; Lee et al., 2012). MET inactivation in vagal neurons (Freem et al., 2010) could also increase severity of DSS-induced injury (Mazelin et al., 1999; Ghia et al., 2006, 2007; Van Der Zanden et al., 2009) since some vagal nuclei express Met (Caton et al., 2000; Wu and Levitt, 2013). Distinguishing between these possibilities is not straightforward, but these data fit with an emerging literature suggesting neuronal activity regulates intestinal epithelial progenitor proliferation and barrier function (Bjerknes and Cheng, 2001; Nezami and Srinivasan, 2010; Hitch et al., 2012; Sharkey and Savidge, 2014).
It is tempting to speculate that neurogenic control underlies high rates of enterocolitis in children with Hirschsprung disease, a birth defect where the ENS is absent from distal bowel (Frykman and Short, 2012; Heuckeroth, 2013). ENS damage in inflammatory bowel disease or necrotizing enterocolitis may also perpetuate bowel inflammation (Margolis and Gershon, 2009; Zhou et al., 2013). Indeed, many ENS transmitters affect bowel inflammation and injury, including CGRP (Eysselein et al., 1992; Wang et al., 2006; Ramachandran et al., 2013), serotonin (Bischoff et al., 2009; Gershon, 2012), neuropeptide Y, VIP (Chandrasekharan et al., 2013), and substance P (Landau et al., 2007), as do enteric glia (Bush et al., 1998; Savidge et al., 2007), enteric neuron density (Margolis et al., 2011), and toll-like receptor 2 (Brun et al., 2013). Our data reinforce this literature and suggest that new therapeutic strategies to treat or prevent intestinal motility or bowel inflammatory diseases may be targeted to the nervous system instead of the immune system.
Footnotes
-
This work was supported by the Irma and Norman Braman Endowment (R.O.H.); the Suzi and Scott Lustgarten Center Endowment (R.O.H.); the Children's Hospital of Philadelphia Research Institute (R.O.H.); the Children's Discovery Institute of Washington University and St. Louis Children's Hospital (Grants CH-II-1008-123, CH-II-2010-390, MD-II-2013-269; R.O.H.); National Institutes of Health Grants RO1 DK087715 (R.O.H.), RO1 DK34153 (J.R.G.), and RO1 DK071619 (T.S.); the Burroughs Wellcome Fund Clinical Scientist Award in Translational Research (Grant 1008525; R.O.H.); and the Canadian Institutes of Health Research (J.A.H.). We thank Dr. Snorri S. Thorgeirsson, Dr. Silvia Arber, and Dr. Kenneth Murphy for generously sharing mice; the Mouse Genetics Core (http://mgc.wustl.edu) for mouse line maintenance; the Washington University Digestive Disease Research Core (P30DK052574) for human tissues; and Karen Carraro, Research Scientific Glass blower in the University of Pennsylvania Glass Shop.
-
The authors declare no competing financial interests.
- Correspondence should be addressed to Robert O. Heuckeroth, MD, PhD, Professor of Pediatrics, Perelman School of Medicine at the University of Pennsylvania, The Children's Hospital of Philadelphia, Research Institute, Abramson Research Center, 1116i, 3615 Civic Center Blvd, Philadelphia, PA 19104. Heuckerothr{at}email.chop.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}