

Abstract
The understanding of brain diseases requires the identification of the molecular, synaptic, and cellular disruptions underpinning the behavioral features that define the disease. The importance of genes related to synaptic function in brain disease has been implied in studies describing de novo germline mutations and copy number variants. Indeed, de novo copy number variations (deletion or duplication of a chromosomal region) of synaptic genes have been recently implicated as risk factors for mental retardation or autism. Among these genes is GRIK4, a gene coding for a glutamate receptor subunit of the kainate type. Here we show that mice overexpressing grik4 in the forebrain displayed social impairment, enhanced anxiety, and depressive states, accompanied by altered synaptic transmission, showing more efficient information transfer through the hippocampal trisynaptic circuit. Together, these data indicate that a single gene variation in the glutamatergic system results in behavioral symptomatology consistent with autism spectrum disorders as well as in alterations in synaptic function in regions involved in social activity. Autistic features of these mice represent powerful tools for improving diagnosis and testing of specific treatments targeting abnormalities in glutamatergic signaling related to autism spectrum disorders.
SIGNIFICANCE STATEMENT A genetic overlap exists between autism spectrum disorders (ASD), currently thought to represent a continuum of the same disorder with varying degrees of severity, and other neurodevelopmental and neuropsychiatric endophenotypes. We show that the duplication of a single gene coding for a high-affinity kainate receptor subunit (i.e., grik4) in a limited area of the brain recapitulates behavioral endophenotypes seen in humans diagnosed with autism (anhedonia, depression, anxiety, and altered social interaction), including some humans with GRIK4 duplications. Therefore, it should be possible to use mice overexpressing grik4 to directly address circuit dysfunctions associated with ASDs and test specific treatments of autism-related behaviors.
Introduction
During development, the human brain is shaped by sensory inputs and cognitive experiences. The shape and strength of the synapses, essential for brain performance, may be altered by changes in their molecular configuration. In the glutamatergic system, the hyperactivity or hypoactivity induced by several factors may be responsible for the pathophysiology of several major mental disorders. For instance, genes directing synapse function appear to influence the risk of autism, schizophrenia, and other diseases (Gilman et al., 2011; Levy et al., 2011; De Rubeis et al., 2014). These genes include those coding for glutamate receptor subunits of the kainate type, such as GRIK2 and GRIK4 (Poot et al., 2010; Griswold et al., 2012). The importance of these particular genes in brain disease is demonstrated in two recent reports, one that found a chromosomal abnormality that disrupts GRIK4 in an individual with schizophrenia and mental retardation (Pickard et al., 2006), and another that found that the 11q23.3–q24.1 cytoband, where GRIK4 maps, was duplicated de novo in a case of autism (Griswold et al., 2012). In addition, the region of chromosome 11, where GRIK4 maps, seems to be prone to undergo translocations, most often giving rise to a gain of function of the encompassed genes (https://decipher.sanger.ac.uk/search?q=GRIK4#consented-patients/results). Importantly, recent studies indicate that de novo germline mutations and copy number variants represent more significant risk factors for diseases, such as autism spectrum disorders (ASDs), than was previously recognized (Iafrate et al., 2004; Sebat et al., 2007; Ebert and Greenberg, 2013), but functional analyses for these cases are largely absent.
An understanding of brain diseases requires the identification of the molecular, synaptic, and cellular alterations underpinning the behavioral features that define the disease. It seems that kainate receptors (KARs) may have a role in above-mentioned syndromes that are to be delineated. The role played by KARs in brain physiology is much more poorly understood than that of other glutamate receptors, yet it is now clear that KARs play significant roles in synapses and in the maturation of neural circuits during development (Lerma and Marques, 2013; Marques et al., 2013). KARs are formed by five different subunits, GluK1-5 (genes GRIK1-5), of which GluK1-3 can coassemble as homomers or heteromers and give rise to functional receptors (Cui and Mayer, 1999; Paternain et al., 2000). The high-affinity subunits, GluK4 and GluK5, however, must coassemble with GluK1-3 subunits to form functional receptor channels. KAR proteins are expressed strongly in the cortex and hippocampus and it has been established that heteromeric assembly is necessary for KARs to be targeted to the synapse (Fernandes et al., 2009; Palacios-Filardo et al., 2014).
To delineate the role played by the GluK4 subunit (i.e., GRIK4) in human brain disease, we assessed the physiological and behavioral consequences of overexpressing grik4 in the mouse forebrain under the control of the CaMKII promoter, which drove expression of exogenous grik4 only in the forebrain (Mayford et al., 1996). We found altered synaptic transmission in these animals in vivo and in slices, showing more efficient information transfer through the hippocampal trisynaptic circuit. Importantly, these mice displayed anhedonia, enhanced anxiety, and depressive states, as well as impaired social interaction, common endophenotypes associated with ASDs. These data determine that increased expression of a single KAR gene may contribute to ASDs, identifying a critical role for excess of function of KARs in human disease.
Materials and Methods
Animals.
Experimental procedures involving use of live mice were performed in accordance with Spanish and European Union regulations (2010/63/EU), and bioethical committees at both the Instituto de Neurociencias and at the Consejo Superior de Investigaciones Científicas approved them. Animals were housed in ventilated cages in a temperature-controlled, standard pathogen-free environment (23°C), at humidity between 40 and 60%, and on a 12 h light/dark cycle. Mice had ad libitum access to food and water and the cages were changed weekly. The experiments were performed on postnatal day (P) 18–P20 mice for electrophysiological analysis and on ≥4-week-old mice for biochemical characterization. Male 8–12-week-old transgenic and littermate mice were used for behavioral studies and in vivo electrophysiology. All the procedures were done after extensive backcrossing to C57BL/6J. All analyses were performed on mice whose genotype was unknown to the experimenter.
Generation of mouse lines.
A tag containing 5 myc epitopes was added to the cDNA of rat grik4, just after the signal peptide (first 20 aa). The functionality of the pcDNA3 myc grik4 plasmid was assayed through electrophysiological recordings when expressed in HEK293 cells. The myc-grik4 construct to generate the transgenic mice was cloned in two consecutive steps. First, myc-grik4 was cloned as an EcoRV-SmaI fragment into the pNN265 plasmid that contains an intron sequence necessary for the correct functioning of the CaMKII promoter. Subsequently, a NotI fragment containing myc-grik4 and the intronic sequence was introduced into the PMM403 plasmid that has the CaMKII promoter inserted in pBluescript (Mayford et al., 1996). The construct, cut with SfI I restriction enzyme to remove the pBluescript backbone, was injected into the pronucleus of fertilized eggs (performed at the Unitat Animals Transgènics Centre de Biotecnologia Animal i Teràpia Gènica, Universitat Autònoma de Barcelona).
Of the nine founders obtained, one was discarded due to integration into the Y chromosome. The levels of myc-grik4 expression were analyzed in the first generation by in situ hybridization, immunohistochemistry, and Western blots. Two different lines with similar expression levels were chosen for further studies.
Western blots.
Hippocampal and cortical protein lysates were analyzed in Western blots probed with the following primary antibodies: mouse anti-α-tubulin (1:1000; Abcam), mouse anti-myc (1:500; Santa Cruz Biotechnology), rabbit anti-GluR6/7 and rabbit anti-KA2 (1:1000; Millipore), rabbit anti-GluR2/3 (1:1000; Novus Biological), and rabbit anti-KA1 (1:1000; a generous gift from Dr. Melanie Darstein; Darstein et al., 2003). Antibody binding was detected on a luminescent image analyzer (LAS-1000PLUS, Fuji) and quantified with Quantity One 1D Analysis Software (Bio-Rad Laboratories). For quantifications, all densities were normalized to the respective tubulin signal.
Immunofluorescence.
Brains were fixed as described previously (Schneider Gasser et al., 2006) and sectioned in 20 μm slices, which were incubated overnight at 4°C with rabbit anti-myc (1:500; Abcam) and mouse anti-PSD95 (1:500; NeuroMab) in blocking solution (PBS, 0.2% Triton X-100, and 10% NGS). Afterward, sections were incubated with secondary antibodies Alexa 555-conjugated anti-rabbit and Alexa 488-conjugated anti-mouse, both used at 1:1000. Evaluation of immunofluorescence staining where performed using a Leica DMLFSA confocal microscope and the image analysis software Imaris (Bitplane). A low-pass filter was applied to subtract background.
Deglycosylation.
EndoH/PNGaseF was assayed as described previously (Fernandes et al., 2009), whereby 500 units of EndoH (P0702, New England Biolabs) were used to remove high-mannose-containing oligosaccharides, and an equal amount of PNGaseF (P0704, New England Biolabs) was used to remove all N-linked oligosaccharides. After a 1 h digestion, the lysates were analyzed by Western blots probed with mouse anti-myc (1:500; Santa Cruz Biotechnology) and rabbit anti-KA1 (1:1000).
In situ hybridization.
In situ hybridization was performed with 35S-labeled specific oligonucleotide probes as described previously (Wisden and Morris, 2002). As a control for specificity, two independent oligonucleotides for the grik4 gene were hybridized in parallel, producing identical results. The oligonucleotide sequences used were as follows: Grik4 a: 5′-CTTGTAGTTGAACCGTAGGATCTCAGCCAACTCCTTGAGCATGTC-3′; Grik4 b: 5′-TAGCCCGGTCTGCGTCCCATATGAACTCTGTAAAGAATACTA-3′; Myc: 5′-CTCCATTTCATTCAAGTCCTCTTCAGAAATGAGCTTTTGCTCCAT-3′.
Slice preparation and electrophysiological recording.
Parasagittal brain slices (300 μm) were prepared as described previously (Christensen et al., 2004). Electrophysiological recordings were performed from CA3 neurons visually identified by infrared-differential interference contrast microscopy using a 40× water-immersion objective. All experiments were performed at room temperature (22–25°C). Slices were continuously perfused with a solution consisting of the following (in mm): 124 NaCl, 3 KCl, 1.25 KH2PO4, 1 MgSO4, 2 CaCl2, 26 NaHCO3, and 10 glucose, equilibrated with 95% O2/5% CO2, pH 7.3 (300 mOsm), supplemented with antagonists as required. Patch pipettes were filled with the following intracellular solution (in mm): 130 CsMeSO3, 4 NaCl, 10 HEPES, 0.2 EGTA, 10 tetraethylamonium, 2 Na2ATP, 0.5 Na3GTP, 5 QX314, pH 7.3 (287 mOsm). Drugs were applied by gravity, switching between four perfusion lines. To evoke mossy fiber AMPA EPSCs, electric shocks were applied through a monopolar electrode made from a glass pipette placed in the mossy fiber pathway (see Fig. 3A). Tight-seal whole-cell recordings were obtained from the cell bodies of neurons situated in the CA3 pyramidal layer. Mossy fiber responses were identified by the large paired-pulse facilitation. The specificity of mossy fiber stimulation was further assessed by looking at the depression induced by (2S-1′S-2′S)-2-(carboxycyclopropyl)glycine (10 μm), a group II mGluR agonist, which specifically depresses mossy fiber-induced synaptic responses. The perfusion solution was supplemented with picrotoxin (100 μm), APV (50 μm), and LY303070 (25 μm) to isolate spontaneous KAR-mediated EPSCs. To record mEPSCs, tetrodotoxin (1 μm) was added to the solution and LY303070 omitted. All electrophysiological analyses were performed on mice whose genotype was unknown to the experimenter.
Recording in vivo local field potentials.
Two-to-3-month-old male mice were anesthetized with urethane (1.8 g/kg in saline, i.p.) and placed in a stereotaxic device. Small craniotomies (2.1 mm from bregma, 1.2 mm lateral) were performed and a linear Michigan probe of 32 recording contacts, 50 μm apart, were implanted (2.2 mm deep) to record all the way along the cornus ammonis (CA) 1–dentate gyrus (DG) region. The probes were soaked in DiI (Invitrogen) before insertion for postmortem evaluation of their placement in histological sections. A bipolar concentric electrode was implanted to activate the Perforant Pathway (−4.3 mm from bregma, 2.5 mm lateral, and 1.5 mm deep). Signals were amplified and acquired using a 32-channel amplifier (MultiChannel Systems). At the end of the experiment, animals were perfused transcardially with 4% paraformaldehyde, and 100 μm vibratome sections obtained and counterstained with DAPI to visualize the position of the recording electrodes. Analysis of the data was done by Spike2 software.
Behavioral tests.
Behavioral tests were performed on 8–12-week-old mice, using three different cohorts of 18–22 mice each. All the procedures and analyses were conducted in a blind manner, although sometimes the phenotype was evident to the experimenter. To detect changes statistically significant in the phenotype, the number of animals (sample size) to be used was chosen according to the estimated measurement's variability and a prespecified effect (≥2 SD) with a p ≤ 0.05 and a power of 90%, according to the equation provided by the Grants Evaluation Committee of the Universidad Miguel Hernández. All animals were included for analysis, except on one occasion where one animal was killed due to skin damage induced by repetitive scratch. Animals were randomly studied independently of their genotype.
Open field.
The open-field test was conducted in a 50 × 50 cm arena over 30 min, scoring behavior with a video tracking system (Panlab). Differences in the distance traveled, resting time, and the time in the center and periphery of the arena were calculated for GluK4+/+ and GluK4over mice in relation to the total time.
Elevated plus maze.
A metal maze, elevated 50 cm above ground, with two dark and enclosed arms and two open and lit arms, was used to examine anxiety-like behaviors. The arms were 50 × 10 cm in size with a 10 × 10 cm central area. The walls of the closed arms were 30 cm high. Individual mice were placed in the center of the maze, tracked for 10 min with a video camera and then returned to their home cage. The plus maze was wiped clean between trials with a 70% alcohol solution. The time and frequency of visits to the different zones of the maze were scored using Smart Video Tracking System (Panlab).
Y maze.
A Y maze was used to investigate spatial memory. Mice were placed in the center of the “Y” and allowed 10 min to freely explore the three arms of the maze. The alternations were determined by counting the number of successive entries into the three arms divided by the total number of entries.
Forced swimming test.
Transparent Plexiglas cylinders, 50 cm high and with a 25 cm diameter, were filled to ∼25 cm with tap water at 22–24°C such that the mice were not able to touch the floor or escape. Individual mice were placed in the water for a 6 min session and videotaped for later analysis. At the end of each session, the mice were dried with a paper towel and returned to their home cage, and the water was replaced for the next trial. The time of immobility, defined as a lack of active swimming, was scored.
Light/dark test.
The testing apparatus consisted of two rectangular Plexiglas boxes (20 × 20 × 20 cm) connected by a small corridor (6 cm). One was open and lit, while the other compartment was dark. Each mouse was placed in the dark compartment and kept in the arena for 10 min. The time to leave the dark compartment for the first time and the time spent in the lit compartment were measured by video recording and analysis using Smart software (Panlab).
Sucrose-intake test.
Anhedonia-like behavior was evaluated by monitoring sucrose intake. Over a week, animals were given two bottles of drinking water, one with tap water and the other with a 2% solution of sucrose. Following this acclimatization, the mice had the choice of drinking from either the sucrose solution or tap water for a period of 4 d. Water and sucrose solution intake was measured daily, and the position of the two bottles was switched daily to reduce side bias. Sucrose preference was calculated as a percentage of the volume of sucrose intake over the total volume of fluid intake and averaged over the 4 d of testing.
Rotarod.
We used the rotarod from Ugo Basile to test for motor coordination. One day before testing, the animals were trained on the rotarod set at 16 rpm until they could stay on it for 30 s. For testing, the rotarod was set to accelerate from 4 to 40 rpm over the course of 5 min, and the latency to fall off the rod was recorded. The test was performed on 4 consecutive days, three times a day.
Three-chamber social-interaction test.
This test was conducted as described previously (Nadler et al., 2004; Smith et al., 2011; Yang et al., 2011). Briefly, the social apparatus consisted of a rectangular three-chambered box, each chamber measuring 20 cm long × 40.5 cm wide × 22 cm high. The dividing walls were of clear Plexiglas, with small openings (10 × 5 cm) that allowed access to each chamber. The test consisted of four phases. Phase I was a habituation period of 5 min where the mouse moved freely in the central chamber. Phase II consisted of habituating the mice to all three chambers for 5 min with two empty round grid cylinders placed in the corner of each of the lateral rooms. In Phase III, mouse sociability was determined over 5 min by measuring the time spent by the freely moving mouse in proximity to and in the chamber of the grid box enclosure containing a first stranger mouse. Phase IV was the test of preference for social novelty when a second novel mouse was placed inside the previously empty grid cylinder while the first novel mouse remained inside its grid cylinder. Preference for social novelty was defined as the time in the chamber with the novel mouse and the time spent sniffing the novel mouse compared with the time spent sniffing the familiar mouse. Tracks were videotaped and analyzed with Smart software.
Drug administration.
Tianeptine sodium and fluoxetine hydrochloride (Abcam Biochemical) were dissolved in normal saline and administrated at a dose of 10 mg/kg intraperitoneally once daily over 30 d.
Results
We generated mice overexpressing grik4 gene coding for a high-affinity KAR subunit, GluK4, in the forebrain because this gene was found duplicated in a case of autism and may be involved in schizophrenia and other diseases. For that purpose, rat grik4 cDNA was tagged with five myc epitopes and inserted into the PMM403 plasmid that had the CaMKII promoter. Of nine mice lines in which different levels of grik4 expression was evident (GluK4over lines), we selected two with similar level of expression for further work [C57BL/6J-Tg(camk2-grik4)2 and C57BL/6J-Tg(camk2-grik4)3]. Results were identical in both lines and we call them generically GluK4over. In situ hybridization (data not shown) and myc immunocytochemistry (Fig. 1A) reported that, as expected, the transgene was expressed in the forebrain, particularly in the neocortex and hippocampus, but also in the striatum. Also, the glycolytic processing of exogenous GluK4 protein in GluK4over mice was entirely normal (Fig. 1B). Western blots for the GluK4 protein encoded by grik4 revealed that this protein was overexpressed by 40–50% in the hippocampus and ∼100% in the neocortex and striatum (Fig. 1C). Other glutamate receptor subunits were either unaltered in the striatum, neocortex, and hippocampus (e.g., GluK2/3 and GluA2/3) or they were repressed in the hippocampus (e.g., GluK5; Fig. 1D), probably reflecting a process to compensate for the excess of GluK4. Immunocitochemical studies revealed that exogenous GluK4 was expressed in the hippocampus and at both sides of the synapse. Indeed, myc immunoreactivity was not only detected in PSD95-labeled structures (Fig. 2A–D) but also in GFP-labeled mossy fiber terminals, after crossing GluK4-overexpressing mice with transgenic mice expressing GFP in DG granule cells (Feng et al., 2000; Fig. 2E–H). Synaptic transmission was evaluated in mice overexpressing GluK4 in the hippocampal CA3, an area where KARs are expressed abundantly at both the presynaptic and postsynaptic sides. The KAR-mediated EPSCs (EPSCKARs) were isolated by blocking NMDA and AMPA receptors (Fig. 3). Spontaneous EPSCKARs in CA3 pyramidal cells had 38% larger amplitude than in normal mice (5.5 ± 0.2 pA and 7.5 ± 0.3 pA for GluK4+/+ and GluK4over, respectively, n = 41 and 32 neurons; p < 0.001), they were more frequent (0.22 ± 0.04 Hz and 0.59 ± 0.11 Hz for GluK4+/+ and GluK4over, respectively; p = 0.013), and they presented faster deactivation rates (103.3 ± 3.7 ms and 89.7 ± 2.9 ms for GluK4+/+ and GluK4over, respectively; p = 0.035; Fig. 3C,D). This result confirmed that overexpressed GluK4 protein participated in functional KARs at the synapse.

grik4 transgenic mice show increased expression of GluK4 protein. A, Expression of GluK4 transcribed from the transgene in the forebrain detected by anti-myc antibody. B, Normal glycolytic processing of exogenous GluK4 protein in GluK4over mice. C, Quantification by Western blots (top) of native (GluK4; Nat) and transgene-derived (myc-GluK4) protein in the hippocampus (Hpc), neocortex (Ncx), and striatum (Str) of both transgenic mice (Tgm) and their littermates not carrying the transgene (WT). Relative levels of expression were estimated by referring GluK4 band densities to the density of tubulin in each structure. D, Expression levels of kainate receptor subunits (GluK5 and GluK2/3) and AMPA receptor subunits (GluA2/3) in the GluK4over transgenic mice and their wild-type littermates in the cortex, hippocampus, and striatum. Histograms at the bottom reflect the mean ± SEM of measurements from four (Tgm) and three (WT) independent samples run in duplicate (*p < 0.05, Student's t test).

Distribution of myc-GluK4 subunit in hippocampal mossy fiber synapses. A–C, myc-GluK4 (A, B) and PSD95 (C) expression in the hippocampus. D, Overlay of B and C. Colocalization of both proteins (yellow) in the stratum lucidum (Lu) suggests the presence of GluK4 in postsynaptic structures. E, F, Cofocal sections through the hippocampus of a mouse expressing GFP in the granule cells of the dentate gyrus (E) and therefore in mossy fiber boutons (F). G, Overlay of GFP-expressing mossy fibers (F) and GluK4 red immunoreactivity (similar section as in B). H, A detail at high-magnification from G showing myc-GluK4 immunoreactivity inside the synaptic boutons (arrows). The projections in three orthogonal planes at the level indicated by the dotted lines are also shown (right and bottom edges). Or, Stratum oriens; Py, pyramidal cell layer; Ra, stratum radiatum. Scale bars: A, E, 100 μm; B–D, F, G, 20 μm; H, 3 μm.

EPSCKARs are enhanced in mossy fiber to CA3 synapses in GluK4over mice. A, Hippocampal slice scheme with recording (Rec) electrode. MF, Mossy fibers. B, Spontaneous KAR-mediated EPSCs in CA3 pyramidal cells recorded at −75 mV of membrane potential have been aligned and superimposed (gray traces). Resulting averages of all responses are included as thick traces. C–E, The mean values for the amplitude (C), frequency (D), and decay times (E) of these responses (scaled and superimposed in the inset) are shown for each genotype (n = 32 neurons/11 slices and n = 41 neurons/10 slices from 8 GluK4over and 6 GluK4+/+ mice, respectively; ***p < 0.001, **p = 0.013, *p = 0.035 Mann–Whitney rank-sum test).
Behavioral abnormalities
The effect of GluK4 overexpression on behavior was assessed using a comprehensive battery of behavioral tests, which demonstrated significant differences between GluK4over mice and their GluK4+/+ siblings. To exclude any influence of motor dysfunction in these analyses, we subjected mice to a rotarod test. Transgenic mice presented no evidence of motor deficits since they performed perfectly well over multiple sessions (Fig. 4A).

Overexpression of GluK4 causes anhedonia and depression. A, Transgenic mice and control littermates performed equally well in the rotarod test over time, not showing any indication of impaired locomotion. Points are mean ± SEM from 10 mice in each case, repeated three times per day. B, GluK4over mice showed anhedonia as their preference for water with sucrose was lower than that of normal mice (n = 22, ***p < 0.001, Student's t test). C, A treatment with the antidepressant tianeptine (TNP; 10 mg/kg, daily, 30 d) abolished anhedonia in these mice. D–F, In forced swimming tests, GluK4over mice remained immobile for longer periods than normal mice, compatible with depressive behaviors. E, F, This behavior was inverted in mice deficient for GluK4 (E, n = 9 mice from each genotype, *p < 0.05, ***p < 0.001, Student's t test) and abolished after treatment with tianeptine (F; TNP, same treatment as in C; 7 wild type and 6 GluK4over for saline; 9 wild type and 10 GluK4over for tianeptine; values are the mean ± SEM; 2-way ANOVA post hoc Bonferroni's test: *p < 0.05, **p < 0.01, ***p < 0.001).
Children with ASDs consistently demonstrate some degree of anhedonia (Chevallier et al., 2012), i.e., failure to experience pleasure in normally enjoyable life experiences, such as eating. Interestingly, GluK4over mice reflected significant anhedonia, as assessed by the lower intake of sucrose compared with normal mice (Fig. 4B). Anhedonia is a clinical symptom of depression. For this reason, we treated transgenic GluK4over mice and their wild-type siblings with well known antidepressants. On one hand, administration of fluoxetine (Prozac), a serotonin reuptake inhibitor, had no effect on sucrose intake (data not shown). On the other hand, tianeptine, an antidepressant structurally similar to tricyclic antidepressants but with a different pharmacological profile (McEwen et al., 2010), abolished any sign of anhedonia in these mice (Fig. 4C).
We then subjected GluK4over mice to the forced-swimming test to assess degree of depression. These transgenic mice demonstrated a larger index of immobility than normal mice in this test (Fig. 4D,E), indicating a large degree of depression. Linking particular behavioral phenotypes to alterations in synaptic receptor levels is not always straightforward. To check for the direct involvement of the GluK4 protein levels, we compared this depressive behavior in mice deficient for GluK4. Knock-out mice spent significantly more time swimming than did control (Catches et al., 2012) and GluK4-overexpressing mice (Fig. 4E), suggesting a direct role of GluK4 protein in this behavioral abnormality. Interestingly, fluoxetine was totally ineffective in the forced-swimming test (data not shown), while tianeptine again totally reverted signs of depression as assessed by this test (Fig. 4F). We also observed that GluK4over mice might experience anxiety as they avoided open areas. Anxiety was tested directly in three independent tests commonly used to evaluate such behavior in animals (Lalonde and Strazielle, 2008): the elevated plus maze, the open-field arena, and a dark–light box. In all these tests, GluK4over mice displayed strong signs of anxiety. For example, they spent significantly less time in the open arms of the plus maze than their siblings (Fig. 5A), they were much less willing to explore the center of an open field (Fig. 5B), and they spent more time in the dark area in the light–dark test (Fig. 5C). In general, GluK4over mice spent less time in motion than their siblings, traveling shorter distances in all the tests. However, this was not due to any motor impairment, since transgenic and wild-type animals performed equally well in rotarod tests (Fig. 4A). Rather, the increased sedentary periods of mice overexpressing GluK4 could reflect a lack of motivation. Indeed, the treatment with tianeptine normalized the distance traveled by transgenic mice during these trials (data not shown). We further explored the possibility of a deficit in spatial memory as the cause of this behavior. However, GluK4over mice performed equally well in the Y maze spontaneous alternation test, a test that measures the willingness of rodents to explore new environments (Fig. 5D), suggesting a lack of deficit in spatial memory formation in GluK4over mice.

Mice overexpressing GluK4 show indications of severe anxiety. A–C, Representative tracks (top) of normal and GluK4over mice during exploration of elevated plus maze (A; thicker lines denote closed arms), open-field arena (B), and dark–light box (C). Shaded area in B denotes the area considered to be not the center of the arena. The clear box outlines the center of the arena. D, Performance in the Y maze was entirely normal. All histograms at the bottom are quantifications (mean ± SEM) of several parameters of each behavioral test from 9 (A, B) and 11 animals (C, D) from each phenotype. ***p < 0.001, Student's t test.
Since the diagnosis of autism is based on deficits in social behavior, we then looked at social interactions. In the three-chamber social-interaction test (Nadler et al., 2004), both GluK4over and wild-type mice interacted equally with two novel objects (Fig. 6A) and both did it more strongly with a mouse than with an object, indicating a correct level of discrimination of congeners from inanimate objects. However, in contrast to normal mice, GluK4over interacted equally with novel and familiar mice when confronted with a novel mouse (Fig. 6B,C), indicating a deficit in their social interactions. Tracking trajectories of GluK4over revealed a tendency to remain in the chamber occupied by familiar mice, albeit far from them (Fig. 6C). Therefore, we measured the time spent by transgenic mice in the corner at a distance of familiar and novel peers. This analysis revealed a clear preference of GluK4over mice to rest after exploration in the far corner of the chamber occupied by familiar mice, a behavior never observed in wild-type siblings (Fig. 6D, top). This prompted us to determine the exact time that mice spent actively interacting with peers (i.e., sniffing). These data confirmed that GluK4over mice had a deficit of social interaction (Fig. 6D, bottom).

Excess of GluK4 produces deficits in social interaction. A–C, A schematic representation of the three-chambered apparatus is shown at the top with superimposed representative examples of GluK4over and normal mice performance (tracks) in the social-interaction test. Histograms at the bottom are quantifications of data from 11 mice for each genotype. GluK4over mice discriminate well between an inanimate object and a congener (A) but, contrary to what happens in normal mice (B), GluK4over mice spent similar time in zones occupied by the novel and familiar mice (C), indicating impaired social interaction. D, GluK4over mice, while remaining for longer periods in the chamber occupied by familiar animals, stayed far away (top histogram; C, circle) and also demonstrated reduced direct interaction with novel mice (lower histogram; n = 11 mice from each genotype, ***p < 0.001, **p < 0.01, *p < 0.05, Student's t test).
Altered functional connectivity
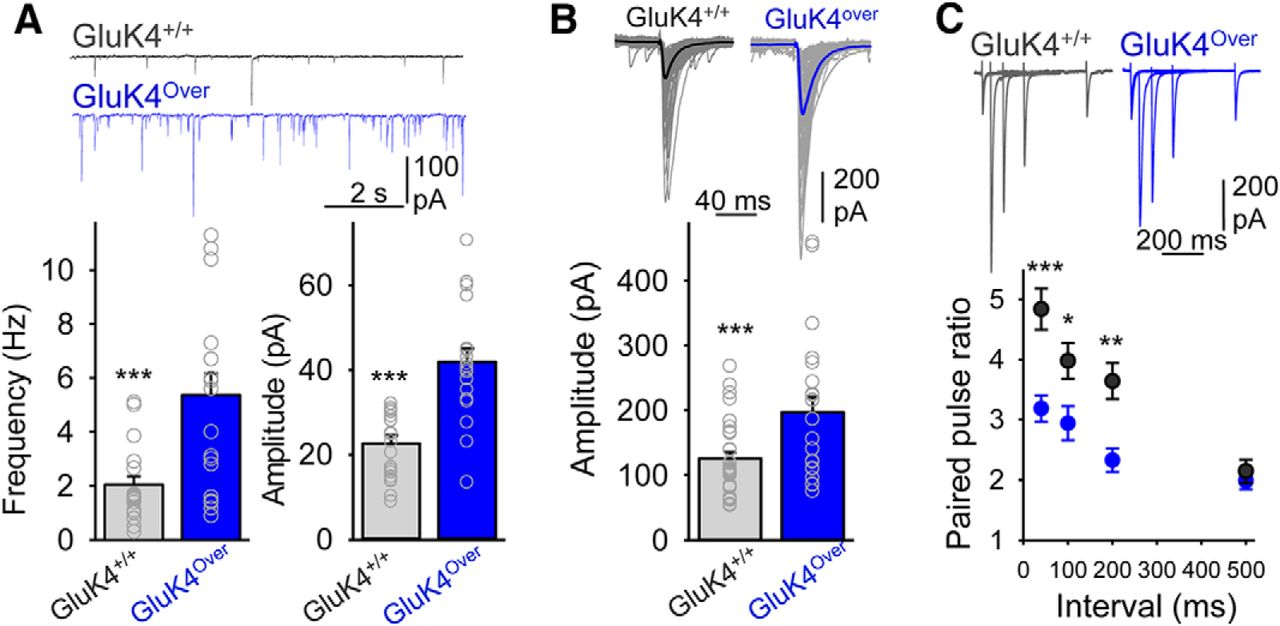
In the light of the above, we wondered whether overexpression of this GluK4 receptor subunit might influence normal information transfer in brain structures related to these behaviors. In addition to memory, the hippocampus has long been associated with aspects of emotional processing, in particular with anxiety (Bannerman et al., 2014). Therefore, we evaluated information flow through the hippocampal trisynaptic circuit, obtaining in vivo recordings using a vertical array of electrodes in anesthetized animals. Electrical stimulation delivered to the perforant pathway, the entry of the trisynaptic circuit, evoked similar responses at the level of the first synapse in the DG, and no differences were observed in input–output curves at the dendritic or somatic levels (Fig. 7). However, the field EPSP evoked at the CA1 through trisynaptic activation appeared to be significantly larger in GluK4over than in normal mice. Indeed, plotting the CA1 EPSC amplitude versus DG population spike (i.e., the output and input to the trisynaptic circuit, respectively) further revealed a change in the gain of this circuit. Neuronal KARs have been best studied in the CA3 field of the hippocampus, where they are localized at presynaptic and postsynaptic sites (Darstein et al., 2003; Fernandes et al., 2009). We, therefore, evaluated synaptic transmission from the mossy fiber to CA3 pyramidal neurons, the second synapse in the circuit, where GluK4 is strongly expressed in normal animals. Examination of the AMPA receptor-mediated mEPSC normally generated at this synapse but also at synapses from association-commissural inputs, revealed a larger frequency and amplitude of mEPSCs in GluK4over mice (Fig. 8A), and the mossy fiber-evoked EPSCs were also consistently larger (Fig. 8B). By contrast, pair-pulse facilitation, a class of short-term plasticity that is a hallmark of these synapses, was dampened significantly (Fig. 8C). Together these data imply that information transfer at mossy fiber synapses critical to behavior is altered in mice overexpressing the GluK4 subunit of KARs.

Synaptic transfer through hippocampal trisynaptic circuit is altered in GluK4over mice. The inset at the center of the figure shows the recording and stimulating electrode arrangement in the in vivo hippocampus from normal and GluK4over animals. A, Stimulus–response curve for fEPSP recorded from electrodes at the stratum moleculare of DG in normal (gray) and overexpressing (blue) mice. B, Stimulus–response curves for population spikes recorded from a place just below the DG granule cell layer. Same color codes apply. C, Stimulus–response curve for the fEPSP evoked trisynaptically in the stratum radiatum of CA1, where the Schaffer collateral pathway ends. Curves were significantly different (2-way ANOVA post hoc Bonferroni's test, p = 0.002). Insets in A–C are illustrative field-averaged responses (n = 10). D, Input–output curve was constructed using amplitudes of population spikes recorded from the DG as the inputs and the trisynaptic EPSPs from CA1 as the circuit's outputs. Different slopes denote a change in circuit's gain. Nine animals were analyzed for GluK4+/+. Eight animals were analyzed for GluK4over.

Synaptic transmission from mossy fiber to CA3 pyramidal cells shows increased gain in mice overexpressing GluK4. A, AMPA-receptor-mediated mEPSCs (Vm = −65 mV) in CA3 pyramidal cells in each genotype (top) and quantification of frequency and amplitude (bottom) from 20 neurons/7 slices (2 mice) and 20 neurons/6 slices (2 mice) from GluK4over and GluK4+/+ mice, respectively (***p < 0.001, Student's t test). B, Mossy fiber-evoked EPSCs in CA3 neurons (top) and their quantification (bottom) for each genotype (22 neurons/20 slices from 11 GluK4over and 32 neurons/30 slices from 12 GluK4+/+ mice; ***p < 0.001, Student's t test). C, Pair-pulse facilitation of CA3 synaptic responses over several intervals for each genotype. Points are the mean ± SEM from 10 cells/6 slices (5 GluK4over mice) and 13 cells/10 slices (5 GluK4+/+ mice; 2-way ANOVA post hoc Bonferroni's test, ***p < 0.001; **p < 0.01; *p < 0.05).
Discussion
Present results indicate that an excess of GluK4 protein content just doubling endogenous levels in such structures as the neocortex or the striatum, and slightly increasing them in the hippocampus, has a profound functional penetrance, altering information transfer and integration in neuronal circuits of the hippocampus and likely in other brain networks. This agrees well with the observation that a genetic variant of the GRIK4 gene, associated with bipolar disorder (Pickard et al., 2006) in humans and shown to increase GluK4 expression in the human hippocampus (Pickard et al., 2008), is also associated with increased hippocampal activation as assessed by functional magnetic resonance imaging (Whalley et al., 2009).
Consequently, larger doses of GluK4 in the forebrain could indeed be significant in human disease since the chromosome region where GRIK4 maps (11q23.3–q24.1 cytoband) has been recently found to be duplicated de novo in a case of autism (Griswold et al., 2012). The affected region harbors 16 genes, most of which are not related to neural functions. Our results likely recapitulate such a case of autism showing that a gain of function of one of those genes, grik4, provokes drastic alterations in animal behavior, including those consistent with endophenotypes associated with ASDs. Indeed, the relevant feature of autism (i.e., a deficit in social interactions) is clearly reproduced in mice with more copies of grik4. Although these animals distinguished perfectly well between a peer and an inanimate object, they showed a tendency to avoid unfamiliar relations. GluK4over mice, compared to wild-type mice, interacted with novel mice for a shorter time and always preferred familiar over novel environments and kept their distance from peers. The meaning of this stereotyped behavior is unknown but is entirely consistent with ASD symptoms.
Restricted interest is another characteristic in ASDs. GluK4over mice, without presenting any motion disability, move less than wild-type animals. This less-active behavior could be understood as a lack of interest or motivation, rather than an impairment of locomotion. Depressive and anxiety syndromes also represent comorbidity for many children with ASD (White et al., 2009; Magnuson and Constantino, 2011). Indeed, anxiety-related conditions are among the most common problems for school-age children and adolescents with ASD (Ghaziuddin, 2002). Behavioral tests revealed that GluK4over mice experience strong symptoms of both syndromes together with some degree of anhedonia, another characteristic commonly observed in ASD patients (Chevallier et al., 2012).
De novo germline mutations and copy number variants have been recognized to indicate significant risk factors for ASDs (Gilman et al., 2011; Levy et al., 2011). De novo mutations appearing in ASDs and schizophrenia cluster to genes related to synaptic function (Ghaziuddin et al., 2002; Baudouin, 2014; De Rubeis et al., 2014; Fromer et al., 2014), implying that there may be a continuum in diverse psychiatric disorders and that integrity of synaptic function is critical for brain performance. However, in only a few cases have these genomic alterations been linked to functional proofs of disease. The remarkable symptoms described here in mice expressing larger doses of GluK4 in the forebrain, recapitulate symptoms of autism demonstrating that de novo duplication of the chromosome region where GRIK4 maps (11q23.3–q24.1 cytoband; Poot et al., 2010; Griswold et al., 2012) may effectively result in a disease within autism spectrum.
The phenotype of the GluK4over mice provides a model for the mechanistic analysis of ASDs. However, a common problem that this kind of study faces is whether the phenotypic change and gene alteration are linked or are just epiphenomena. In the present study, the behavioral data obtained in GluK4-deficient and GluK4-overexpressing mice appear totally contrasting. While a gain of function of this gene (i.e., GluK4over mice) induced severe depression and anxiety, loss of GluK4 function (i.e., GluK4 knock-out) caused anxiolytic and antidepressant-like behaviors (Fig. 4; Catches et al., 2012). This antagonistic effect strongly indicates a link between behavior and GluK4 levels in the forebrain.
As examined in the hippocampus, the presence of more GluK4 protein induced a prominent modification of synaptic properties, likely underlying a change in synaptic gain. Two aspects of our results lead to the conclusion that increased synaptic transfer may arise from either a change in release probability (p) or in number of release sites (N) at the mossy fibers. The increase in frequency of both AMPAR-mediated and KAR-mediated events is compatible with a larger p or a larger N (e.g., more synapses). However, the dampening of frequency facilitation, a fingerprint of these synapses, in GluK4over mice advocates for an enhancement of release probability rather than an alteration in synapse number, although it could not be excluded. KAR localization is limited to the mossy fiber synapses, and the absence of myc-GluK4 immunoreactivity outside the stratum lucidum ruled out a change of distribution of synaptic KARs in GluK4over mice, indicating that most of these events are of mossy fiber origin. An additional phenomenon observed in GluK4over mice was the larger amplitude of elementary AMPA-mediated EPSCs at mossy fiber synapses. This implies higher AMPAR density at the postsynaptic membrane, which would lead to increasing information transfer at the circuit level, as found in the in vivo hippocampus.
What is special in this subunit that may not be shared by other KAR subunits? We do not believe that this subunit is the only one critical to affect circuit performance and therefore animal behavior. We are tempted to propose that what makes this subunit a key element is its position in neuronal circuits. The alteration of synaptic transmission at these points may unbalance circuit behavior leading to behavioral phenotypes. On one hand, the overproduction of this subunit may alter the impact not only of canonical signaling of KARs but also of the noncanonical signaling of KARs. In fact, it seems that grik4 overexpression conveys synaptic changes beyond a change in KAR-mediated synaptic currents (e.g., it also induces more frequent and larger mEPSCAMPAs). The impact of more GluK4 subunits ultimately depends on where and when this subunit is overexpressed. It would be interesting, then, to design experiments where grik4 overexpression might be limited to specific pathways to find out which ones are responsible for the observed variety of ASD-linked phenotypes.
Autism is considered a developmental brain disease (DiCicco-Bloom et al., 2006; Chang et al., 2015) and known genetic variations associated with ASDs and other psychiatric disorders converge on several biological networks that are involved in neurogenesis and synaptic function (Gilman et al., 2011; van Bokhoven, 2011). A significant number of genes associated with ASDs are related to the development of neuronal projections and actin cytoskeleton (Hua and Smith, 2004; van Bokhoven, 2011). There is strong evidence indicating that KARs influence the development of neuronal networks (Marques et al., 2013). For instance, immature network activity appears to influence the development of synaptic connectivity (Ben-Ari, 2002; Lauri et al., 2005) and KARs play a critical role in pacing network activity in both the developing and adult hippocampus (Lauri et al., 2005, 2006). Furthermore, KAR tonic activity influences the maturation and elongation of neurites (Tashiro et al., 2003; Marques et al., 2013). Therefore, altered synaptic transfer through neuronal networks may arise as a consequence of an excess of GluK4 function at particular glutamatergic synapses during network formation and maturation, perturbing the development of neuronal projections and likely resulting in a permanent change in synaptic gain. Several studies in patients of ASD and schizophrenia support the notion of both diseases involve biological pathways engaged in synaptogenesis and glutamate neurotransmission (Kenny et al., 2014). Particularly relevant for ASD-linked behaviors may be the overexpression of GluK4 and the effects that it could induce in striatum. Recently it has been demonstrated that enhanced acquisition of repetitive behaviors, commonly seen in autism, is recapitulated by altering synaptic transfer in a ventral striatal subregion that interfaces with limbic and motor systems (Rothwell et al., 2014).
A typical case of copy number variation of genes belonging to chromosome 11, encompassing GRIK4, is the Emanuel syndrome (Online Mendelian Inheritance in Man #609029). This syndrome is characterized by multiple congenital anomalies and significant cognitive handicap (Emanuel et al., 1976; Carter et al., 2009). Affected individuals have an unbalanced chromosome complement as a result of 3:1 malsegregation during meiosis of a parental balanced translocation between chromosomes 11 and 22, which is very common. One hundred percent of children with Emanuel syndrome have global developmental delay and intellectual disability (Carter et al., 2009). However, the segments involved are large (e.g., 18 Mb; Kurahashi and Emanuel, 2001) and too many genes are affected, producing a very severe phenotype that makes genotype–phenotype correlations uncertain.
It is quite remarkable that the duplication of a single gene in a limited area of the brain reproduces endophenotypes seen in autism, somehow mimicking the situation in humans diagnosed with autism. ASDs are currently thought to represent a continuum of the same disorder with varying degrees of severity, and there is genetic overlap between ASDs and other neurodevelopmental and neuropsychiatric endophenotypes (Silverman et al., 2010). For instance, ASDs are associated with clinical indications of anxiety (Gillot et al., 2001; Simonoff et al., 2008; White et al., 2009; Magnuson and Constantino, 2011). These alterations in behavior in mice may recapitulate abnormal behaviors in human patients with GRIK4 duplications. If so, it should be possible to use this mouse to directly address circuit dysfunctions associated with ASDs and test specific treatments of autism-related behaviors.
Footnotes
This work was supported by grants to J.L. from the Spanish Ministry of Science and Innovation (BFU2011-24084), Consolider (CSD2007-00023) Generalitat Valenciana GVPRE/2008/023, Prometeo/2011/086, and PrometeoII/2015/012. The Santiago Grisolia fellowship program from the Generalitat Valenciana supports V.P. The Instituto de Neurociencias is a Centre of Excellence Severo Ochoa. We dedicate this paper to the memory of Dr. Steve F. Heinemann (Salk Institute). We thank Dr. A. Contractor (Northwestern University) for kindly providing the GluK4−/− mouse and Dr. A. Barco (Instituto de Neurociencias) for making available GFP mice (GFP-M). We also thank Dr. H. van Bokhoven (Radboud University Nijmegen Medical Centre) for advice and patient information and Dr. M. Dominguez (Instituto de Neurociencias) for critical reading of the manuscript.
The Consejo Superior de Investigaciones Científicas requested a patent covering the commercial use of the transgenic mouse generated for this work. The authors declare no other competing financial interests.
- Correspondence should be addressed to Juan Lerma, Instituto de Neurociencias, CSIC-UMH, 03550 San Juan de Alicante, Spain. jlerma{at}umh.es





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}