

Abstract
Macrophages are recruited into the cochlea in response to injury caused by acoustic trauma or ototoxicity, but the nature of the interaction between macrophages and the sensory structures of the inner ear remains unclear. The present study examined the role of fractalkine signaling in regulating the injury-evoked behavior of macrophages following the selective ablation of cochlear hair cells. We used a novel transgenic mouse model in which the human diphtheria toxin receptor (huDTR) is selectively expressed under the control of Pou4f3, a hair cell-specific transcription factor. Administration of diphtheria toxin (DT) to these mice resulted in nearly complete ablation of cochlear hair cells, with no evident pathology among supporting cells, spiral ganglion neurons, or cells of the cochlear lateral wall. Hair cell death led to an increase in macrophages associated with the sensory epithelium of the cochlea. Their numbers peaked at 14 days after DT and then declined at later survival times. Increased macrophages were also observed within the spiral ganglion, but their numbers remained elevated for (at least) 56 d after DT. To investigate the role of fractalkine signaling in macrophage recruitment, we crossed huDTR mice to a mouse line that lacks expression of the fractalkine receptor (CX3CR1). Disruption of fractalkine signaling reduced macrophage recruitment into both the sensory epithelium and spiral ganglion and also resulted in diminished survival of spiral ganglion neurons after hair cell death. Our results suggest a fractalkine-mediated interaction between macrophages and the neurons of the cochlea.
SIGNIFICANCE STATEMENT It is known that damage to the inner ear leads to recruitment of inflammatory cells (macrophages), but the chemical signals that initiate this recruitment and the functions of macrophages in the damaged ear are unclear. Here we show that fractalkine signaling regulates macrophage recruitment into the cochlea and also promotes the survival of cochlear afferents after selective hair cell lesion. Because these afferent neurons carry sound information from the cochlea to the auditory brainstem, their survival is a key determinant of the success of cochlear prosthetics. Our data suggest that fractalkine signaling in the cochlea is neuroprotective, and reveal a previously uncharacterized interaction between cells of the cochlea and the innate immune system.
Introduction
Resident macrophages are present in most somatic tissues, where they play critical roles in innate immunity, homeostasis, and injury response (Stefater et al., 2011). The mammalian cochlea contains a population of resident macrophages, and increased numbers of macrophages are recruited into the cochlea after acoustic trauma or ototoxic injury (Fredelius and Rask-Andersen, 1990; Hirose et al., 2005; Sato et al., 2010). Still, the precise role of macrophages in the lesioned cochlea is not known and the signals that recruit macrophages into injured ear have not been identified. Prior studies of injury-evoked macrophage recruitment in the cochlea have used either noise or ototoxic antibiotics to kill sensory hair cells. However, both of these methods can damage other cochlear tissues, and it is not clear whether the loss of hair cells alone can result in macrophage activation and recruitment. To determine whether the loss of cochlear hair cells (without any other direct tissue injury) was sufficient to recruit macrophages into the cochlea, we used a novel transgenic mouse model, in which the gene for the human diphtheria toxin receptor (huDTR) was inserted under regulation of the promoter for Pou4f3 (a hair cell-specific transcription factor). In these mice, a single injection of diphtheria toxin (DT) kills nearly all cochlear hair cells but does not affect the nonsensory tissues of the inner ear (Tong et al., 2015). The Pou4f3-huDTR mice were crossed with a second transgenic line, in which one or both alleles of the fractalkine receptor CX3CR1 was replaced with the gene for GFP. CX3CR1 is expressed by macrophages, monocytes, microglia, and related cells. As such, the CX3CR1-GFP mouse line expresses GFP in all macrophages, permitting us to characterize the numbers and location of macrophages within the injured cochlea.
Use of the CX3CR1-GFP mouse line also permitted us to investigate the role of fractalkine signaling in cochlear pathology. Fractalkine (also known as CX3CL1) is a transmembrane glycoprotein that is widely expressed on neurons (Harrison et al., 1998), endothelial cells (Bazan et al., 1997), and epithelial cells (Lucas et al., 2001), and is involved in two distinct processes: Soluble fractalkine can act as macrophage chemoattractant, whereas membrane-bound fractalkine can serve as an adhesion molecule between macrophages and adjoining cells. In the CNS, fractalkine signaling mediates interactions between neurons and microglia (Cardona et al., 2006; Bhaskar et al., 2010; Paolicelli et al., 2011). However, the possible role of fractalkine in neural-immune interactions outside of the CNS has not been explored. To examine the role of fractalkine signaling after hair cell injury, we used mice in which both alleles of CX3CR1 had been replaced with GFP.
We found that DT-evoked hair cell death led to increased macrophage numbers within the cochlea and spiral ganglion. The numbers of cochlear macrophages peaked at 14 d after DT injection, whereas macrophage numbers within the spiral ganglia remained elevated as late as 56 d after DT. In addition, we found that interruption of fractalkine signaling after hair cell death resulted in decreased macrophages associated with both the sensory epithelium and the spiral ganglion. Furthermore, deletion of CX3CR1 led to reduced survival of spiral ganglion neurons (SGNs) after hair cell injury. Our findings point to an unexpected interaction between cells of the innate immune system and the afferent neurons of the cochlea, and imply that fractalkine signaling may protect SGNs after hair cell loss.
Materials and Methods
Animals.
All studies used transgenic mice generated on a C57BL/6J background. One mouse line expressed the gene for the human DT receptor (huDTR; heparin-binding epidermal growth factor receptor) under regulation of the Pou4f3 promoter. Generation and characterization of these mice have been previously described (Tong et al., 2011, 2015; Golub et al., 2012), and systemic treatment with DT results in specific ablation of hair cells in the inner ear. Heterozygous Pou4f3-huDTR mice were crossed to CX3CR1GFP/GFP mice (Dan Littmann, New York University, New York). These mice express GFP in all macrophages, monocytes, and microglia (Jung et al., 2000). In one set of experiments, double heterozygotes (CX3CR1+/GFP:Pou4f3DTR/+) were used as experimental mice and single heterozygotes (CX3CR1+/GFP:Pou4f3+/+) served as controls. A second series of experiments examined the effects of CX3CR1 deletion and used CX3CR1+/−:Pou4f3DTR/+ and CX3CR1−/−:Pou4f3DTR/+ as experimental mice and CX3CR1+/−:Pou4f3+/+ and CX3CR1−/−:Pou4f3+/+ as controls. Mice were housed in the animal facility at the Central Institute for the Deaf (Washington University) and were maintained on a 12 h/day-night light cycle with open access to food and water. All experimental protocols involving animals were approved by the Animal Studies Committee of the Washington University School of Medicine (St. Louis, MO).
Genotyping.
Identification of Pou4f3DTR/+ and Pou4f3+/+ mice used methods similar to those described by Tong et al. (2015). Briefly, tail biopsies were collected, and DNA was extracted using ethanol precipitation. The targeted allele was amplified using PCR (Quick-Load Taq 2× Master Mix, New England Biolabs) using the following primers (0.4 μm): Pou4f3 (WT) forward, 5′ CAC TTG GAG CGC GGA GAG CTA G; Pou4f3 (mutant) reverse, 5′ CCG ACG GCA GCA GCT TCA TGG TC. PCRs were run using the following conditions: 95°C for 5 min; 95°C for 30 s, 59°C for 30 s, 72°C for 1 min, 30 cycles; 72°C for 7 min; 4°C infinity. PCR products (expected band ∼150 bp) were separated on 2% agarose gel containing 1 μl/ml SYBR safe DNA gel stain (Invitrogen). Identification of CX3CR1+/GFP (GFP heterozygous) and CX3CR1GFP/GFP (GFP homozygous) followed previously described methods (Jung et al., 2000).
Hair cell ablation.
Both experimental and control mice (6–8 weeks old; either sex) received a single intramuscular injection of DT (25 ng/g, Sigma-Aldrich). Body weights were recorded daily, and mice also received daily intraperitoneal injections of 0.5 ml of lactated Ringer's solution, at days 3–6 after DT or until body weights had stabilized. Mouse food was also supplemented with high-calorie gel (Tomlyn from Nutri-cal). At 1–56 d after DT injection, mice were deeply anesthetized with Somnasol and perfused with phosphate-buffered 4% PFA (Electron Microscopy Sciences). Temporal bones were removed and postfixed for 1 h in 4% PFA, rinsed in PBS, and placed in 0.1 m EDTA, to allow decalcification for whole-mount dissections and sectioning.
BrdU administration.
To determine whether resident cochlear macrophages undergo proliferation in response to hair cell lesions, both CX3CR1+/GFP:Pou4f3DTR/+ and CX3CR1+/GFP:Pou4f3+/+ adult mice (6–8 weeks) received a single intramuscular injection of DT (25 ng/g, Sigma-Aldrich) and were allowed to survive for a total of 24 or 48 h. At 4 h before death and perfusion, the mice then received a single intraperitoneal injection of BrdU (100 mg/kg body weight, catalog #B5002, Sigma Aldrich). Fixed cochleae were processed for BrdU immunohistochemistry.
Auditory brainstem response (ABR).
Before and 1 week after DT injection, ABRs to tone pips were quantified for both experimental and control subjects. Mice were first anesthetized via intraperitoneal injections of ketamine (100 mg/kg) and xylazine (20 mg/kg). Subcutaneous electrodes were placed behind the right pinna (inverting) and vertex (active). A ground electrode was placed near the tail of the mouse. Stimuli were 5 ms tone pips (0.5 ms cos2 rise-fall), delivered at 40/s with alternating stimulus polarity. Recorded electrical responses were amplified (∼10,000×), filtered (100 Hz to 3 kHz), and averaged, using BioSig software (Tucker-Davis Technologies). The sound level was decreased in 5 dB steps from 99 dB SPL down to 15 dB SPL. At each sound level, 1024 responses were averaged, and response waveforms were discarded as artifacts if the peak-to-peak voltage exceeded 15 μV. Thresholds at 5.6, 8, 11, 16, 22.63, 32, 45.2, and 64 kHz were determined by a single observer, who noted the lowest sound level at which a recognizable waveform could be obtained. Waveforms were confirmed as auditory-evoked responses by their increasing latency and decreasing amplitude as the intensity of the stimulus was lowered. If a hearing threshold at a particular stimulus frequency could not be detected at a sound level of <99 dB, a threshold of “100 dB” was assigned for that frequency. These threshold values (actual or assigned) were then used to calculate the mean ABR thresholds at each stimulus frequency.
Distortion product otoacoustic emissions.
Mice were anesthetized as described above. Stimuli were presented at 5–40 kHz and delivered to the right ear by a custom-coupling insert. Distortion product otoacoustic emissions (DPOAEs) were elicited for primary tones f1 and f2, with a frequency ratio f2/f1 of 1.2 and f1 typically presented at a level 10 dB above f2). Recordings were performed using EMAV software (Boystown National Research Hospital).
Endocochlear potential recording.
Endocochlear potentials were measured at 3 d after DT injection. Measurements were made from both experimental (Pou4f3DTR/+) and control (Pou4f3+/+) mice, using techniques described previously (Ohlemiller et al., 2006). Briefly, a fine drill was used to make a hole in the left cochlear capsule, directly over scala media of the lower basal turn. Glass capillary pipettes filled with 0.15 m KCl (40–80 MOhms) were mounted on a hydraulic microdrive (Frederick Haer) and advanced until a stable positive potential that did not change with increased electrode depth was noted. The signal from the recording electrode was fed to an AM Systems model 1600 intracellular amplifier.
Histological methods.
After perfusion, cochleae were isolated and processed for immunohistochemical labeling. Proteins were detected in both cochlear whole mounts and frozen mid-modiolar sections using standard immunofluorescence methods. Briefly, tissue was rinsed with PBS (3×) and incubated at room temperature for 2 h in blocking solution (5% normal horse serum in 0.2% Triton X-100 in PBS). Cochleae were incubated overnight at room temperature with combinations of the following primary antibodies: Hair cells were labeled with an antibody against anti-myosin VIIa (catalog #25–6790, Proteus Biosciences, 1:500). The apical surfaces of the sensory epithelia were stained using Phalloidin (catalog #A12379, Invitrogen; 25 μl/ml). Neuronal peripheral processes and ganglion cell bodies were labeled using a combination of anti-Neurofilament (mouse monoclonal, clone #2H3, Developmental Studies Hybridoma Bank, University of Iowa, 1:100) and anti-β-III tubulin antibodies (mouse monoclonal, catalog #MMS-435P, Covance, 1:500). Nuclei of supporting cells were labeled using an antibody against SOX2 (catalog #SC-17320, Santa Cruz Biotechnology, 1:200). Visualization of GFP-expressing macrophages was enhanced using rabbit anti-GFP (catalog #A11122, Invitrogen, 1:500), and active macrophages were labeled using mouse anti-CD68 (catalog #MCA 1957, AbD Serotec, 1:50). Two different antibodies against the chemotactic domain of CX3CL1 were used to verify the expression pattern of CX3CL1: Abcam (catalog #ab25088, 1:100) and R&D Systems (catalog #AF537, 5 μg/ml). Proliferating cells were labeled using mouse anti-BrdU (catalog #347580, BD Biosciences, 1:50). Following incubation in primaries, specimens were rinsed 5× in PBS and treated for 2 h in secondary antibodies, conjugated to either AlexaFluor-488, -546, -555, or -647 (1:500; Invitrogen). The secondary antibody solutions also contained DAPI (catalog #D9542, Sigma-Aldrich, 1 μg/ml), to label cell nuclei. Proliferating (S-phase) cells were identified via BrdU immunohistochemistry (Warchol et al., 2012). Macrophages that were both BrdU- and GFP-labeled were considered to be undergoing proliferation. All specimens were coverslipped in glycerol:PBS (9:1) before microscopic imaging.
For plastic embedding, decalcified cochleae were osmicated (1% OsO4 in dH2O) for 60 min, dehydrated in serial ethanols and propylene oxide, embedded in Araldite resin, and incubated for 3 d at 60°C. The cochleae were serial sectioned with a carbide steel knife into 20 μm sections.
Cellular imaging and analyses.
Fluorescence imaging was performed using a Zeiss LSM 700 confocal microscope. For all cochleae, Z-series images (10×, 20×, 40×, or 60×) were obtained, beginning at apical surface of hair cells and extending through the neuronal innervation. Image processing and quantitative analysis were performed using Volocity 3D image analysis software (version 6.1.1, PerkinElmer).
Cochlear hair cell and supporting cell counts.
The time course of hair cell loss was assessed at 1, 3, 7, and 14 d after DT, in both Pou4f3+/+ (WT) and Pou4f3DTR/+ mice (n = 5 or 6 per group). Mice were terminally anesthetized with Somnasol (50 mg/kg) and transcardially perfused with 4% PFA. Temporal bones were isolated, the stapes was removed from the oval window, and tissue was removed from the round window. The temporal bones were kept in 4% PFA for an additional 1 h at room temperature. After fixation, the tissue was rinsed three times with PBS and then immersed in 0.1 m EDTA to allow decalcification for cochlear whole-mount dissections. The cochlea was microdissected into apical, middle, and basal region. The stria vascularis was trimmed, and the tectorial membrane was removed with #55 forceps. Whole-mount preparations were then processed for immunolabeling, as described above. Hair cells were identified by their immunoreactivity for myosin VIIA, whereas supporting cells were identified by nuclear immunoreactivity for SOX2. Both cell types were counted from the apical, middle, and basal region of the cochlea, as recorded in 40× objective images. Data were expressed as either hair cell survival (% control) or SOX2-positive cells/10,000 μm2.
Macrophage counts.
The time course of macrophage numbers was assessed at 1, 3, 7, 14, 28, and 56 d after DT injection in both adult Pou4f3+/+ (WT) and Pou4f3DTR/+ mice (n = 6 or 7 per group). To assess macrophages per 100 μm of sensory epithelium, GFP-labeled macrophages were counted from 20× images taken from the middle region of cochlear whole mounts. To assess macrophages in the spiral ganglia, GFP-labeled macrophages were counted from 20× images taken from the lower basal (base), upper basal (middle), and apical portions of the Rosenthal's canal of sectioned specimens. Macrophages in spiral ganglia were counted from at least 4 or 5 sections per cochlea and normalized to the cross-sectional area of the Rosenthal's canal of the respective cochlear turn and averaged and reported as number per 1000 μm2.
SGN counts.
Two different methods were used to assess SGN survival. The first method used immunofluorescence labeling of SGNs. One temporal bone (right) from each animal was used for whole-mount microdissection, whereas the second (left) temporal bone was decalcified in 0.1 m EDTA and frozen-sectioned along the mid-modiolar plane. Serial floating sections (30 μm) were immunolabeled using standard immunofluorescence methods. Briefly, tissue was rinsed with PBS (3×) and incubated at room temperature for 2 h in blocking solution (5% normal horse serum in 0.2% Triton X-100 in PBS). Sections were then incubated overnight at room temperature, and neurons were labeled with a combination of anti-Neurofilament and anti-β-III tubulin (as described above). Specimens were then rinsed 5× in PBS and incubated for 2 h in secondary antibody (AlexaFluor-546 anti-mouse IgG; Invitrogen; 1:500). Specimens were coverslipped in glycerol:PBS (9:1), and fluorescent imaging was performed using a Zeiss LSM 700 confocal microscope. Z-series images at 0.3 μm intervals were obtained using a 20× objective. Image processing and quantitative analysis were performed using Volocity 3D image analysis software (version 6.1.1, PerkinElmer). To assess the numbers of spiral ganglion cell bodies, Neurofilament and β-III tubulin-labeled somata within Rosenthal's canal were counted from the maximum intensity projections of each section. Cell bodies counted from 4 or 5 sections per cochlea were normalized to the cross-sectional area of Rosenthal's canal per cochlear turn and averaged and reported as SGN density (cells per 1000 μm2).
The second method for assessing SGN survival was by counting spiral ganglion cell bodies in osmicated plastic-embedded sections. Images were obtained from 4 or 5 mid-modiolar sections (20 μm each) per specimen, using an Axiovert 135 inverted microscope (Zeiss) that was equipped with Retiga 1300 camera (Q imaging) and a 63× NA oil-immersion objective. All images were captured using IP lab software (Scanalytics, version 3.9) the apical, middle, and basal turns of Rosenthal's canal. The images were then exported as TIFF files onto ImageJ 1.42a (National Institutes of Health), and spiral ganglion cell bodies were counted from 100 μm × 100 μm area per turn of the cochlea. From these counts, neuronal profile density (somata per 10,000 μm2 area of Rosenthal's canal) was calculated for apical, middle, and basal turns.
Statistical analysis.
All the data analysis and statistics were performed using GraphPad Prism version 6.0d. Data are presented as mean ± SD. Student's t tests or ANOVA was applied as appropriate. Significance main effects or interactions were followed using Bonferroni's or Tukey's post hoc tests. Results were considered statistically significant when p < 0.05.
Results
Single DT injection results in complete hair cell death and profound hearing loss
Previous studies have characterized the Pou4f3DTR/+ transgenic mouse model and have shown that injection of DT leads to the selective death of inner ear hair cells (Tong et al., 2011, 2015; Golub et al., 2012). Because those previous studies used the Pou4f3-huDTR transgene on a CBA/CaJ background, our initial experiments first characterized cochlear pathology when that same transgene was expressed on C57BL/6J background. Both Pou4f3+/+ and Pou4f3DTR/+ mice (6 weeks old) were injected with DT (25 ng/g), and temporal bones were isolated at different postinjection time points. DT treatment had no apparent effect on of Pou4f3+/+; healthy-appearing hair cells remained present throughout the cochlea (Fig. 1A,A′,A″). In contrast, DT treatment of Pou4f3DTR/+ mice resulted in the rapid death of both inner and outer hair cells. Intact hair cells were observed at 1 and 2 d after DT (Fig. 1B,B′,B″; and data not shown for 2 dpi). However, by 3 d after DT, almost all hair cells had been ablated, with the exception of a few inner hair cells in the middle and basal turn of the cochlea (Fig. 1C,C′,C″). By 7 d after DT, virtually all hair cells had been lost, although remnants of dead hair cells were observed in the basal turn of the organ of Corti (Fig. 1D,D′,D″). Quantification of hair cell numbers at various survival times (Fig. 1E) confirmed significant hair cell loss at 3 and 7 d after DT compared with earlier time points or with the cochleae of Pou4f3+/+ mice that had also been injected with DT (p < 0.0001). Examination of plastic sections revealed that organ of Corti in Pou4f3+/+ mice retained a normal appearance at 8 d after DT (Fig. 1F), but that the cochleae of Pou4f3DTR/+ mice had lost both inner and outer hair cells (Fig. 1G, arrows).

Pou4f3DTR/+ mice show extensive loss of hair cells after a single DT injection. A–A″, Cochlear whole mounts labeled for Myosin VIIa (red, hair cells) from Pou4f3+/+ mice at 7 d following DT injection. Hair cells are intact in all the regions of the cochlea, suggesting that DT treatment does not affect the inner ear. B–B″, Myosin VIIa immunolabeling (red) of cochleae of Pou4f3DTR/+ revealed no clear hair cell pathology at 1 d after DT. C–C″, Severe loss of hair cells was first evident in the cochleae of Pou4f3DTR/+ mice at 3 d after DT injection. D–D″, Loss of cochlear hair cells in Pou4f3DTR/+ mice was nearly complete at 7 d after DT injection. Note the complete ablation of hair cells in the apical and middle region of the cochlea of Pou4f3DTR/+ mice, with some remnants of dead hair cells in the basal region. E, Quantification of the percentage of surviving hair cells in Pou4f3DTR/+ mice at 1, 3, 7, and 14 d after DT injections (dpi). Complete loss of hair cells was observed at 14 d after DT. Data are mean ± SD; n = 5 or 6 cochleae per time point. ****p < 0.0001, 1 dpi versus all other time points at respective turn of the cochlea (two-way ANOVA followed by Tukey's multiple comparison post hoc tests). F, Plastic-embedded section from DT-treated Pou4f3 +/+ mouse cochlea (lower apical turn) displaying well-defined nuclei of inner and outer hair cells, and as nuclei of Dieter cells and inner and outer pillar cells. G, Plastic-embedded section from DT-treated Pou4f3 DTR/+ mouse cochlea (lower apical turn) showing the loss of both outer and inner hair cells (arrows). OHC, Outer hair cell; IHC, inner hair cell; D1–3, Deiter's cells; OPC, outer pillar cell; IPC, inner pillar cell. Scale bars: A–A‴, B–B‴, C–C‴, D–D‴, 30 μm; F–G, 55 μm.
A single DT injection in Pou4f3DTR/+ mice resulted in profound hearing loss, as assessed by both ABRs and DPOAEs. ABR thresholds of DT-treated mice typically exceeded 90 dB SPL at all test frequencies (data not shown). In many animals, we were unable to obtain clear ABR responses, even at the highest sound intensities that could be generated by our test apparatus. Similarly, a single DT treatment reduced the amplitude of DPOAEs so that they were indistinguishable from the “noise floor” of the measurement system (data not shown). Prior studies have shown that this functional deficit persists for at least 3 months (Mahrt et al., 2013; Tong et al., 2015). Finally, we measured the endocochlear potential (EP) from the basal cochlear turn in both Pou4f3+/+ and Pou4f3DTR/+ mice at 3 d after DT injection. Control (Pou4f3+/+) mice had an average EP of 101.7 ± 0.6 mV (n = 3), which is normal for mature C57BL/6 mice (Ohlemiller et al., 2009). In contrast, Pou4f3DTR/+ mice had an average EP of 78.3 ± 7.4 mV (n = 4) at 3 d after DT treatment. Notably, the EP recovered to 102.33 ± 1.2 mV (n = 3) after 1 month recovery from hair cell death. We used light microscopy to examine the stria vascularis in the same Pou4f3DTR/+ mice following EP measurements and observed no obvious structural changes (data not shown). These data on C57BL/6J mice confirm and extend the data presented earlier by Tong et al. (2015).
DT administration has no direct effect on other cell types in the cochlea
Although the expression of Pou4f3 in the ear is restricted to hair cells, we sought to further verify that DT administration did not damage other cells types in the cochlea. Representative photomicrographs from the cochleae of both Pou4f3DTR/+ and Pou4f3+/+ mice, taken at 7 d (Fig. 2B), 14 d (Fig. 2C), and 56 d (Fig. 2D) after DT, showed complete loss of hair cells in Pou4f3DTR/+ mice, but no signs of supporting cell loss (as identified by SOX2 immunolabeling). The average number of supporting cells/10,000 μm2 in Pou4f3+/+ and Pou4f3DTR/+ mice is shown in Figure 2E. Although the spatial distribution of supporting cell nuclei appears to have undergone a moderate degree of rearrangement after hair cell loss, we observed no significant differences between the numbers of supporting cells in DT-treated Pou4f3+/+ mice versus Pou4f3DTR/+ (p = 0.9126, one-way ANOVA). In addition, a single DT injection in 6-week-old Pou4f3DTR/+ mice did not cause a significant loss of SGN (Fig. 2F–J). The average densities of spiral ganglion neurons in the middle cochlear turn of Pou4f3+/+ mice are compared in Figure 2J. No significant difference in SGN numbers was noted at any time point (Fig. 2J; p = 0.8353, one-way ANOVA). Finally, examination of other cochlear regions, such as spiral limbus and stria vascularis in the DT-treated Pou4f3DTR/+ mice (at 3 and 56 dpi) showed no obvious morphological changes at any time point (data not shown). These results are consistent with those reported for Pou4f3DTR/+ mice on a CBA/CaJ background (Tong et al., 2015) and suggest that a single DT injection in mice that possess the Pou4f3DTR/+ transgene results in the rapid and complete loss of hair cells without any apparent effect on other cell types in the cochlea. Moreover, administration of DT to mice that lack the huDTR transgene (Pou4f3+/+) does not lead to hair cell loss or other obvious morphological changes.

Cochlear supporting cells and SGNs are not affected by DT-mediated hair cell ablation. Top row, Cochlear whole mounts immunolabeled for SOX2 (green, supporting cells) from (A) Pou4f3+/+ and Pou4f3DTR/+ mice at (B) 7 d, (C) 14 d, and (D) 56 d after DT injection. We observed no evidence for the loss of supporting cells, despite the complete ablation of both inner and outer hair cells. Supporting cell nuclei change their shape and frequently redistribute after hair cell loss. E, Average number of SOX2-positive supporting cells at various times after DT injection. Bottom row, Mid-modiolar sections labeled for neurofilament and β-III tubulin (red, neurons) from (F) Pou4f3+/+ and (G–I) Pou4f3DTR/+ mice at 7, 14, and 56 d following DT injection. No loss of spiral ganglion cell bodies was evident at any survival time. J, Average SGN density from the middle region of the cochlea at various survival times. Data are mean ± SD; n = 4–6 per group. Scale bar, 30 μm.
DT-induced hair cell ablation recruits macrophages to the damaged sensory epithelium
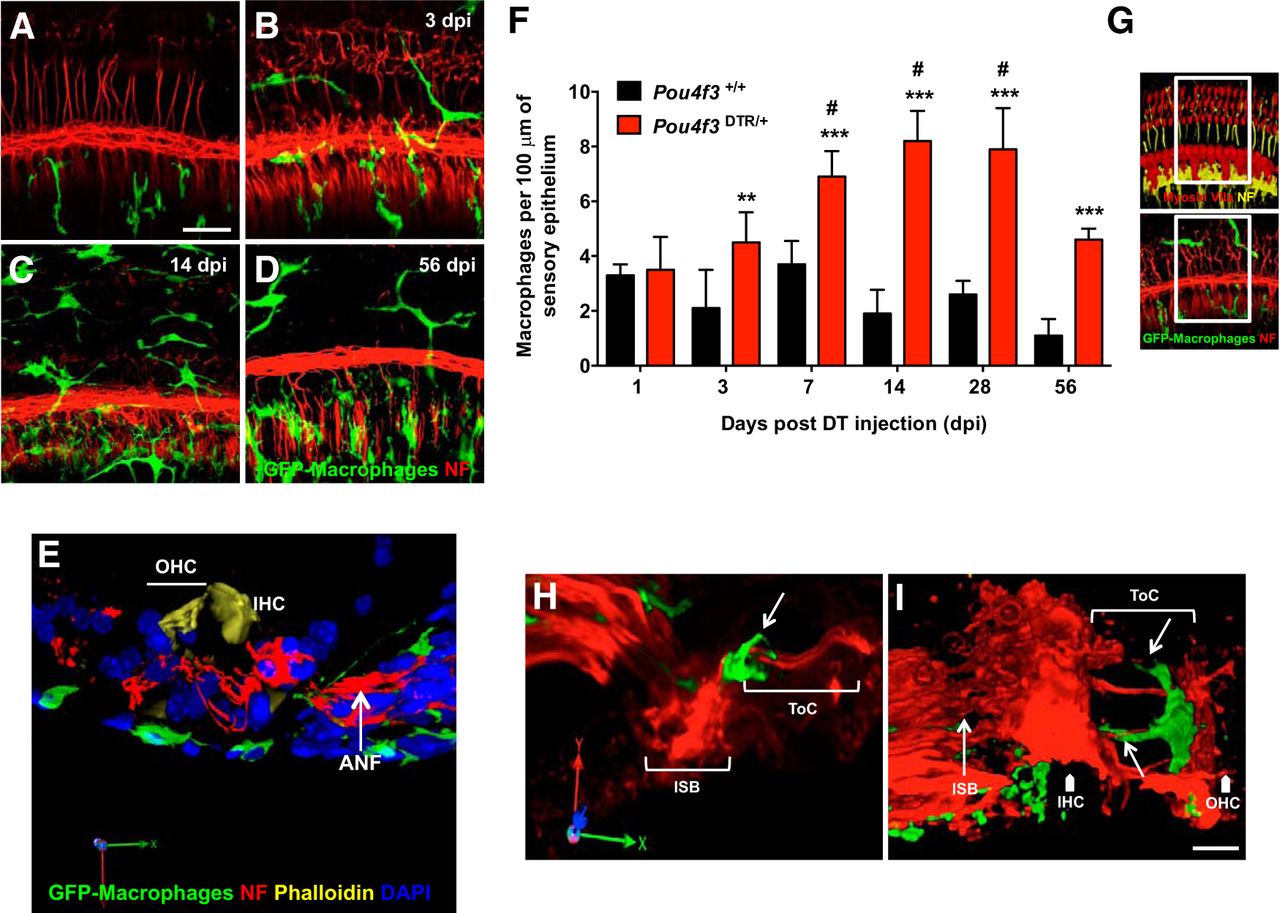
Prior studies have demonstrated that macrophages are recruited into the cochlea after acoustic trauma (Fredelius and Rask-Andersen, 1990; Hirose et al., 2005) or aminoglycoside ototoxicity (Sato et al., 2010). However, it is unclear whether this increase in macrophages was entirely attributable to hair cell death or whether other tissue injury caused by noise or ototoxic insults might also attract macrophages. Data above demonstrate that treatment of Pou4f3DTR/+ mice with a single injection of DT results in complete loss of hair cells, without any apparent effect on the other cells of the cochlea. As such, this mouse model can be used to study the role of hair cell death as an initiator of immune cell recruitment into the injured cochlea. Pou4f3DTR/+ mice were crossed to CX3CR1GFP/GFP mice, in which all macrophages, monocytes, microglia, and NK cells express GFP (Jung et al., 2000). Double heterozygotes (Pou4f3DTR/+:CX3CR1+/GFP) were used as experimental mice, whereas single heterozygotes (Pou4f3+/+:CX3CR1+/GFP) served as controls. Both experimental and control mice (n = 6 in each group) were injected with a single dose of DT (25 ng/g) and allowed to recover for 1, 3, 7, 14, 28, and 56 d after injection. At these points, temporal bones were harvested and processed for immunolabeling of nerve fibers and GFP. Macrophage numbers along the sensory epithelium were quantified and expressed as macrophages/100 μm of basilar membrane length. Very few macrophages (0–4/100 μm) were present in sensory epithelium of uninjured (control) cochleae (Fig. 3A), and those macrophages were usually located in the osseous spiral lamina and only rarely observed below the sensory epithelium. Treatment with DT resulted in enhanced numbers of GFP-expressing macrophages in the Pou4f3DTR/+ mice, but not in the Pou4f3+/+ mice (Fig. 3B–D). This increase in macrophages was first noted at 3 d after DT, the time when hair cell death is first evident (see above). Macrophages in the lesioned sensory epithelia of Pou4f3DTR/+ mice were typically located below the basilar membrane among the tympanic mesothelial cells and in the osseous spiral lamina next to the inner sulcus region (Fig. 3E). The numbers of macrophages associated with the sensory epithelium peaked at 14 d after DT and then declined at later survival times (Fig. 3F). Macrophage numbers in Pou4f3DTR/+ mice at 3, 7, 14, 28, and 56 d after DT were significantly different from those in Pou4f3+/+ mice (black bars) at respective time points (p < 0.01; p < 0.0001) and from 1 d after survival (p < 0.001, analyzed by two-way ANOVA followed by Tukey's multiple comparisons post hoc tests). Proliferation assays using BrdU incorporation (S-phase) and phosphohistone H3 (M-phase) immunolabeling indicated that these macrophages had not undergone DNA replication within 48 h of hair cell ablation, suggesting that the macrophage increase was due to infiltration of circulating monocytes, rather than proliferation of resident macrophages (data not shown). Together, these results suggest that the death of hair cells, without any other apparent otic pathology, is sufficient to recruit macrophages into the inner ear.

Loss of hair cells is sufficient to recruit macrophages toward the cochlear sensory epithelium. Pou4f3+/+ (control) and Pou4f3DTR/+ mice received a single DT injection, and cochleae were examined at 1, 3, 7, 14, 28, and 56 d recovery. A–D, Cochlear whole mounts showing GFP-labeled macrophages (green) and neurons (Neurofilament and β-III tubulin, red) demonstrate increased numbers of macrophages within the sensory regions of lesioned (e.g., Pou4f3DTR/+) mice (B–D), compared with controls (A). Elevated macrophage numbers were first observed at 3 d after DT. E, The 3D renderings of confocal image stacks of lesioned cochleae show that macrophages (green) are typically located below the basilar membrane among the tympanic mesothelial cells and in the osseous spiral lamina. F, Quantification of macrophages per 100 μm of sensory epithelium. Macrophage numbers near the sensory epithelium peak at 14 dpi and diminish by 56 dpi. G, The area in the sensory epithelium (white box) of the cochlea from which macrophages were counted. H, I, The 3D renderings obtained from confocal image stacks of lesioned cochleae show that macrophages (green) are often in close contact with neurons (red) crossing the tunnel of Corti (white arrows) and extended processes toward hair cells (position shown with white arrowheads). Data are mean ± SD; n = 6 per group. dpi, days post injection; NF, neurofilament; IHC, inner hair cell; OHC; outer hair cell; ISB; inner spiral bundle; ToC; tunnel of Corti Statistical significance was computed using two-way ANOVA followed by Tukey's multiple comparison tests. **p < 0.01 (Pou4f3+/+ vs Pou4f3DTR/+ mice at 3 dpi). ***p < 0.0001 (Pou4f3+/+ vs Pou4f3DTR/+ mice at 7, 14, 28, and 56 dpi). #p < 0.001 (1 dpi vs all others). Scale bar, 30 μm.
Injury-evoked changes in peripheral processes of cochlear neurons
We qualitatively assessed the integrity of the peripheral neuronal processes terminating in the inner and outer hair cell regions, using antibodies against neurofilament and β-III tubulin (Fig. 3A–D, red: nerve fibers). In DT-injected Pou4f3DTR/+ mice, the processes within the organ of Corti appeared normal until 2 weeks after injection (Fig. 3B). GFP-expressing macrophages were often observed in close proximity with nerve fibers that contacted both inner and outer hair cells as early as 7 d after injection (Fig. 3H,I, indicated by arrows).
DT-induced hair cell ablation recruits cochlear macrophages into the spiral ganglion
Our results, which are consistent with those of Tong et al. (2015), indicate that SGNs can survive without hair cell targets for (at least) 56 d after DT treatment (Fig. 2F–J). Nevertheless, we found that DT-mediated hair cell loss led to increased macrophage numbers within the Rosenthal canal region of the cochlea and associated with the SGN cell bodies. Enhanced numbers of macrophages in the spiral ganglion were observed, beginning at 7 d after DT. However, unlike the pattern observed in the sensory region of the cochlea, the number of macrophages within the spiral ganglion remained elevated at long recovery times (Fig. 4A–E). Quantification of macrophages in spiral ganglia at 7, 14, 28, and 56 after DT revealed significant differences between Pou4f3DTR/+ and Pou4f3+/+mice (Fig. 4E; p < 0.001, by two-way ANOVA followed by Bonferroni's multiple comparisons post hoc tests).

Loss of hair cells leads to increased numbers of macrophages in the spiral ganglion. Pou4f3+/+ and Pou4f3DTR/+ mice were injected with DT, and cochleae were examined at 1, 3, 7, 14, 28, and 56 d recovery. A, Mid-modiolar frozen section from a DT-treated Pou4f3+/+ (control) mouse, showing GFP-expressing macrophages (green) and neurons (Neurofilament/TUJ-1, red). B–D, Comparable images taken from mid-modiolar sections of DT-treated Pou4f3DTR/+ mice at various recovery times. E, Quantification of macrophage numbers per 1000 μm2 of spiral ganglia (middle region of the cochlea) of control and Pou4f3DTR/+ mice. Increased numbers of macrophages were observed in mice that had DT-induced hair cell lesions. Notably, macrophage numbers remained elevated as late as 56 d after DT. Data are mean ± SD; n = 3–7 per group. **p < 0.01, 1 dpi vs 7, 14, and 28 dpi (two-way ANOVA followed by Bonferroni's multiple comparisons post hoc tests). ***p < 0.001, 1 dpi vs 56 dpi (two-way ANOVA followed by Bonferroni's multiple comparisons post hoc tests). #p < 0.001, Pou4f3+/+ and Pou4f3DTR/+ mice at respective days after DT injection (two-way ANOVA followed by Bonferroni's multiple comparisons post hoc tests). dpi, days post DT injection. Scale bar, 30 μm.
CX3CL1-CX3CR1 signaling modulates macrophage recruitment to the injured cochlea
Although the death of hair cells is apparently sufficient to recruit macrophages into the injured cochlea, the chemical signals that mediate this signal are not known. Studies of injury to other tissue types indicate that fractalkine (CX3CL1) can stimulate macrophage chemotaxis (Truman et al., 2008; Fuhrmann et al., 2010), but the cochlear expression of CX3CL1 has not been characterized. Using antibodies directed against the chemotactic domain of fractalkine, we observed immunoreactivity for CX3CL1 on mature spiral ganglion cell bodies (Fig. 5B–D). This labeling was specific to SGN soma, and we did not observe any immunoreactivity in the surrounding Schwann cells and satellite cells (Fig. 5E–G). Within the cochlear sensory epithelium, we also observed immunolabeling for CX3CL1 on Deiter's, pillar cells, and on inner hair cells, but we did not observe CX3CL1 immunoreactivity on outer hair cells (Fig. 5A).

Fractalkine (CX3CL1) is expressed in the adult mouse cochlea. A, Mid-modiolar section from mature C57BL/6 mouse immunolabeled for fractalkine (red, antibody against the chemotactic domain of CX3CL1) and neurons (green) showing expression of CX3CL1 in certain cell types within the sensory epithelium. B–D, Mid-modiolar section, showing the spiral ganglion and labeled for fractalkine (red) and neurons (green). Immunoreactivity for CX3CL1 was observed on SGNs. E–G, Higher-magnification image reveals that CX3CL1 immunoreactivity was confined to SGN somata, and was not present in surrounding Schwann and satellite cells. OHC, Outer hair cell; IHC, inner hair cell; D1–3, Dieter cell; OPC, outer pillar cell; IPC, inner pillar cell; BC, Boettcher cells. Scale bars: A, 55 μm; B–D, 20 μm; E, F, G, 15 μm.
We next used a genetic approach to characterize the role of fractalkine signaling after hair cell injury. Mice were crossed to produce four distinct genotypes: either Pou4f3+/+ or Pou4f3DTR/+ as well as either CX3CR1GFP/+ or CX3CR1GFP/GFP. Mice that were CX3CR1GFP/+ possessed a single copy of CX3CR1 and retained normal fractalkine signaling, whereas mice that were CX3CR1GFP/GFP lacked the ability to respond to fractalkine. All mice received DT, and cochleae were fixed and processed after 14 d survival. Consistent with previous results, there was an increase (∼4-fold) in the numbers of macrophages associated with the cochlear sensory epithelia in Pou4f3DTR/+: CX3CR1GFP/+ mice (Fig. 6A′). However, fewer macrophages (∼60%) were observed in the DT-lesioned mice that lacked CX3CR1 (CX3CR1GFP/GFP; Fig. 6B′). Deletion of CX3CR1 (CX3CR1GFP/GFP) did not affect the numbers of resident macrophages in (undamaged) cochleae of Pou4f3+/+ mice (Fig. 6B), which were similar to those in undamaged CX3CR1+/− mice (Fig. 6A). Complete quantitative data are shown in Figure 6C.

Genetic deletion of CX3CR1 led to a reduction in macrophages near the sensory epithelium after hair cell ablation. The effects of CX3CR1 on macrophage recruitment were assessed using mice of four genotypes: Pou4f3DTR/+ and either heterozygous or homozygous for CX3CR1 (Pou4f3DTR/+:CX3CR1+/− and Pou4f3DTR/+:CX3CR1−/−), and Pou4f3+/+ and either heterozygous or homozygous for CX3CR1 (Pou4f3+/+:CX3CR1+/− and Pou4f3+/+:CX3CR1−/−). Cochlear whole mounts and mid-modiolar sections were processed to image macrophages (CX3CR1+/GFP, green) and neurons (neurofilament, red). We observed a significant reduction in the numbers of macrophages near the sensory region of the cochlea (B′) and in the spiral ganglion (E′) in CX3CR1−/− mice, compared with CX3CR1+/− mice (A′, D′) at both 14 d (top) and 56 d (bottom) after DT injection. C, F, G, Quantitative data on macrophages in sensory epithelium at 14 dpi (C) and in the spiral ganglia at 56 dpi (F). Quantification of macrophages within the spiral ganglia in the middle region of the cochlea at all time points after DT-induced hair cell lesion (G). Data are mean ± SD; n = 3–7 for 7 dpi per group; n = 6–7 for 14 dpi per group; n = 3–5 for 28 dpi per group; and n = 9–11 for 56 dpi per group. dpi, days post DT injection; NF, Neurofilament. Statistical significance was computed using two-way ANOVA followed by Tukey's multiple comparison tests. *p < 0.0001 (Pou4f3+/+:CX3CR1+/− vs Pou4f3DTR/+:CX3CR1+/−). #p < 0.0001 (Pou4f3DTR/+:CX3CR1+/− vs Pou4f3DTR/+:CX3CR1−/−). p < 0.05 (Pou4f3+/+:CX3CR1−/− vs Pou4f3DTR/+:CX3CR1−/− in C, for middle region of the cochlea in G, and at 56 d post DT in F. Scale bar, 30 μm.
Disruption of fractalkine signaling also reduced macrophage numbers in the spiral ganglia of Pou4f3DTR/+ mice (Fig. 6D–G). Cochlear mid-modiolar sections (obtained from specimens that were fixed at 56 d after DT) showed a reduction in the number of macrophages in the spiral ganglia of CX3CR1GFP/GFP mice (Fig. 6E′) versus CX3CR1GFP/+ mice (Fig. 6D′). Quantification of macrophages revealed a significant reduction in macrophages in CX3CR1GFP/GFP mice versus CX3CR1GFP/+ in both the mid- and basal cochlear turns (p < 0.0001, Fig. 6F). Notably, deletion of CX3CR1 did not affect the macrophage numbers in Pou4f3+/+ mice (Fig. 6E), which were similar to those in Pou4f3+/+:CX3CR1GFP/+ mice (Fig. 6D). Finally, genetic deletion of CX3CR1 appeared to reduce cochlear macrophage numbers in the spiral ganglia at all time points after DT-induced hair cell lesion (Fig. 6G).
Deletion of CX3CR1 leads to diminished survival of SGNs following hair cell death
Our results, consistent with those of a previous study, indicate that DT-induced hair cell lesions in mature Pou4f3DTR/+ mice do not cause a significant loss of SGNs (Fig. 2F–J) (see also Tong et al., 2015). To determine whether fractalkine signaling plays a role in neuronal survival, we compared the numbers of spiral ganglion cells in the lesioned cochleae of Pou4f3DTR/+:CX3CR1GFP/GFP mice (i.e., fractalkine signaling disrupted) with those in Pou4f3DTR/+:CX3CR1GFP/+ mice (i.e., fractalkine signaling intact) at 56 d after DT. We found that genetic deletion of the fractalkine receptor resulted in reduced numbers of SGNs in all regions of the cochlea (Fig. 7). Cochlear mid-modiolar sections that were immunolabeled for neurofilament and β-III-tubulin show significant loss of spiral ganglion cell bodies in the CX3CR1GFP/GFP mice (Fig. 7B′,C), compared with CX3CR1GFP/+ mice (Fig. 7A′,C). No reduction in SGNs was observed in Pou4f3+/+:CX3CR1GFP/GFP mice, suggesting that the loss of neurons occurred only after hair cell death. Examination of spiral ganglion cell bodies in plastic-embedded sections confirmed these findings (Fig. 7D′,E′). At 56 d after DT, the average density of SGNs in CX3CR1GFP/GFP mice was 10.19 ± 1.17 per 10,000 μm2, which was significantly different from that observed in CX3CR1GFP/+ mice (20.75 ± 1.78; p < 0.0001, Student's t test). This loss of neurons was only observed at the 56 d recovery time point; we did not see evidence for SGN loss in CX3CR1−/− mice at 7, 14, and 28 d after DT (Fig. 7F). These results suggest that fractalkine signaling may participate in those processes that maintain the survival of SGNs at long periods after hair cell death.

Genetic deletion of CX3CR1 results in significant loss of SGNs after hair cell ablation. Cochlear mid-modiolar sections labeled for neurofilament (neurons, red) revealed significant loss of spiral ganglion cell bodies in the CX3CR1−/− mice (B′), compared with CX3CR1+/− (A′) mice, at 56 d after DT-induced hair cell lesion. We observed no evidence for SGN loss in Pou4f3+/+ mice that were either CX3CR1−/− (B) or CX3CR1+/− (A). C, Quantitative data on SGN density (per 1000 μm2) for the four genotypes. D, E, Plastic sections from the basal cochlear turn of Pou4f3+/+mice at 56 d after DT injection. Both CX3CR1+/− (D) and CX3CR1−/− (E) possessed normal numbers of SGNs. D′, E′, Plastic sections from the basal cochlear turn of Pou4f3 DTR/+ mice at 56 d after DT injection. Spiral ganglia of CX3CR1+/− mice (D′) contained normal numbers of auditory neurons after hair cell lesions, whereas the ganglia of CX3CR1−/− mice (E′) showed diminished neuronal survival. F, Density of SGNs (per 1000 μm2) from the basal cochlear turn at all time points after DT-induced hair cell lesion. Data are mean ± SD; n = 5–10 per group. dpi, days post-DT injection. Statistical significance was computed using two-way ANOVA followed by Tukey's multiple comparison post hoc tests. *p < 0.05. **p < 0.01. ***p < 0.001 (Pou4f3DTR/+:CX3CR1+/− vs Pou4f3DTR/+:CX3CR1−/−). #p < 0.05 (Pou4f3+/+:CX3CR1−/− vs Pou4f3DTR/+:CX3CR1−/− and Pou4f3+/+:CX3CR1+/− vs Pou4f3DTR/+:CX3CR1−/−). Scale bars: A, A′, B, B′, 30 μm; D, D′, E, E′, 10 μm.
Discussion
Our results confirm that the mammalian cochlea contains a resident population of macrophages and that their numbers are increased after cochlear injury. Because the Pou4f3-huDTR model permits the selective ablation of hair cells without damaging other cells types, our data further suggest that loss of hair cells alone is sufficient to recruit macrophages into the cochlea. Hair cell injury also resulted in enhanced macrophage numbers within the spiral ganglion, and those numbers remained elevated for 2 months or longer. Finally, genetic deletion of CX3CR1 reduced macrophage recruitment and also impaired the survival of SGNs after the loss of their hair cell targets. These findings point to a previously unreported interaction between macrophages and the afferent neurons of the cochlea.
The Pou4f3DTR/+ mouse model enables selective hair cell lesion in the mature cochlea
The Pou4f3-huDTR transgenic mouse line has been characterized in several previous publications, some of which involved mice with a different genetic background than in the present study (Golub et al., 2012; Tong et al., 2015). We observed nearly identical results to those prior studies. Treatment with DT led to hair cell death, which was first noted at 3 d after DT injection, and the loss of nearly all inner hair cells and outer hair cells was apparent after 7 d. We also observed no DT-induced hair cell death in mice that lacked the Pou4f3-huDTR transgene. Functional analysis (ABRs and DPOAEs) of the DT-treated Pou4f3-huDTR mice revealed profound and irreversible hearing loss. In addition, we observed an acute reduction in the endocochlear potential after DT-induced hair cell lesions, which is likely attributable to disruption of the reticular lamina following hair cell death (Ahmad et al., 2003). These observations suggest that DT specifically targets hair cells in this transgenic line, without causing injury to other otic cell phenotypes.
Loss of cochlear hair cells is sufficient for macrophage recruitment
Macrophages are recruited into the cochlea after acoustic trauma (Fredelius and Rask-Andersen, 1990; Hirose et al., 2005) and aminoglycoside ototoxicity (Sato et al., 2010). However, because both noise and ototoxins can injure multiple cell types within the ear, it was unclear whether the injury-evoked increase in macrophages was caused by hair cell death or by damage to nonsensory tissues of the cochlea. In the present study, Pou4f3DTR/+ mice were crossed to CX3CR1GFP/GFP mice, yielding offspring that expressed GFP in all macrophages and also displayed specific hair cell death in response to DT treatment. We observed increased macrophage numbers after DT-induced lesions, particularly near and below the injured sensory epithelium. In contrast, DT-induced hair cell death did not cause any significant changes in the numbers of macrophages in the cochlear lateral wall, spiral ligament, or spiral limbus. This outcome is quite different from that observed after cochlear injury caused by noise or aminoglycoside antibiotics, where enhanced numbers of macrophages are observed in many nonsensory regions of the cochlea (Hirose et al., 2005; Sato et al., 2010). The present results suggest that hair cell death without any accompanying cochlear pathology is sufficient to attract macrophages into the organ of Corti.
Function of macrophages in maintaining the cochlear sensory epithelium
Macrophages are known to act as “professional” phagocytes at sites of tissue injury, removing the corpses of dead cells. Similar function has been observed within the vestibular organs of DT-treated Pou4f3-huDTR mice (Kaur et al., 2015). However, we did not observe macrophages engulfing apoptotic hair cell debris in the organ of Corti of DT-treated Pou4f3-huDTR mice. Macrophage-mediated phagocytosis is likely to occur quickly, so we cannot exclude the possibility that we simply missed such events in our samples. We also noted an increase in macrophage volume and expression of CD68 (component of lysosome-associated membrane proteins, phagocytic marker) on cochlear macrophages after DT-induced hair cell death, which is suggestive of phagocytic activity (data not shown). Nevertheless, the relative paucity of observed phagocytosis by macrophages, combined with the recent demonstration that dying outer hair cells are engulfed by surrounding Deiter's cells (Anttonen et al., 2014), suggests that macrophages may play a relatively minor role in the removal of debris from the injured cochlea. In addition to clearing debris, macrophages and their secretory products can also promote cellular and tissue repair (Stefater et al., 2011). The possible involvement of macrophages in cochlear epithelial repair will require further experiments.
Fractalkine signaling influences macrophage numbers after hair cell death
Resident macrophages often remove dying cells from injured tissues, so as to minimize subsequent inflammation (e.g., Gregory and Devitt, 2004). Apoptotic cells recruit macrophages via the release of a number of different chemoattractants. Among these, fractalkine (CX3CL1) has been shown to promote activation and recruitment of monocytes and microglia (Cardona et al., 2006; Auffray et al., 2007; Ransohoff et al., 2007; Fuhrmann et al., 2010). We observed CX3CL1 immunoreactivity on inner hair cells, cochlear supporting cells, and spiral ganglion cell bodies. We also found that genetic deletion of CX3CR1 (the receptor for CX3CL1, expressed by macrophages and monocytes) resulted in diminished macrophage accumulation following cochlear injury. These results suggest that fractalkine signaling plays an important role in the regulation of macrophages after injury to cochlear hair cells. However, the precise relationship between fractalkine signaling and macrophage recruitment may be complex. A prior study has shown that genetic deletion of CX3CR1 caused increased macrophage recruitment into certain regions of the cochlea after kanamycin ototoxicity (Sato et al., 2010). This enhancement of macrophage numbers was particularly evident in the cochlear lateral wall, a structure that is damaged by aminoglycosides (Schmitz et al., 2014), but not by the DT treatment used in the present study. Finally, genetic deletion of fractalkine signaling does not affect macrophage recruitment or behavior in the vestibular organs after similar hair cell lesions (Kaur et al., 2015).
Interaction between macrophages and SGNs
Our finding that SGNs survive for (at least) 2 months after DT-mediated hair cell loss is consistent with a previous study that used the same mouse model (Tong et al., 2015) and supports the idea that mature SGNs do not require ongoing trophic support from cochlear hair cells. Slow secondary degeneration of SGNs can occur after hair cell injury caused by acoustic trauma and ototoxicity (e.g., Oesterle and Campbell, 2009). Despite the lack of SGN degeneration, we still observed a significant increase in macrophages within the spiral ganglion after DT injection. Moreover, the macrophage population within the ganglion remained elevated for (at least) 56 d after DT. Such observations suggest a long-term interaction between macrophages and SGNs after cochlear injury.
Previous studies have shown that cochlear supporting cells are critical for promoting long-term survival of SGNs in the absence of hair cells (Stankovic et al., 2004; Sugawara et al., 2005; Zilberstein et al., 2012). Our findings indicate that macrophages and/or fractalkine signaling also play an important role in maintaining viable SGNs after the loss of their target hair cells. The complimentary expression of CX3CL1 on SGNs and CX3CR1 on macrophages suggests that cochlear afferents may communicate with macrophages via fractalkine release. The downstream effects of fractalkine signaling are complex and can lead to several distinct outcomes. Once synthesized by expressing cells, fractalkine is trafficked to the cell membrane, where it is anchored and extends from the extracellular surface (Pan et al., 1997; Wong et al., 2014). Activation of ADAM-family metalloproteases results in cleavage of the fractalkine protein, thereby releasing the soluble fragment into the surrounding fluid. It is possible that hair cell injury and/or the accompanying loss of afferent activity may trigger the activation of metalloproteases, leading to the release of soluble fractalkine and recruitment of macrophages. Such a mechanism would account for the reduction in SGN macrophages that was observed in the lesioned cochleae of CX3CR1GFP/GFP mice. However, direct interaction between membrane-bound CX3CL1 and CX3CR1 can also mediate cell adhesion, so it is possible that binding of fractalkine (on SGNs) to receptors (on macrophages) serves to stabilize the presence of macrophages in the vicinity of the ganglion cell bodies. Elimination of CX3CR1 would disrupt this process and result in fewer macrophages within the SGN. Either mechanism is consistent with the present data, and these mechanisms are not mutually exclusive.
Our data further indicate that deletion of CX3CR1 leads to reduced SGN numbers after hair cell injury. How might macrophages and/or fractalkine signaling affect the survival of cochlear afferents? Studies of the CNS have revealed that signaling via fractalkine can regulate the activation status of microglia and affect the production and release of several cytokines (Biber et al., 2007). If similar signaling processes were present in cochlear macrophages, then fractalkine signaling may regulate certain neuroprotective or neurotoxic properties of those macrophages after injury. It is possible, for example, that recruited macrophages produce a neurotrophic factor that enhances SGN survival after hair cell loss. This notion is supported by prior studies, which suggest that macrophages and microglia can promote neuronal survival via the production and release of neurotrophic factors, such as BDNF and IGF (Lauro et al., 2010; Parkhurst et al., 2013). In this case, the reduction in SGN survival observed in CX3CR1-knock-out mice might be attributed to a deficit in the recruitment of neurotrophin-expressing macrophages in those animals. However, additional studies of the CNS have shown that deletion of CX3CR1 can also lead to a dysregulation of microglial behavior, resulting in neurotoxicity (Cardona et al., 2006; Bhasker et al., 2010). Such pathology has been attributed to enhanced microglial expression of cytotoxic cytokines, such as interleukin-1β. A similar mechanism in cochlear macrophages might explain the loss of SGNs reported here. Even though genetic deletion of CX3CR1 resulted in fewer macrophages within the SGN, it is possible that the remaining macrophages were releasing cytotoxic factors that were inducing the death of nearby ganglion cells. Additional experimentation will be necessary to resolve these issues.
Footnotes
This work was supported by the National Institutes of Health Grants R01DC006283 to M.E.W., R01DC003829 to E.W.R., and P30DC004665 and P30DC004661. D.Z. was supported by National Institutes of Health T35 Grant DC008765.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Mark E. Warchol, Department of Otolaryngology, Washington University School of Medicine, 660 South Euclid Avenue, Box 8115, St. Louis, MO 63110. Warcholm{at}ent.wustl.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}