

Abstract
Preconditioning (PC) using a preceding sublethal ischemic insult is an attractive strategy for protecting neurons by inducing ischemic tolerance in the brain. Although the underlying molecular mechanisms have been extensively studied, almost all studies have focused on neurons. Here, using a middle cerebral artery occlusion model in mice, we show that astrocytes play an essential role in the induction of brain ischemic tolerance. PC caused activation of glial cells without producing any noticeable brain damage. The spatiotemporal pattern of astrocytic, but not microglial, activation correlated well with that of ischemic tolerance. Interestingly, such activation in astrocytes lasted at least 8 weeks. Importantly, inhibiting astrocytes with fluorocitrate abolished the induction of ischemic tolerance. To investigate the underlying mechanisms, we focused on the P2X7 receptor as a key molecule in astrocyte-mediated ischemic tolerance. P2X7 receptors were dramatically upregulated in activated astrocytes. PC-induced ischemic tolerance was abolished in P2X7 receptor knock-out mice. Moreover, our results suggest that hypoxia-inducible factor-1α, a well known mediator of ischemic tolerance, is involved in P2X7 receptor-mediated ischemic tolerance. Unlike previous reports focusing on neuron-based mechanisms, our results show that astrocytes play indispensable roles in inducing ischemic tolerance, and that upregulation of P2X7 receptors in astrocytes is essential.
Introduction
Neuroprotection and repair after brain injury such as cerebral ischemia remains a major medical challenge, and pharmacological treatments are only partially effective. A brief episode of sublethal ischemia, referred to as preconditioning (PC), induces resistance to a subsequent lethal ischemic insult. This phenomenon, known as ischemic tolerance, is an endogenous process that provides robust neuroprotection (Kirino, 2002). Consequently, PC may be a powerful strategy for the treatment of brain ischemia. Indeed, PC is being evaluated in a number of randomized clinical trials (Dirnagl et al., 2009). Furthermore, there are numerous studies investigating ischemic tolerance and PC, and various mechanisms and molecules have been implicated in these processes.
Ischemic tolerance consists of a rapid phase and a delayed phase; the former occurs immediately after PC (within a couple of minutes) and the latter occurs after a delay of 1–3 d and is dependent on protein synthesis (Dirnagl et al., 2009). In the rapid phase, adenosine, ATP, and other chemical transmitters that inhibit excitatory synaptic transmission appear to play key roles (Heurteaux et al., 1995). In the delayed phase, various molecules have been identified as effectors of ischemic tolerance (Kirino, 2002; Stenzel-Poore et al., 2003). These include hypoxia-inducible factor (HIF)-1α and hundreds of its target genes, including erythropoietin (EPO; Prass et al., 2003), vascular endothelial growth factor (Bernaudin et al., 2002), and adrenomedullin (Tixier et al., 2008). In addition, heat shock protein (Liu et al., 1993; Beere et al., 2000) and several neuroprotective growth factors, such as brain-derived neurotrophic factor (Terasaki et al., 2010), also play critical roles. These molecules are thought to be responsible for neuron-based ischemic tolerance. Almost all studies on ischemic tolerance and PC have focused exclusively on neurons, despite the fact that the brain consists of a much higher numbers of glial cells, which have crucial roles in both CNS injury and recovery (Hatten et al., 1991; Kreutzberg, 1996). In particular, astrocytes, the most abundant glial cells, surround synapses, express various neurotransmitter receptors and transporters, and release gliotransmitters, such as ATP, in response to various stimuli (Haydon and Carmignoto, 2006). However, the contribution of astrocytes to brain ischemic tolerance remains unknown.
In the present study, we reveal an essential role for astrocytes in the induction of ischemic tolerance using an in vivo middle cerebral artery occlusion (MCAO) model, and provide insights on the underlying molecular mechanisms involved.
Materials and Methods
Animals.
All procedures were performed in accordance with the Guiding Principles for the Care and Use of Animals in the Field of Physiologic Sciences published by the Physiologic Society of Japan and with the previous approval of the Animal Care Committee of Yamanashi University (Chuo, Yamanashi, Japan; Approval No. A23-9). C57BL/6J mice, 8–10 weeks old, were purchased from CLEA Japan. P2X7 receptor knock-out mice (Solle et al., 2001; P2X7−/− mice, on a C57BL/6 background) were kindly provided by Drs. Hiroshi Enaida (Saga University, Saga, Japan) and Shoji Notomi (Kyushu University, Fukuoka, Japan). P2X7-EGFP mice were generated from the stock Tg (P2rx7-EGFP) FY174Gsat/Mmucd purchased from KOMP repository (Mutant Mouse Regional Resource Centers; www.mmrrc.org) as frozen sperm (ID No. 011959-UCD) and recovered at the RIKEN BioResource Center. Male mice were used in all experiments.
Brain ischemic tolerance model.
Unilateral transient focal ischemia in mice was induced by intraluminal filament occlusion of the right middle cerebral artery, as previously described (Ikeda-Matsuo et al., 2006). The animals were anesthetized with 4% (v/v) isoflurane and maintained on 1.2% (v/v) isoflurane with a facemask. After a neck incision was made, the common carotid artery, internal carotid artery (ICA), and external carotid artery (ECA) were exposed by dissection. Subsequently, the ECA was ligated, and a 12 mm length of 6-0 silicone-coated monofilament suture was inserted via the proximal ECA into the ICA and then into the circle of Willis, thereby occluding the MCA. For PC, the MCA was occluded for 15 min, then the suture was carefully withdrawn to allow reperfusion of the ischemic region. One day, 3 d, or 6 d later, the same suture was inserted to occlude the MCA again for 1 h (i.e., for severe MCAO). The suture was then carefully withdrawn, and the mice were allowed to survive for 3 d. Sham-operated animals were subjected to similar surgical procedures without occlusion of the MCA.
Drug administration.
Fluorocitrate (FC) solution for intrastriatal injection was prepared as follows: 8 mg of dl-fluorocitric acid and Ba2+ salt (Sigma-Aldrich) was dissolved in 1 ml of 0.1 mmol/L HCl. Two to three drops of 0.1 mmol/L Na2SO4 were added to precipitate the Ba2+. Two milliliters of 0.1 mmol/L Na2HPO4 was added, and then the suspension was centrifuged at 1000 g for 5 min. The supernatant was diluted with saline to the final concentration, and the pH was adjusted to 7.4. The FC solution was microinjected stereotaxically into the striatum (anterior: −0.22 mm; lateral: 2.5 mm from bregma; depth: 4 mm from the skull surface) 2 d after PC. FC solution (1 pmol in 1 μl) was injected continuously at a rate of 0.5 μl/min through a stainless steel cannula (26 gauge) connected to a 10 μl syringe. For control mice, the same volume of saline was injected into the striatum. Minocycline (45 mg/kg; Sigma-Aldrich) was administered intraperitoneally just before and 12 and 24 h after PC. For the control, saline was intraperitoneally administered.
Analysis of cerebral infarct size.
Mice were anesthetized with pentobarbital (100 mg/kg, i.p.) and perfused transcardially with saline. The brains were removed, and the forebrain was sliced into 2-mm-thick coronal sections (Brain Matrices). The sections were stained with 2% (w/v) 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich) saline solution at 37°C for 10 min. Slices were digitally scanned after staining using a scanner (GT-X750; Epson). The infarct volume of the brain slice was measured using Scion Image software. Infarct size was calculated as infarct volume of cortex or striatum/volume of whole brain × 100. For experiments using FC injections, we measured the infarct area instead of volume, because the administration of small amounts of FC affected only a limited area around the injected site.
Immunohistochemistry.
Mice were anesthetized with pentobarbital (100 mg/kg, i.p.) and perfused transcardially with saline, followed by 4% (w/v) paraformaldehyde in PBS. The brains were removed, postfixed overnight in a solution containing 4% (w/v) paraformaldehyde and 4% (w/v) sucrose in PBS, and cryoprotected in solutions containing 10 and 20% (w/v) sucrose in PBS for 1 d each. The brains were frozen in an embedding compound (Sakura Finetek) on dry ice, and coronal sections (20 μm) were cut on a cryostat (Leica CM 1100). The sections were fixed with 4% (w/v) paraformaldehyde for 30 min, permeabilized with 0.3% (v/v) Triton X-100 for 10 min, and treated with 3% (v/v) bovine serum albumin for 30 min in PBS to block nonspecific binding. The sections were incubated overnight at 4°C with the following primary antibodies: mouse anti-GFAP (1:1000; Millipore), rabbit anti-Iba1 (1:1000; Wako), chicken anti-GFP (1:500; Invitrogen), rabbit anti-NeuN (1:500; Millipore), or mouse anti-HIF-1α (1:250; Novus Biologicals). The anti-GFP antibody was used to label the P2X7 receptor. The sections were washed and then incubated for 1 h at room temperature with secondary antibodies: Alexa 488- or Alexa 546-conjugated mouse, chicken, or rabbit IgGs (Invitrogen). Fluorescence images were obtained using a confocal laser microscope system (FluoView1000; Olympus). For quantitative analysis of cell type-specific fluorescent signals, we used Adobe Photoshop CS4 (Adobe Systems) and ImageJ (NIH). As shown in Figure 1D, to calculate the number of NeuN (green)-positive neurons, we randomly chose four ipsilateral striatal fields (each 4 × 105 μm2) per mouse (n = 3–4), then counted the number of NeuN-positive neurons. For the quantitative analysis of the immunofluorescence intensity of GFAP (green) or Iba1 (red) seen in Figures 2 and 3, we randomly chose four fields (each 4 × 104 μm2) per mouse (n = 3–4). The intensity was calculated and expressed as the average pixel density (an 8-bit scale). Background fluorescence was also measured and was subtracted from each signal. In Figure 6, to calculate the percentage of HIF-1α-positive cells, we randomly chose four ipsilateral striatal fields (each 9 × 104 μm2) per mouse (n = 3–6), then counted the number of HIF-1α (red)-positive neurons (NeuN: green) and astrocytes (GFAP: green), as well as the total number of neurons and astrocytes. The percentage of HIF-1α-positive cells was calculated by dividing the number of HIF-1α-positive cells by the total number of cells. Similarly, in Figure 4, we also investigated the number of P2X7 receptor (green)-positive microglia (Iba1: red) and astrocytes (GFAP: red), as well as the total number of microglia and astrocytes. For analysis of P2X7 receptor expression, we randomly chose three ipsilateral striatal fields (each 4 × 105 μm2) per mouse (n = 3–4).
Quantitative RT-PCR.
RT-PCR amplifications were performed using the One Step PrimeScript RT-PCR Kit (Takara Bio). RT-PCR amplifications and real-time detection were performed using an Applied Biosystems 7500 Real-Time PCR System. The thermocycling parameters were as follows: 5 min at 42°C for reverse transcription, 10 s at 95°C for inactivation of RT enzyme, and 40 cycles of denaturation (5 s at 95°C) and annealing/extension (34 s at 60°C). All primer probe sets and reagents were purchased from Applied Biosystems (P2rx7: Mm00440578_m1; Aif1: Mm00479862_g1; GFAP: Mm01253033_m1; EPO: Mm01202755_m1). Sense and antisense primers and probes for GAPDH were also obtained from Applied Biosystems (Rodent GAPDH Control Reagents).
Cell culture and treatment.
Primary cultures of astrocytes were prepared from cerebral cortices of P0–P1 wild-type C57BL/6J or P2X7−/− mice as described previously (Koizumi et al., 2003). Dissected cerebral cortices were dissociated in Dulbecco's PBS (Gibco). The cells were plated in 75 cm2 flasks (two brains per flask) coated with poly-l-lysine (10 μg/ml; Sigma-Aldrich) in DMEM (Gibco) containing 5% (v/v) fetal bovine serum, 5% (v/v) horse serum, and 0.1% (v/v) penicillin/streptomycin. To purify astrocytes from postnatal brain primary cell cultures, the cells were subjected to 4 h of continuous shaking 7 d after plating to remove detached cells. For Western blotting, cells were seeded on 6-well cell culture plates coated with poly-l-lysine (10 μg/ml) at a density of 3 × 104 cells/cm2 and used at 12 d after plating. Approximately 96% of the cultured cells expressed abundant GFAP. Before treatment with BzATP (Sigma-Aldrich), a P2X7 receptor agonist, cells were serum starved for 24 h.
Western blot analysis.
Astrocytes were prepared as described above. After BzATP-stimulation, cells were lysed and the lysates were resolved on a 12.5% (w/v) SDS polyacrylamide gel and transferred to polyvinylidene fluoride membranes. The membranes were blocked for 1 h in Tris-buffered saline containing 0.1% (v/v) Tween 20 (TBS-T) and 4% (w/v) skim milk at room temperature. Membranes were incubated with the following primary antibodies: mouse anti-HIF-1α (1:250), mouse anti-β-actin (1:5000; Sigma-Aldrich), or rabbit anti-P2X7 receptor (1:200; Alomone Labs) for 3 h at room temperature. After three washes with TBS-T, the membranes were incubated with horseradish peroxidase-conjugated anti-mouse antibody (1:10,000; GE Healthcare) or anti-rabbit antibody (1:10,000; GE Healthcare) for 1 h at room temperature. The membranes were washed with TBS-T three times, and the proteins were visualized using the Super Signal West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific). Images were obtained using the LAS-4000 (Fujifilm).
Statistical analysis.
Results are expressed as the mean ± SEM. Statistical significance was evaluated with one-way ANOVA, followed by Tukey's multiple comparison. A value of p < 0.05 was considered statistically significant.
Results
PC-induced ischemic tolerance
We used the mouse MCAO model throughout the study. We occluded the MCA for different time periods (from 15 min to 1.5 h), then assessed brain damage by TTC staining 3 d later. Figure 1A shows the time-dependent brain damage in mice. A 15 min MCAO period caused no damage, whereas a 1 h MCAO period caused severe damage, with an infarct size similar to that induced by the 1.5 h MCAO period. We chose a 15 min MCAO period for PC and a 1 h MCAO period to cause severe ischemic injury (Fig. 1A). We combined PC with severe MCAO to generate a model of ischemic tolerance. As shown in Figure 1B, the MCA was occluded transiently for 15 min for PC, and then 1 d (P1), 3 d (P3), or 6 d (P6) later, it was occluded again for 1 h (severe MCAO). In all cases, brain damage was assessed by TTC staining 3 d after severe MCAO. PC significantly decreased brain damage induced by severe MCAO (P3 and 6), suggesting that this paradigm can be used to model ischemic tolerance (Fig. 1B,C). We also used immunohistochemical staining against NeuN to assessed MCAO-evoked neuronal damage, and found that TTC staining data were well correlated with the NeuN data (Fig. 1D). Interestingly, PC-induced ischemic tolerance was observed earlier in the striatum (3 d after PC) than in the cortex (Fig. 1C). No ischemic tolerance was observed 1 d after PC. Thus, these results suggest that the time interval between PC and severe MCAO (at least 3 d) is important for the induction of ischemic tolerance.

Brain ischemic tolerance model using mouse MCAO. A, The MCA was occluded for various periods (from 15 min to 1.5 h), and 3 d later brain damage was assessed by TTC staining of coronal brain sections. A 15 min MCAO period caused no damage, whereas a 1 h MCAO period induced severe injury, mainly in the striatum and cortex. The 15 min MCAO was used for PC, while the 1 h MCAO represented severe MCAO. Scale bar, 2 mm. Sham, sham-operated mice. B, As shown in the experimental protocols, mice received PC 1 d (P1), 3 d (P3), or 6 d (P6) before severe MCAO. The images on the right depict a typical infarct evoked by severe MCAO, showing the effects of PC, which are summarized in C. The severe MCAO-evoked damage in the striatum was significantly reduced when mice received PC 3 d or 6 d earlier (P3 and P6), whereas damage in the cortex was significantly reduced only 6 d after PC (P6). PC 1 d prior (P1) had no effect on brain damage induced by severe MCAO in either brain region. Values are expressed as means ± SEM; *p < 0.05, **p < 0.01 versus severe MCAO alone; n = 4–9. C, Control. D, There was no difference in number of NeuN (green)-positive neurons between Sham and PC groups. Similar to TTC staining, the reduction in number of NeuN-positive neurons after severe MCAO was significantly inhibited by PC (P3). Scale bar, 50 μm. Values are shown as means ± SEM; **p < 0.01; n = 3–4.
Spatiotemporal pattern of astrocytic activation correlates with ischemic tolerance
To examine the role of glial cells in brain ischemic tolerance, PC-induced morphological changes in both astrocytes and microglia were analyzed by immunohistochemical staining. Figure 2 shows changes in the spatiotemporal profile of astrocytes and microglia stained with the anti-GFAP and anti-Iba1 antibodies, respectively. Ipsilaterally, GFAP immunoreactivity was significantly increased 3 and 6 d after PC in the striatum and 6 d after PC in the cortex (Fig. 2C,D). PC-induced astrocytic activation lasted at least 8 weeks (data not shown). In comparison, Iba1 immunoreactivity was increased 1 d after PC in the striatum, but there was no change in the cortex. Neither GFAP immunoreactivity nor Iba1 immunoreactivity was upregulated in the contralateral striatum or cortex (Fig. 2B). A comparison with the results in Figure 1 suggests that the spatiotemporal pattern of astrocyte activation correlates well with the spatiotemporal pattern of ischemic tolerance.

PC-induced activation of glial cells. A, Open squares show the area in the striatum and cortex used for microscopic examination. Immunostaining with anti-GFAP (green) and anti-Iba1 (red) antibodies in the contralateral (B) and ipsilateral (C) hemispheres. Scale bars: main images, 100 μm; insets, 10 μm. D, The immunofluorescence intensity was quantified. Values are shown as means ± SEM; **p < 0.01 versus contralateral side (Contra); n = 3–4. GFAP-positive and Iba1-positive signals were not affected by PC in the contralateral cortex or striatum (B). Ipsilaterally, PC induced the activation of astrocytes (i.e., upregulation of GFAP immunoreactivity) 3 and 6 d after PC in the striatum, and 6 d after PC in the cortex. Activation lasted for at least 8 weeks (data not shown). PC induced the activation of microglia (i.e., upregulated Iba1 immunoreactivity and promoted the appearance of amoeboid morphology) 1 d after PC, which lasted at least 6 d after PC in the striatum. PC caused no activation in the cortex.
Astrocytic activation induces ischemic tolerance
To elucidate whether astrocytic activation is required for ischemic tolerance, we investigated the effect of FC, a metabolic inhibitor of astrocytes (Paulsen et al., 1987), on ischemic tolerance. As shown in Figure 3, A–C, FC was injected into the striatum 2 d after PC or sham operation. Intrastriatal injection of FC suppressed PC-induced activation of astrocytes, but not microglia (Fig. 3E) and, importantly, abolished PC-induced ischemic tolerance; i.e., the PC-induced decrease in infarct size was no longer detected (Fig. 3C,D). FC administration following PC alone, i.e., without severe MCAO, produced no brain damage (Fig. 3A). FC did not affect the size of the infarct induced by severe MCAO alone, i.e., without PC (Fig. 3B). These results suggest that astrocytic activation has a major role in PC-induced ischemic tolerance. Our results also suggest that the time interval between PC and severe MCAO is important for astrocyte activation after PC.
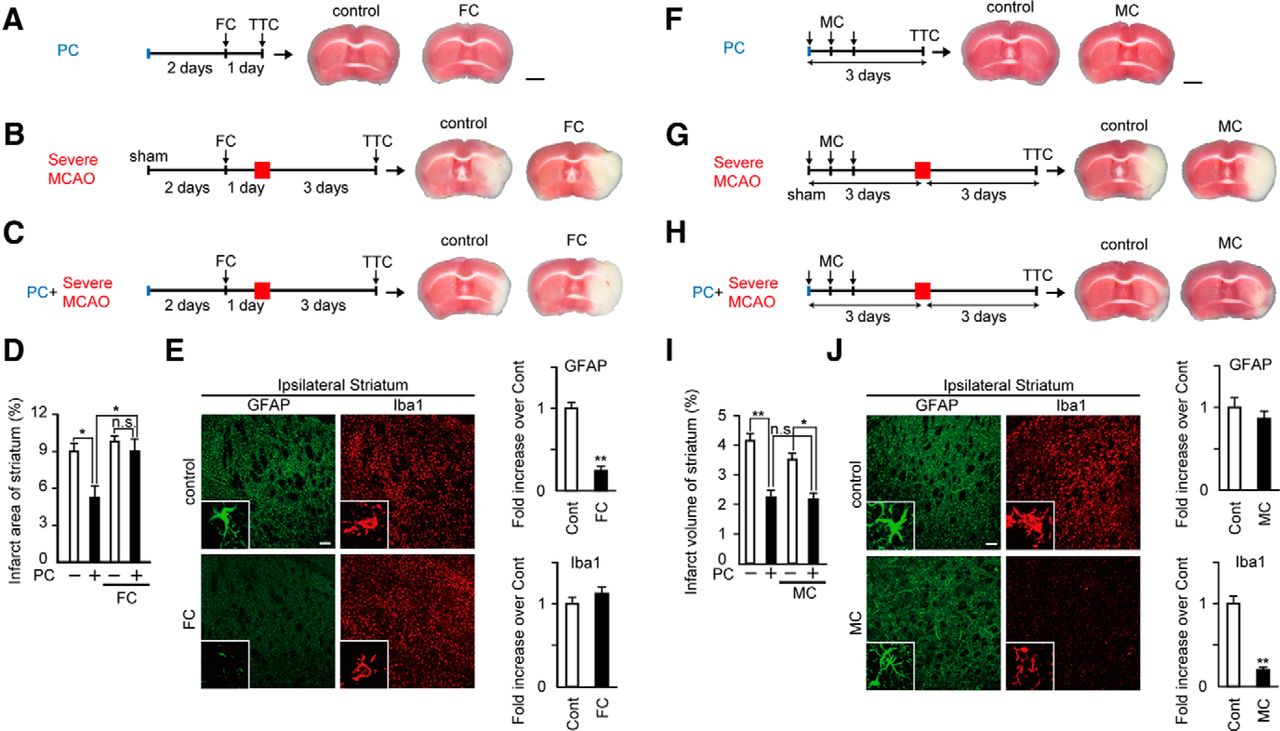
PC-induced activation of astrocytes, but not microglia, is essential for ischemic tolerance. A, As shown in the experimental protocol, after PC, mice received an intrastriatal injection of FC (1 pmol/site). Brain damage was assessed by TTC staining. FC injection after PC caused no damage. Scale bar, 2 mm. B, FC had no effect on severe MCAO (1 h MCAO)-induced ischemic injury, but FC abolished PC-induced ischemic tolerance (C; please compare with results in Fig. 1). Results are summarized in D. Values are shown as means ± SEM; *p < 0.05; n = 4–6. E, The morphology of astrocytes (GFAP: green) and microglia (Iba1: red) in the ipsilateral striatum were analyzed by immunohistochemical staining 3 d after PC. The immunofluorescence (IF) intensity was quantified. PC-induced activation of astrocytes was suppressed by FC injection, whereas that of microglia was not affected. Scale bars: main images, 100 μm; insets, 10 μm. Values are shown as means ± SEM; **p < 0.01 versus control (Cont); n = 3. F, As shown in the experimental protocols, after PC, mice received three intraperitoneal injections of MC (45 mg/kg). MC injections after PC caused no damage. Scale bar, 2 mm. MC affected neither severe MCAO (1 h MCAO)-induced ischemic injury (G) nor PC-induced ischemic tolerance (H). Results are summarized in I. Values are shown as means ± SEM; *p < 0.05, **p < 0.01; n = 5–11. J, The morphology of microglia (Iba1: red) and astrocytes (GFAP: green) in the ipsilateral striatum was assessed by immunohistochemical staining 3 d after PC and the IF intensity was quantified. PC-induced activation of microglia was suppressed by MC injection, whereas that of astrocytes was not affected. Scale bars: main images, 100 μm; insets, 10 μm. Values are shown as means ± SEM; **p < 0.01 versus Cont; n = 3.
Microglial activation is not involved in ischemic tolerance
We also investigated the effect of minocycline (MC), an inhibitor of microglial activation (Tikka and Koistinaho, 2001), on ischemic tolerance. MC was injected intraperitoneally three times as shown in Figure 3F–H. Intraperitoneal injection of MC suppressed PC-induced activation of microglia, but not astrocytes (Fig. 3J). MC administration following PC alone, i.e., without severe MCAO, produced no brain damage (Fig. 3F). In addition, MC did not affect the size of the infarct induced by severe MCAO alone, i.e., without PC (Fig. 3G). In contrast to FC, MC had no effect on ischemic tolerance (Fig. 3H,I). These findings suggest that PC-induced activation of microglia does not have a substantial role in ischemic tolerance.
The P2X7 receptor is expressed in activated astrocytes
ATP plays a central role in the interaction between neurons and astrocytes. The P2X7 receptor, an ATP-gated cation channel, mediates glia–neuron communication, especially in pathophysiological conditions (Abbracchio et al., 2009). As shown in Figure 4C, P2rx7 mRNA was upregulated after PC. To determine the localization of P2X7 receptors after PC, we performed immunohistochemical staining using P2rx7-EGFP transgenic mice. Several reports have shown that P2X7 receptors are expressed in both resting and activated microglia after MCAO (Collo et al., 1997; Yanagisawa et al., 2008). Our immunohistochemical staining also showed that P2X7 receptors were present in microglia in both sham-operated and PC-treated mice (Fig. 4A,B, Table 1). Although P2X7 receptors were not expressed in astrocytes in sham-operated control mice or 1 d after PC, a significant upregulation in astrocytes was observed in the ipsilateral striatum 3 d after PC (Fig. 4A,B, Table 1). This result is in agreement with a previous report (Franke et al., 2004). The upregulation of P2X7 receptors was dependent upon the activation of astrocytes, because FC, an inhibitor of astrocyte activation, suppressed P2X7 receptor upregulation (Fig. 4D). These results indicate that rapid (1 d after PC) and slow (3 d after PC) upregulation of P2X7 receptor mRNA levels are mainly involved in microglia and astrocytes, respectively. Thus, this result suggests that the upregulation of the temporal pattern of astrocytic P2X7 receptors correlates with that of ischemic tolerance.

P2X7 receptors are upregulated in activated astrocytes by PC. A, The localization of P2X7 receptors was analyzed by double immunohistochemical staining for P2rx7-EGFP (green), GFAP (red), and Iba1 (red) in the ipsilateral striatum after PC or sham operation. Scale bars: main images, 60 μm; insets, 27 μm. Results are summarized in B. Values are shown as means ± SEM; **p < 0.01 versus sham operation, n = 3–4. C, The time dependency of upregulation of P2rx7 mRNA in the ipsilateral striatum, assessed by quantitative RT-PCR (n = 3–4). P2X7 receptors were significantly upregulated 1, 3, and 6 d after PC. Values are shown as means ± SEM, **p < 0.01 versus naive mice (nv). sh, Sham-operated mice. D, The upregulation of P2rx7 mRNA by PC was significantly suppressed by FC, a metabolic inhibitor of astrocytes. Values are shown as means ± SEM; *p < 0.05, **p < 0.01, n = 3.
Time course of the changes in number of GFAP- and Iba1-P2X7 receptor-positive cells in the ipsilateral striatum after PC
The P2X7 receptor is essential for PC-induced ischemic tolerance
Using WT and P2X7 receptor knock-out (P2X7−/−) mice, we investigated the involvement of the P2X7 receptor in brain ischemic tolerance. There was no significant difference in the degree of severe MCAO-induced brain damage (the size of the infarct) between WT and P2X7−/− mice (Fig. 5A,B). However, PC-induced ischemic tolerance was absent in P2X7−/− mice. These findings suggest that the P2X7 receptor is essential for ischemic tolerance. To clarify whether the P2X7 receptor is involved in PC-mediated astrocyte activation, we compared PC-evoked glial activation in WT and P2X7−/− mice. PC itself did not cause any visible TTC staining in either mouse line (Fig. 5A). Immunohistochemical staining and quantitative RT-PCR analysis showed that there was no significant difference in the PC-evoked upregulation of GFAP immunoreactivity or GFAP mRNA expression in the striatum between WT and P2X7−/− mice (Fig. 5C,D). Similarly, there was no difference in microglial activation between WT and P2X7−/− mice. These results suggest that the upregulation of P2X7 receptor expression is an event downstream of astrocyte activation, and that it is essential for the induction of ischemic tolerance.

Absence of PC-induced ischemic tolerance in P2X7−/− mice. A, PC itself had no effect on TTC staining in either WT or P2X7−/− mice. There was no significant difference in severe MCAO-evoked brain damage (the size of the infarct) between WT and P2X7−/− mice. PC-induced ischemic tolerance was abolished in P2X7−/− mice. Results are summarized in B. Scale bar, 2 mm. Values are shown as means ± SEM; *p < 0.05, **p < 0.01; n = 7–8. C, D, There was no difference in PC-evoked glial activation between WT and P2X7−/− mice. Both WT and P2X7−/− mice showed similar morphologies for astrocytes (GFAP: green) and microglia (Iba1: red) in the ipsilateral striatum 3 d after PC. Scale bars: main images, 100 μm; insets, 10 μm. The PC-evoked upregulation of mRNAs for GFAP and Iba1 (Aif1) in P2X7−/− mice was almost identical to that in WT mice. Values are shown as means ± SEM; *p < 0.05, **p < 0.01; n = 3–5.
PC-induced upregulation of HIF-1α and EPO is dependent on the P2X7 receptor
To elucidate the molecular mechanisms involved in astrocytic P2X7 receptor-mediated ischemic tolerance, we focused on HIF-1α and EPO, well known mediators of ischemic tolerance. First, we investigated the spatiotemporal pattern of HIF-1α protein expression by immunohistochemical staining. Similar to previous reports (Baranova et al., 2007), HIF-1α was transiently increased mainly in neurons (1 d after PC; Fig. 6B,C); this was observed in both WT and P2X7−/− mice. Thereafter, in addition to neurons, HIF-1α was also upregulated in activated astrocytes. Interestingly, this upregulation was dependent on the P2X7 receptor as it was observed only in WT mice, and not in P2X7−/− mice (3 and 6 d after PC; Fig. 6A,C). Next, we investigated the temporal profile of EPO mRNA expression. Three and six days after PC, mRNA levels of EPO, a major target of HIF-1α, were significantly upregulated in WT mice, but not in P2X7−/− mice (Fig. 6D). One day after PC, no changes in EPO mRNA levels were seen. Thus, both the temporal pattern of EPO mRNA upregulation and dependency of P2X7 receptors on EPO mRNA upregulation correlated well with those of ischemic tolerance.

PC-evoked upregulation of astrocytic HIF-1α and EPO require the P2X7 receptor. A–C, Immunohistochemical analysis of HIF-1α in the ipsilateral striatum. Typical pictures and quantitative data are shown in A–C, respectively. Representative immunohistochemical pictures stained with anti-HIF-1α (red), anti-GFAP (green), and anti-NeuN (green) antibodies in the ipsilateral striatum. At 1 d after PC, HIF-1α expression was increased mainly in neurons, and this upregulation was independent of the activation of P2X7 receptors; i.e., upregulation of HIF-1α was observed in both WT and P2X7−/− mice. In contrast, 3 and 6 d after PC, HIF-1α expression was increased in astrocytes. This upregulation was dependent on the P2X7 receptor; i.e., astrocytic upregulation of HIF-1α on day 3 and 6 after PC was significantly higher in WT mice than in P2X7−/− mice. For quantitative analysis, we used the index of percentage of HIF-1α-positive cells (see the Materials and Methods section for details). Scale bars: A, B, main images, 30 μm; insets, 12 μm. Values are shown as means ± SEM; **p < 0.01 versus WT-d3; ##p < 0.01 versus WT-d6; n = 3–6. sh, Sham-operated mice; D, PC significantly upregulated mRNA for EPO, a well known target molecule of HIF-1α, at 3 and 6 d after PC in WT mice, but not in P2X7−/− mice. Values are shown as means ± SEM; *p < 0.05, **p < 0.01; n = 3–6. nv, Naive mice.
Activation of P2X7 receptors result in upregulation of HIF-1α in astrocytes
Finally, we tested whether activation of P2X7 receptors resulted in the upregulation of HIF-1α in astrocytes. Although P2X7 receptor expression was very low in the sham-operated mouse brain (Fig. 4A), P2X7 receptors were present in primary cortical astrocyte cultures obtained from WT mice, but not P2X7−/− mice (Fig. 7C), and have been reported to be functional (Panenka et al., 2001). Thus, primary cultures of astrocytes obtained from WT and P2X7−/− mice were stimulated with BzATP, an agonist of the P2X7 receptor, and the induction of HIF-1α was quantitatively analyzed by Western blotting. BzATP increased the expression of HIF-1α in a concentration-dependent manner in WT astrocytes, but not P2X7−/− astrocytes (Fig. 7A,B).

BzATP-evoked upregulation of HIF-1α in primary cultures of astrocytes. A, Cortical astrocytes derived from WT or P2X7−/− mice were treated with BzATP, a P2X7 receptor agonist, for 24 h at the indicated concentrations, and then Western blotting was performed. HIF-1α protein was increased by BzATP in WT astrocytes, but not in P2X7−/− astrocytes. Data are representative of four independent experiments. C, control. B, The BzATP-evoked increase in HIF-1α protein. Increase in HIF-1α was normalized to β-actin. Data show the fold increase over control (no treatment). Values are shown as means ± SEM; **p < 0.01 versus WT-control; ## p < 0.01 versus WT-50 μM; n = 4–6. C, The expression of P2X7 receptors in cortical astrocytes was assessed using Western blotting. The P2X7 receptor was expressed in cultured astrocytes obtained from WT mice, but not from P2X7−/− mice.
Discussion
The key findings of our study are as follows. (1) Astrocyte activation has an indispensable role in the induction of brain ischemic tolerance. (2) P2X7 receptor expression is upregulated in astrocytes after PC, and this receptor upregulation is essential for ischemic tolerance. (3) HIF-1α, a master molecule for oxygen homeostasis, is upregulated in a P2X7 receptor-dependent manner in astrocytes, followed by the subsequent induction of neuroprotective molecules, such as EPO, which likely plays a role in ischemic tolerance. Unlike the majority of previous ischemic tolerance studies, which focused on neurons, we demonstrate the necessity of astrocytes in the induction of ischemic tolerance (Fig. 8).

Schematic diagram of the mechanisms underlying astrocyte-mediated brain ischemic tolerance. Severe ischemia induces neuronal death (without PC), whereas PC does not cause ischemic injury. Although neuron-derived HIF-1α is upregulated 1 d after PC, this event has no effect on brain injury by severe ischemia. In contrast, 3 d after PC, the upregulation of P2X7 receptors, followed by the upregulation of HIF-1α and neuroprotective molecules (EPO, etc.), is observed in activated astrocytes. Upregulation of P2X7 receptors provides neuroprotection against severe ischemia and induces ischemic tolerance.
Astrocyte-mediated ischemic tolerance
Astrocytes regulate various neuronal functions. For example, they enhance and inhibit synaptic transmission by releasing glutamate (Araque et al., 1998) and ATP (Koizumi et al., 2003; Pascual et al., 2005), respectively. Furthermore, they produce and release various neurotrophic factors (Ridet et al., 1997), and contribute to energy metabolism (Benarroch, 2005). Thus, astrocytes play crucial roles in the regulation of brain function, likely including the induction of ischemic tolerance. However, to date, almost all reports on ischemic tolerance have focused exclusively on neuron-dependent mechanisms. Recently, Dirnagl et al. (2009) reported that several molecules play key roles in PC, including adenosine, adenosine A1 receptor, Toll-like receptors, HIF, and excitatory amino acid transporters (Dirnagl et al., 2009). Interestingly, all of these molecules are expressed by astrocytes. Thus, for a better understanding of PC and ischemic tolerance, researchers should carefully evaluate the contribution of astrocytes to these phenomena.
P2X7 receptor as an initiator of astrocyte-mediated ischemic tolerance
We showed that (1) the spatiotemporal pattern of astrocytic activation correlated well with that of ischemic tolerance (Figs. 1, 2) and (2) inhibition of astrocytic activation abolished PC-induced ischemic tolerance (Fig. 3), suggesting the importance of astrocytic activation as an initial process of ischemic tolerance. We previously showed that ATP has a central role in mediating neuron-to-glia communication, especially in pathological conditions (Tsuda et al., 2003; Koizumi et al., 2007; Imura et al., 2013). It has been reported that P2Y1, P2X2, P2X4, and P2X7 receptors are upregulated after ischemic injury (Franke and Illes, 2006). In the present study, we found that P2X7 receptor expression emerged after PC (Fig. 4C). Therefore, we focused on the P2X7 receptor and found that the upregulation of this receptor in astrocytes was essential for PC-mediated induction of ischemic tolerance. The P2X7 receptor is an ionotropic P2 receptor channel that regulates various cellular responses through interaction with extracellular signal-regulated kinase, phosphatidylinositol 3-kinase (PI3K)/Akt, and mitogen-activated protein kinases (Duan and Neary, 2006; Sperlágh et al., 2006). In the CNS, the P2X7 receptor has been well studied in microglia, and the ATP/P2X7 receptor has been found to control numerous microglial responses (Chakfe et al., 2002; Suzuki et al., 2004). However, the localization of P2X7 receptors in the CNS remains controversial because immunohistochemical staining patterns are inconsistent between antibodies (Yu et al., 2008). Thus, using P2rx7-EGFP mice, we investigated the precise distribution of the P2X7 receptor after PC, and showed that (1) in the control striatum, P2X7 receptors were mainly present in microglia and no, or only faint, expression was seen in astrocytes and (2) upon PC (3 d after), P2X7 receptors increased in astrocytes. The upregulation of the temporal pattern of astrocytic P2X7 receptors correlated with that of ischemic tolerance (Figs. 1, 4A,B), and inhibiting the upregulation of astrocytic P2X7 receptors by FC administration resulted in the disappearance of ischemic tolerance (Figs. 3, 4D). In addition, ischemic tolerance was absent in P2X7−/− mice (Fig. 5). These findings demonstrate the importance of P2X7 receptors in astrocytes, rather than microglia. Furthermore, the observation that the spatiotemporal pattern of ischemic tolerance correlated well with that of astrocytic activation, but not microglial activation (Figs. 1, 2), also supports this concept.
The PC-mediated activation of astrocytes resulted in the induction of ischemic tolerance. However, although P2X7−/− mice showed astrocytic activation similar to WT mice in response to PC, they failed to exhibit ischemic tolerance (Fig. 5). FC inhibited the PC-evoked upregulation of P2X7 receptors in astrocytes (Fig. 4D). This strongly suggests that astrocytic activation itself is not necessary for the induction of ischemic tolerance, but that the upregulation of P2X7 receptors in activated astrocytes is essential. The findings that FC, known to inhibit astrocytic activation by suppressing mitochondrial metabolism, inhibited both the PC-evoked upregulation of P2X7 receptors in astrocytes and ischemic tolerance, would also support this idea (Figs. 3, 4D). However, the molecular mechanisms underlying astrocytic upregulation of P2X7 receptors remain to be clarified. With regard to the effect of FC, we cannot exclude the possibility that FC might also reduce secretion of ATP by inhibiting mitochondrial function, followed by inhibition of the ATP/P2X7 receptor-mediated signals in astrocytes, leading to disappearance of ischemic tolerance.
Molecular mechanisms of astrocyte-mediated ischemic tolerance
Among effectors responsible for ischemic tolerance, HIF has received the greatest attention and is considered a master molecule that initiates a multitude of downstream signaling cascades (Gidday, 2006). HIF-1α plays an essential role in cellular and systemic oxygen homeostasis by regulating the expression of genes involved in glycolysis, erythropoiesis, angiogenesis, and numerous other processes (Kaelin and Ratcliffe, 2008). Thus, it is considered a master molecule that induces ischemic tolerance (Gidday, 2006; Dirnagl et al., 2009). However, although brain HIF-1α clearly plays a key role in PC-induced ischemic tolerance (Bergeron et al., 2000), a recent study by Baranova et al. (2007) using neuron-specific HIF-1α-deficient mice demonstrated that ischemic tolerance was not dependent on neuronal HIF-1α (Baranova et al., 2007). Because HIF-1α is expressed in both neurons and astrocytes (Chavez et al., 2006), we analyzed the spatiotemporal profile of HIF-1α expression after PC. As shown in Figure 6C, HIF-1α levels rose rapidly in neurons and peaked 1 d after PC. There was no significant difference in neuronal HIF-1α expression between WT and P2X7−/− mice. In comparison, HIF-1α levels rose slowly in astrocytes, peaking 6 d after PC. Interestingly, the increase in astrocytic HIF-1α levels was dependent on the P2X7 receptor because expression was dramatically inhibited in P2X7−/− mice at 3 and 6 d. These findings show that the PC-evoked increase in HIF-1α expression occurs in both cell types and is temporally regulated in a cell type-dependent manner. Baranova et al. (2007) also showed that the PC-evoked increase in HIF-1α protein expression exhibits a biphasic pattern—a quick and transient increase (within 12 h), followed by a slow and sustained increase (after 2 d; Baranova et al., 2007). This raises the possibility that the quick and slow increase in HIF-1α expression may be due to neurons and astrocytes, respectively. In addition, such a slow-onset but long-lasting increase in HIF-1α in astrocytes may be important for inducing ischemic tolerance. Further investigations are required to confirm this possibility.
HIF-1α is rapidly hydroxylated by prolyl hydroxylase in normoxia. This process is inhibited under hypoxic conditions, allowing HIF-1α protein to escape degradation and accumulate within cells (Bruick and McKnight, 2001). However, HIF-1α is also regulated by oxygen-independent transcription pathways such as PI3K/Akt/mTOR (Zhong et al., 2000; Agani and Jiang, 2013). Interestingly, activation of P2X7 receptors leads to activation of PI3K/Akt signaling (Jacques-Silva et al., 2004) and upregulates HIF-1α target genes (Adinolfi et al., 2012). Furthermore, we demonstrated that HIF-1α expression was increased by BzATP, a P2X7 receptor agonist, in primary astrocyte cultures (Fig. 7A,B). Therefore, we hypothesize that the slow increase in HIF-1α expression in astrocytes is likely independent of oxygen status, and is instead dependent on PC-induced P2X7 receptor upregulation.
Microglia make a small contribution to ischemic tolerance
Microglia, another type of glial cell, function as highly sensitive sensors, constitutively surveying the brain microenvironment (Wake et al., 2009). In this study, microglia appeared to have no direct effect on brain ischemic tolerance. The findings that the spatiotemporal pattern of microglial activation did not correlate with that of ischemic tolerance (Figs. 1, 2), and that the inhibition of microglial activation had no effect on ischemic tolerance (Fig. 3) clearly show that microglia are not involved in ischemic tolerance.
Microglia can sense changes in the brain microenvironment, including PC. In the ipsilateral striatum, PC induced the activation of microglia, followed by the activation of astrocytes (Fig. 2C,D). Microglia release various cytokines, which subsequently activate astrocytes (Hanisch, 2002). Such sequential patterns of glial activation are also seen in other pathological models, such as neuropathic pain (Shibata et al., 2011). Thus, microglia may be indirectly involved in ischemic tolerance. However, in the ipsilateral cortex, where ischemic tolerance was induced, astrocytes were activated without prior microglial activation (Figs. 1, 2). Although immunohistochemical staining with the anti-Iba1 antibody may not have been able to detect low levels of microglial activation, our findings suggest that activation of microglia is not necessarily required for the induction of ischemic tolerance in the brain.
Together, our findings indicate that astrocytes upregulate P2X7 receptor expression upon PC stimulation, and that they have an indispensable role in the induction of ischemic tolerance. Although microglial P2X7 receptors are considered to be death receptors under pathological conditions, we demonstrate for the first time that the astrocytic P2X7 receptor is upregulated by PC and is involved in ischemic tolerance. Therefore, when evaluating the role of P2X7 receptors in ischemic tolerance, a cell type-specific consideration is required. In addition, although it is well established that neurons play an important role in the induction of ischemic tolerance, our findings suggest that glial cells may be more important. Thus, a better understanding of ischemic tolerance will require researchers to evaluate the combined contribution of neurons and glial cells. This new insight into the role of glial cells in the cell-autonomous and noncell-autonomous mechanisms that mediate neuroprotection should promote the development of novel therapeutic strategies for brain ischemia.
Footnotes
This study was supported by CREST (to S.K.) and KAKENHI on Innovative Areas (25116512 and 25117003 to S.K.) and on Challenging Exploratory Research (25670622 to S.K.). We thank Koizumi lab members for helpful comments and discussion.
The authors declare no competing financial interests.
- Correspondence should be addressed to Schuichi Koizumi, Department of Neuropharmacology, Interdisciplinary Graduate School of Medicine, University of Yamanashi, 1110 Shimokato, Chuo, Yamanashi 409-3898, Japan. skoizumi{at}yamanashi.ac.jp





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}