

Abstract
Maturation of excitatory drive onto fast-spiking interneurons (FS INs) in the visual cortex has been implicated in the control of the timing of the critical period for ocular dominance plasticity. However, the mechanisms that regulate the strength of these synapses over cortical development are not understood. Here we use a mouse model to show that neuregulin (NRG) and the receptor tyrosine kinase erbB4 regulate the timing of the critical period. NRG1 enhanced the strength of excitatory synapses onto FS INs, which inhibited ocular dominance plasticity during the critical period but rescued plasticity in transgenics with hypoexcitable FS INs. Blocking the effects of endogenous neuregulin via inhibition of erbBs rescued ocular dominance plasticity in postcritical period adults, allowing recovery from amblyopia induced by chronic monocular deprivation. Thus, the strength of excitation onto FS INs is a key determinant of critical period plasticity and is maintained at high levels by NRG–erbB4 signaling to constrain plasticity in adulthood.
SIGNIFICANCE STATEMENT Despite decades of experimentation, the mechanisms by which critical periods of enhanced synaptic plasticity are initiated and terminated are not completely understood. Here we show that neuregulin (NRG) and the receptor tyrosine kinase erbB4 determine critical period timing by controlling the strength of excitatory synapses onto FS INs. NRG1 enhanced excitatory drive onto fast spiking interneurons, which inhibited ocular dominance plasticity in juveniles but rescued plasticity in transgenics with hypoexcitable FS INs. Blocking the effects of endogenous neuregulin via inhibition of erbBs rescued ocular dominance plasticity in adults, allowing recovery from amblyopia induced by chronic monocular deprivation. Thus, in contrast to prevailing views of the termination of the critical period, active maintenance of strong excitation onto FS INs constrains plasticity in adults.
Introduction
The shift in the ocular dominance of binocular neurons induced by monocular deprivation (MD) is the canonical model of receptive field plasticity confined to a developmental critical period. Research performed primarily in mice is beginning to reveal the mechanisms by which critical period plasticity is regulated. It is well appreciated that the perisomatic inhibition mediated by fast-spiking interneurons (FS INs) exerts powerful control over the excitability and plasticity of downstream pyramidal neurons. Accordingly, enhancing the output of FS INs can enable plasticity in precritical period mice, whereas decreasing the output can reactivate plasticity in adults (Huang et al., 1999; Fagiolini and Hensch, 2000; Di Cristo et al., 2007; Kuhlman et al., 2013; Krishnan et al., 2015). However, ocular dominance plasticity persists after the developmental maturation of perisomatic inhibition (Sawtell et al., 2003; Pham et al., 2004; Lehmann and Löwel, 2008; Huang et al., 2010), suggesting that regulatory control is upstream of inhibitory output.
Alternatively, we have proposed that the maturation of excitatory drive onto FS INs determines the timing of the critical period (Huang et al., 2010; Gu et al., 2013). Importantly, several proteins that regulate synaptic strength and/or number, including neuregulin (NRG) and neuronal activity-regulated pentraxin (NARP), are highly enriched at excitatory synapses onto FS INs. Signaling mediated by NRGs has been implicated in the maturation of excitation onto FS INs, and dysregulation of NRG1 is a risk factor for schizophrenia (Rico and Marín, 2011; Penzes et al., 2013). NRGs are synthesized as transmembrane precursors, requiring proteolytic cleavage to release a soluble peptide containing an epidermal growth factor (EGF)-like sequence that activates the receptor tyrosine kinase erbB4 (Mei and Nave, 2014). ErbB4 is a primary target of NRG1, and erbB4 expression is highly enriched in parvalbumin+ (PV+) interneurons of prefrontal cortex, hippocampus and amygdala (Fazzari et al., 2010; Neddens and Buonanno, 2010; Bean et al., 2014; Lu et al., 2014).
Small NRG1-derived recombinant peptides corresponding to the soluble EGF domain common to all NRG1 splice variants have been used to elucidate the role of erbB4 activation in vivo. A single application of a NRG1 peptide acutely regulates the intrinsic properties and ion channel distribution of erbB4+ interneurons (Janssen et al., 2012; Li KX et al., 2012; Mitchell et al., 2013). In contrast, repeated delivery of a NRG1 peptide is thought to recruit ErbB4 to the synapse, enabling a tyrosine kinase-dependent stabilization of PSD-95 and an increase in AMPAR-mediated EPSC amplitudes (Li et al., 2007; Ting et al., 2011). Accordingly, repeated delivery of a NRG1 peptide increases the amplitudes of mEPSCs recorded in FS INs, accompanied by an increase in the amplitudes of AMPAR-mediated excitatory currents and an increase in GluR1 surface expression (Abe et al., 2011). Blocking endogenous NRG signaling, through pharmacological inhibition or genetic ablation of ErbB4, reverses these effects. Specifically, inhibition of erbB4 with a function-blocking peptide reduces the number and size of excitatory synapses on cultured GABAergic interneurons (Ting et al., 2011). Longer-term manipulations of NRG/erbB signaling may also impact the number of excitatory synapses onto FS INs. Indeed, treatment with a NRG1 peptide induces a delayed increase in the frequencies of mEPSCs in FS INs, and a decrease in mEPSC frequencies after erbB4 deletion is revealed only in adults (Abe et al., 2011; Yang et al., 2013).
Changes in FS IN excitability mediated by NRG–erbB4 signaling impact the plasticity of excitatory synapses onto downstream pyramidal neurons. NRG1 treatment of hippocampal slices suppresses LTP of Schaffer collaterals in hippocampus and inhibits activity-dependent enhancement of synaptic NMDAR currents (Pitcher et al., 2011; Tamura et al., 2012). Similarly, transgenic deletion of erbB4 in PV+ interneurons in the amygdala increases LTP of inputs onto pyramidal neurons and impairs fear conditioning (Lu et al., 2014). Together, this suggests that excitatory synapses onto FS INs are the major cellular target of NRG–ErbB4 signaling, and this pathway exerts powerful control over FS IN excitability and plasticity of downstream pyramidal neurons. Here we show that manipulation of this pathway exerts bidirectional control over the strength of excitatory drive onto FS INs in the visual cortex, allowing for reversible regulation of the critical period for ocular dominance plasticity throughout life.
Materials and Methods
Wild-type and NARP−/− mice (Kirkpatrick et al., 2000), equal numbers of males and females, of C57BL/6, 129/SVJII mixed genetic background were raised on a 12 h light/dark cycle, with food and water available ad libitum. PV-Cre/ErbB4−/− mice, obtained by crossing mice expressing Cre recombinase from the PV locus (Pva1btm1(cre)Arbr) with mice expressing a floxed ErbB4 allele, were a generous gift from Dr. A. Buonanno (NIH–NICHD, Bethesda, MD; Shamir et al., 2012). All procedures conformed to the guidelines of the U.S. Department of Health and Human Services and the Institutional Animal Care and Use Committees of the University of Maryland and Johns Hopkins University. MD was performed under ketamine/xylazine anesthesia (50 mg/10 mg/kg, i.p.). The margins of the upper and lower lids of one eye were trimmed and sutured together. The animals were returned to their home cages and disqualified in the event of suture opening or infection. For dark exposure (DE), animals were placed in a light-tight dark room, with animal care provided under far red illumination.
Immunohistochemistry.
Postnatal day 30 (P30) mice were perfused transcardially with 4% paraformaldehyde. Brains were incubated in 4% paraformaldehyde overnight, 30% sucrose for 1 d at 4°C, and cryoprotectant for 1 d at 4°C. Forty-micrometer coronal slices were prepared on a freezing microtome (Leica). Sections were incubated in a blocking solution (4% normal goat serum in PBS) for 30 min at room temperature before incubation with primary antibodies (mouse monoclonal anti-parvalbumin, 1:1000; Millipore) or rabbit polyclonal anti-ErbB4 (1:500; a gift from Dr. A. Buonanno) in a blocking solution overnight at 4°C, followed by secondary antibodies (Alexa 555 goat anti-mouse IgG, 1:300; Invitrogen) or biotinylated goat anti-rabbit IgG (1:300; Vector Laboratories) at room temperature for 4 h. ErbB4 was visualized after incubation in Alexa 488-conjugated streptavidin (1:300; Invitrogen) at room temperature for 4 h. Slices were washed with PBS three times for 10 min between each step. Z-stack of eight images of V1b at 1 μm intervals of 1024 × 1024 pixels were acquired on a Zeiss LSM710 confocal microscope with a 40 × lens.
Quantitative immunoblots.
P30 mice received a single intraperitoneal injection of NRG1 (10 ng/kg in 0.9% sodium chloride solution) or vehicle 90 min before dissection. Primary visual cortices were dissected bilaterally as described previously (Scott et al., 2010) under isoflurane anesthesia (4% in 100% O2; flow rate, 800 cc/min). Tissue was immediately homogenized with a sonic dismembrator (model 100; Thermo Fisher Scientific) in ice-cold lysis buffer (150 mm NaCl, 1% NP-40, 50 mm Tris-HCl, pH 8.0) containing a protease inhibitor mixture (Roche). Protein concentrations were determined using the BCA protein assay kit (Thermo Fisher Scientific). Sample concentrations were normalized in 4 × SDS loading buffer and boiled for 5 min. Fifty micrograms of total protein per lane were separated on polyacrylamide gels, followed by transfer to nitrocellulose. Nitrocellulose was blocked with 4% nonfat dry milk in PBS for 30 min and incubated with rabbit polyclonal pTyr-1284 erbB4 antibody (1:500 in blocking solution; Abcam) overnight at 4°C. Alexa 488-conjugated goat anti-rabbit IgG was used as a secondary antibody (1:500 in blocking solution, incubated at room temperature for 4 h; Life Technologies). Membranes were washed three times for 10 min with Tris-buffered saline with 0.05% Tween 20 between each step. The proteins were detected with Typhoon TRIO Variable Mode Imager (GE Healthcare) and analyzed with ImageQuant TL (GE Healthcare). The membranes were stripped with 100 mm glycine-HCl (pH 3.0) and reprobed with a rabbit polyclonal anti-erbB4 antibody.
Slice electrophysiology.
Visual cortical slices were prepared as described previously (Guo et al., 2012). Slices (300 μm thick) were cut in ice-cold dissection buffer containing (in mm) 212.7 sucrose, 5 KCl, 1.25 NaH2PO4, 10 MgCl2, 0.5 CaCl2, 26 NaHCO3, and 10 dextrose, bubbled with 95% O2/5% CO2, pH 7.4. Slices were transferred to normal artificial CSF (ACSF) for at least 1 h before recording. Normal ACSF was similar to the dissection buffer except that sucrose was replaced by 124 mm NaCl, MgCl2 was lowered to 1 mm, and CaCl2 was raised to 2 mm. Visualized whole-cell recordings were made from layer 2/3 neurons with glass pipettes (4–6 MΩ). Data were filtered at 2 kHz and digitized at 5–10 kHz using Igor Pro (Wavemetrics). Only cells with membrane potentials more negative than −65 mV, series resistance <20 MΩ, and input resistance larger than 100 MΩ were studied. Cells were excluded if input resistance changed >20% over the entire experiment. Miniature EPSC recordings were performed with an intracellular solution containing (in mm) 130 Cs-gluconate, 8 KCl, 1 EGTA, 10 HEPES, 4 (Na)ATP, and 5 QX-314 (pH adjusted to 7.25 with CsOH, 280–290 mOsm) under voltage clamp (vehicle, −70 mV). TTX (1 μm), APV (100 μm), and picrotoxin (10 μm) were included in the bath. Events were detected and analyzed using Mini Analysis (Synaptosoft).
In vivo electrophysiology was performed under light isoflurane anesthesia (∼1.5% in 100% O2 via modified nose cone). The dura covering binocular visual cortex was exposed through a hole (∼3 mm diameter) in the skull. The exposed brain was kept moist with artificial CSF (in mm 124 NaCl, 5 KCl, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, 26 NaHCO3, and 10 dextrose, bubbled with 95% O2/5% CO2, pH 7.4) in a room with supplemental humidity (ZD300Y0; Zenith). Subjects were retained in a stereotax in a darkened room (without visual stimulation) between measurements. Body temperature was maintained at 37°C via a circulating water heating pad (T/PUMP; Gaymar Industries), monitored with a rectal probe (BAT-12; Sensortek). A broadband signal was collected from the lateral aspect (binocular region) of the primary visual cortex [site of the largest ipsilateral eye visually evoked potential (VEP), typically 3 mm lateral to the intersection of lambda and the midline], with a tungsten microelectrode (0.5 MΩ) relative to a ground screw in the frontal bone.
A 50 Hz low-pass filter was used to isolate VEPs in response to 1 Hz reversals of 100% contrast gratings (0.04 cycles/° and 40 cd/m2 luminosity) presented on a computer monitor 25 cm from the eyes. VEPs were averaged in synchrony with the visual stimulus using OpenEX software (TDT). A bandpass filter (700–7000 Hz) was used to isolate multiunit activity, which was sorted into single units based on waveform shape and principal component analysis (OpenEx software; TDT). Individual waveforms were aligned by the most negative point, and amplitudes were normalized from −1 to 1. Single units recorded from layer 2/3 of binocular visual cortex of head-fixed mice sorted into two clusters based on waveform characteristics (the height of the positive peak relative to the initial negative trough and the slope of the waveform 0.5 ms after the initial trough), representing fast-spiking (presumptive PV interneurons) and regular-spiking (RS; predominantly pyramidal neurons; after Niell and Stryker, 2008) neurons. Spontaneous firing rates were measured in response to 100 s of blank screen. One hertz reversals of 100% contrast gratings were presented at nine orientations ranging from 0° (vertical) to 180 °. Evoked spiking rates are reported in response to presentation of the visual stimulus in the preferred orientation.
The recombinant human NRG1-β1/HRG1-β1EGF domain (7.5 kDa; R&D Systems), corresponding to residues 176–246 of the soluble EGF domain of heregulin β1 common to all NRG1 splice variants, was dissolved in 100% saline. PD158780 (Tocris) was dissolved in 10% Tween 80, 20% DMSO, and 70% saline.
Statistics.
An unpaired two-tailed t test was used to determine statistical significance between two experimental groups. A one-way ANOVA was used to determine the statistical significance between three or more independent experimental groups, followed by a subsequent Tukey–Kramer honestly significant difference post hoc test for pairwise comparisons, when appropriate. A Kolmogorov–Smirnov test was used to determine the statistical significance between the distributions of two independent data sets. In all cases, the sample size (n) used for statistical comparison equals the number of subjects.
Results
NRG1 inhibits ocular dominance plasticity during the critical period
The receptor tyrosine kinase erbB4 is a primary target of NRG signaling in the brain and is highly enriched in PV+ interneurons of prefrontal cortex, hippocampus, and amygdala (Fazzari et al., 2010; Neddens and Buonanno, 2010; Bean et al., 2014; Lu et al., 2014). To determine the distribution of this signaling pathway in the mouse visual cortex, we performed immunohistochemistry for erbB4 and parvalbumin, the calcium-binding protein enriched in FS INs in the cortex. Immunoreactivity for erbB4 was highly colocalized with parvalbumin in the visual cortex of postnatal day 30 mice (P30; percentage of observations: PV+EB+, 85.58 ± 0.82%; PV+EB−, 4.54 ± 0.99%; PV−EB+, 9.88 ± −1.39%; 250 cells from 18 slices; six subjects; Fig. 1A,B). Furthermore, peripheral administration of a 71 amino acid (7.5 kDa) peptide corresponding to residues 176–246 of the soluble EGF domain of NRG-1-β1 (hereafter called NRG1; 10 ng/kg, i.p.) significantly increased the phosphorylation of erbB4 receptors in visual cortex (Fig. 1C). To determine the effect of repeated delivery of the NRG1 peptide on the strength of excitatory synapses onto FS INs, whole-cell recordings of pharmacologically isolated mEPSCs were performed in slices of visual cortex prepared from G42 mice in which the expression of GFP is restricted to FS basket cells (Taniguchi et al., 2013). Systemic administration of NRG1 (10 ng/kg, i.p., two times per day for 3 d) to P30 wild-type mice significantly increased the amplitudes, but not the frequencies, of mEPSCs onto PV interneurons (Fig. 1D–F). To confirm that these responses to NRG1 are mediated by erbB4 receptors on cortical FS INs, we repeated these experiments in transgenic mice with a conditional deletion of erbB4 restricted to PV+ interneurons (PV-erbB4−/−; Shamir et al., 2012). The frequency of mEPSCs onto FS INs was significantly reduced in these mice. In addition, the regulation of mEPSC amplitudes by NRG1 treatment was absent in PV-erbB4−/− mice (Fig. 1G).

NRG1 activates cortical erbB4 receptors and increases excitation onto PV+ interneurons. A, Immunohistochemical colocalization of ErbB4 receptor (green) and PV (red) in P30 visual cortex. B, Quantification of colocalization as a percentage of all observations of fluorescence: PV+Erb−, 4.54 ± 0.99%; PV+EB+, 85.58 ± 0.82%; PV−EB+, 9.88 ± 1.39%. n = 250 neurons from 18 slices from six subjects. One-way ANOVA, F(4,17) = 1718.07, p < 0.0001; *p < 0.01, Tukey–Kramer post hoc test. C, Single intraperitoneal injection of NRG1 (10 ng/kg in 0.9% sodium chloride solution) induced a significant increase in phosphorylation of erbB4 receptors in primary visual cortex. Inset, Representative example of immunoblot for phospho-erbB4 and total erbB4. *p < 0.05, t test. D, Representative examples of 2 s current traces of pharmacologically isolated mEPSCs in PV interneurons from layer 2/3 of visual cortex slices after vehicle (black) or NRG1 (10 ng/kg; i.p.; 2 times per day for 3 d; red) treatment. E, Average of mEPSCs recorded in both conditions. Isolated mEPSCs were averaged first by neuron and second by condition. F, NRG1 treatment increased the amplitudes (left; *p = 0.035, t test) but not the frequencies (right; t test, p = 0.201) of mEPSCs in PV INs. G, Absence of response to NRG1 in PV-erbB4−/− mice. The numbers of mice, neurons are indicated within the columns. Vh, Vehicle.
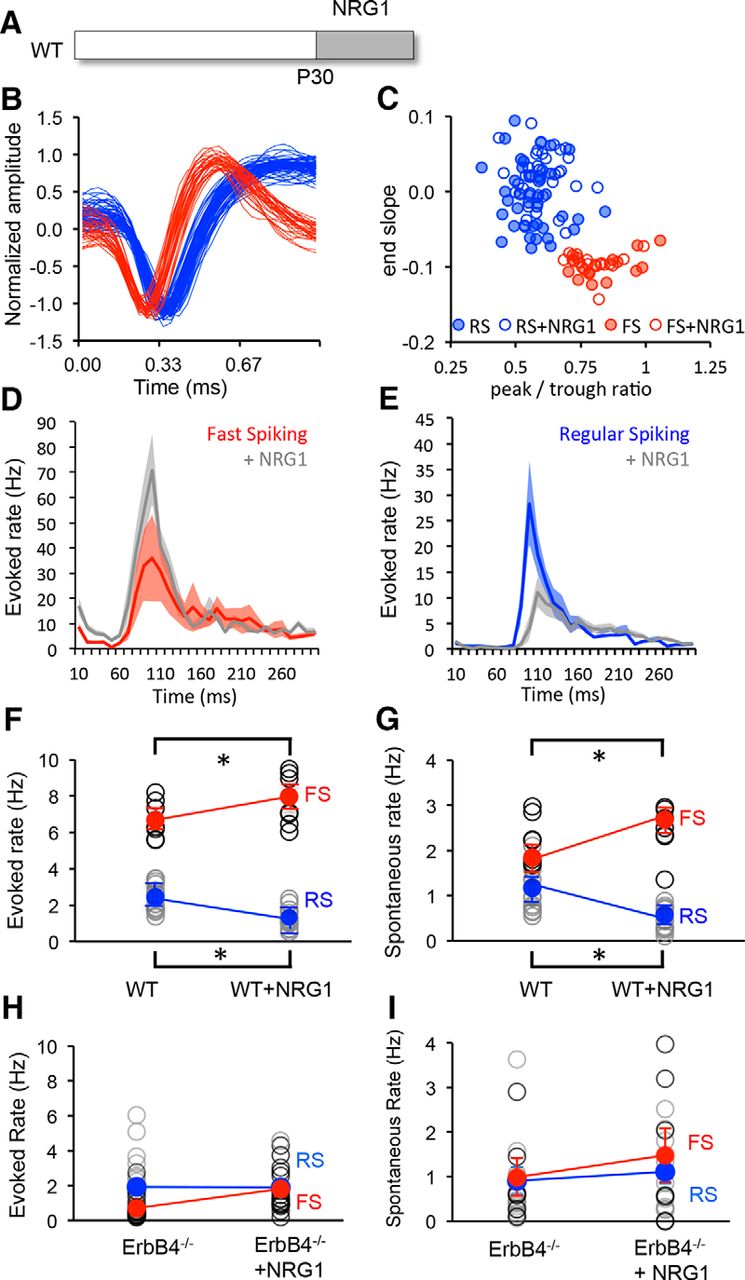
To determine whether NRG1 can be used to control excitation of FS INs in vivo, NRG1 was delivered to wild-type mice at the peak of the critical period (P30; 10 ng/kg, i.p., two times per day for 3 d). Single units recorded from layer 2/3 of binocular visual cortex of head-fixed mice were sorted into two clusters based on waveform characteristics, representing FS (presumptive PV interneurons) and RS (predominantly pyramidal neurons; after Niell and Stryker, 2008) neurons. NRG1 treatment did not impact the waveform characteristics or the clustering of units based on waveform parameters [Fig. 2A,B; FS IN, n = 16, 6; +NRG1, n = 18, 6; RS, n = 40, 6; +NRG1, n = 37, 6 (neurons, mice)]. However, NRG1 treatment significantly increased the excitability of FS INs (average ± SEM evoked, 6.58 ± 0.35; +NRG1, 7.67 ± 0.46; spontaneous, 2.15 ± 0.17; +NRG1, 2.56 ± 0.19) and decreased the excitability of RS neurons (evoked, 2.39 ± 0.21; +NRG1, 1.30 ± 0.14; spontaneous, 0.99 ± 0.13; +NRG1, 0.44 ± 0.07; Fig. 2C–G). Importantly, conditional deletion of erbB4 restricted to PV+ interneurons significantly reduced the excitability of FS INs in the P30 visual cortex. Furthermore, the regulation of neuronal excitability by NRG1 treatment was absent in PV-erbB4−/− mice (average ± SEM FS evoked: 0.71 ± 0.16, n = 17, 3; +NRG1: 1.79 ± 1.05, n = 3, 3; spontaneous: 0.99 ± 0.43, n = 6, 3; +NRG1: 1.48 ± 0.61, n = 7, 3; or RS evoked: 1.94 ± 0.37, n = 19, 3; +NRG1: 1.90 ± 0.3, n = 14, 3; spontaneous: 0.91 ± 0.13, n = 16, 3; +NRG1: 1.10 ± 0.26, n = 10; Fig. 2H,I).
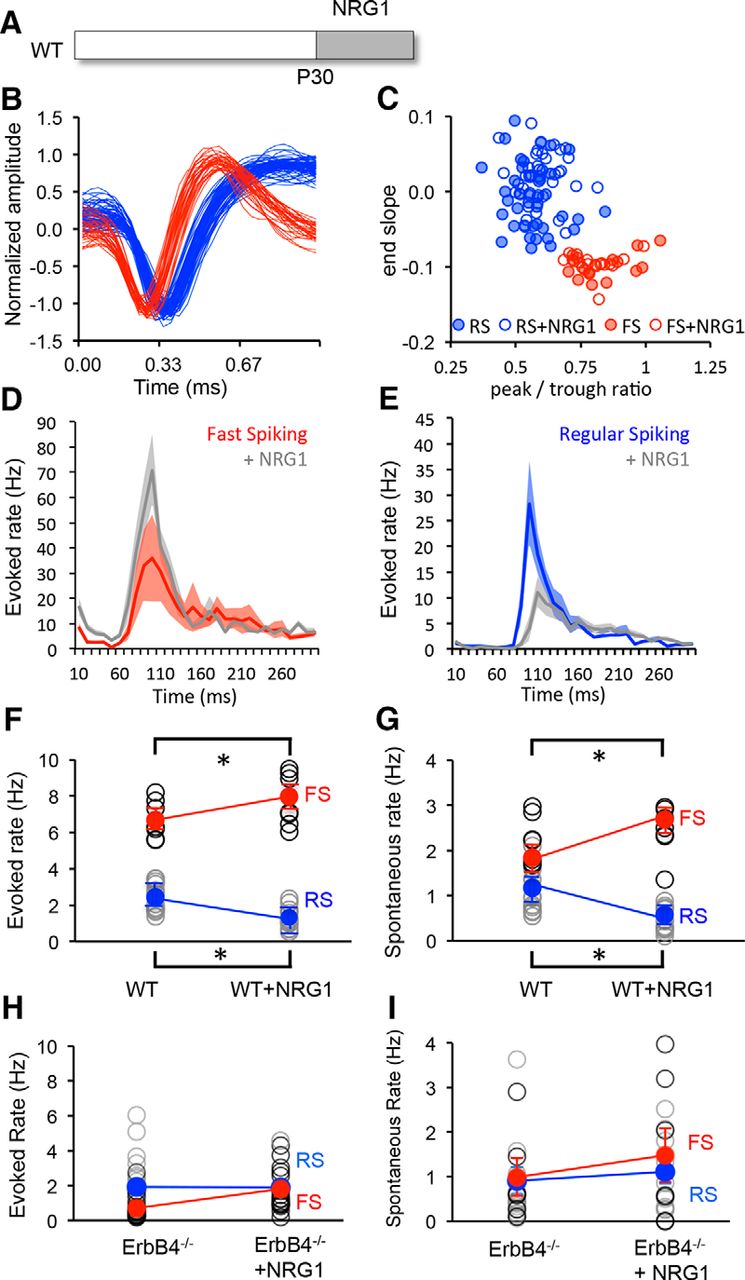
NRG1 increases excitability of FS INs in vivo. A, Experimental schematic. NRG1 treatment was initiated on P30. B, C, The treatment does not change the characteristics of single-unit waveforms (B) or the sorting into two clusters based on waveform characteristics (C; red, FS; blue, RS). D, E, Average ± SEM poststimulus time histograms for fast-spiking (red) and regular-spiking (blue) neurons in binocular visual cortex before and after NRG1 treatment. Activity was evoked with 100% contrast, 0.05 cycles/° gratings reversing at 1 Hz. F, G, Significant difference in average visually evoked (F) and spontaneous (G) spike rates in FS (red) and RS (blue) neurons before and after treatment. H, I, Absence of response to NRG1 in PV-erbB4−/− mouse. Activity was averaged over 500 ms. Lines connect group averages. *p < 0.05, t test.
To determine how enhancing the excitability of FS INs impacted ocular dominance plasticity, we examined the effect of NRG1 treatment on the response to concurrent brief (3 d) monocular deprivation at the peak of the critical period (P30; Fig. 3A). VEPs were recorded in layer 2/3 of the binocular region of the deprived visual cortex in response to contralateral (deprived) and ipsilateral (nondeprived) eye stimulation (Fig. 3B). Brief MD induces a robust shift in ocular dominance at this age, revealed by a significant decrease in the VEP ocular dominance index [ODI = (contralateral eye VEP − ipsilateral eye VEP)/(C + I)]. Treatment with NRG1 inhibited the shift in ocular dominance in response to brief MD [*H(2) = 11.94, p < 0.01, Kruskal–Wallis test, n = 6 per condition; Fig. 3C]. This suggests that the NRG1-dependent increase in the excitability of FS INs induced a precocious closure of the critical period of ocular dominance plasticity.
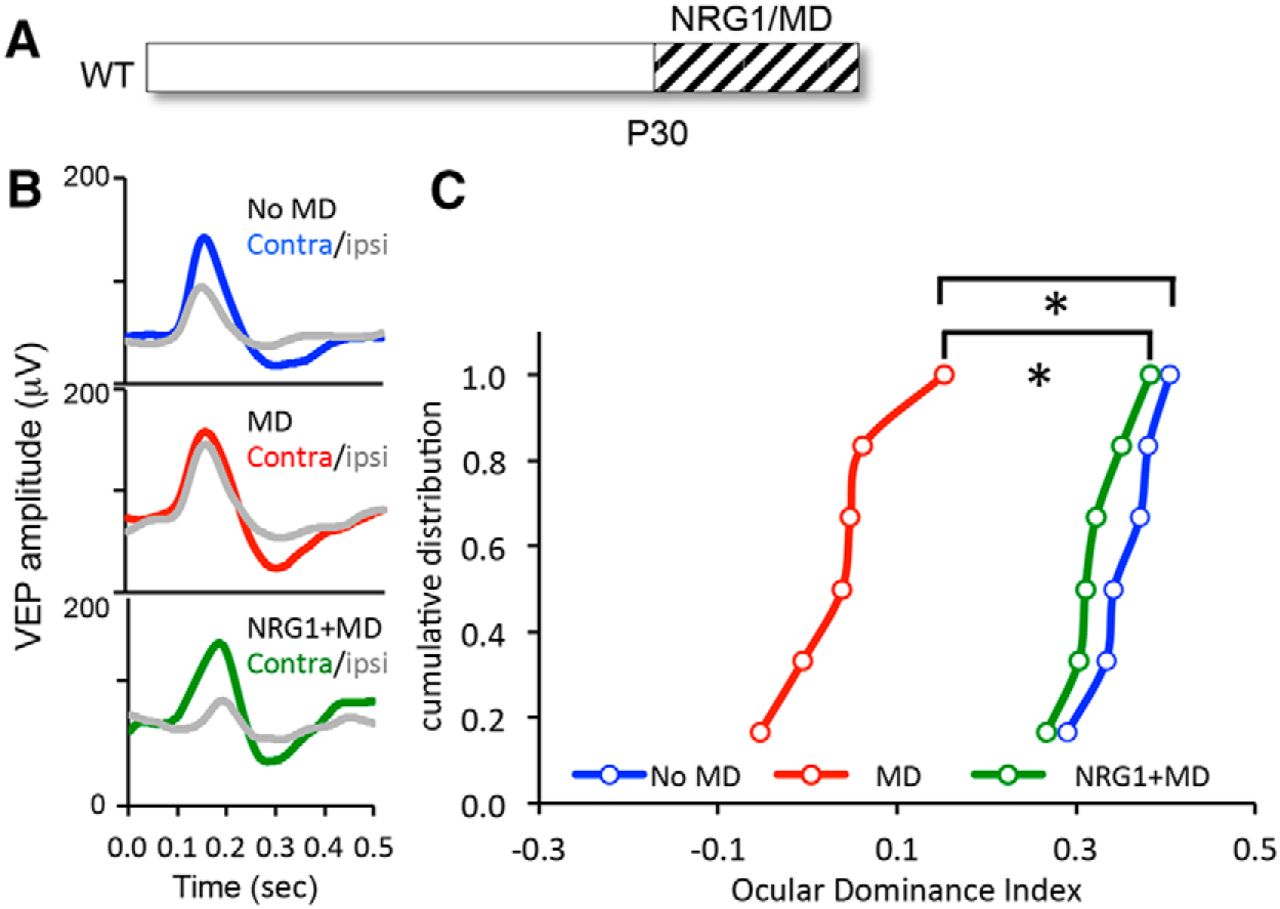
NRG1 inhibits ocular dominance plasticity during the critical period. A, Experimental schematic. NRG1 treatment with concurrent monocular deprivation (3 d) was initiated at P30. B, Average VEP waveforms in response to stimulation of contralateral (Contra) and ipsilateral (Ipsi) eyes in binocular controls (top), after monocular deprivation (middle), and after NRG1 treatment with concurrent monocular deprivation (bottom). C, Cumulative distributions of ODI (contralateral eye VEP − ipsilateral eye VEP)/(C + I). NRG1 treatment inhibited the shift in ocular dominance in response to MD (green). One-way ANOVA, *p < 0.01, Kruskal–Wallis test, H(2) = 11.94.
NRG1 enhancement of FS IN excitability rescues ocular dominance plasticity in NARP−/− mice
We recently demonstrated that a reduction in excitatory drive onto FS INs, mediated by transgenic deletion of NARP, inhibited the initiation of the critical period for ocular dominance plasticity (Gu et al., 2013). To ask whether enhancing excitation onto FS INs can rescue this deficit, NARP−/− mice were treated with NRG1. Single-unit recordings in layer 2/3 of the binocular region of primary visual cortex demonstrate that the hypoexcitability of FS INs and the hyperexcitability of RS neurons in NARP−/− mice are reversed by NRG1 treatment (spontaneous firing rates, FS: NARP−/−, 1.82 ± 0.11; +NRG1, 2.14 ± 0.07; n = 7; RS: NARP−/−, 1.82 ± 0.17; +NRG1, 0.91 ± 0.08; n = 7; Fig. 4A). Furthermore, NRG1 treatment rescued ocular dominance plasticity in NARP−/− mice, revealed by the significant shift in the ODI induced by brief concurrent MD [*H(2) = 18.38, p < 0.001, Kruskal–Wallis test, n = 9 per condition; Fig. 4B]. The ability to rescue the phenotype of the NARP−/− mouse with NRG1 suggests that these two signaling pathways independently regulate excitation onto FS INs.

NRG1 treatment in NARP−/− and dark-exposed mice. A, Experimental schematic. NRG1 treatment was initiated at P30 in NARP−/− mice. Spontaneous spiking rates were increased in FS INs (red) and decreased in RS neurons (blue) after the treatment. Lines connect group averages *p < 0.05, t test. B, Experimental schematic. NRG1 treatment was initiated at P30 with concurrent monocular deprivation (3 d) in NARP−/− mice. Cumulative distributions of ODI. NRG1 enabled a shift in ocular dominance in response to MD (green). Kruskal–Wallis test, H(2) = 18.38, *p < 0.001. C, Experimental schematic. Ten days of dark exposure (from P90 to P100) was followed by NRG1 treatment (from P100 to P103). Spontaneous firing rates were increased in FS INs (red) and decreased in RS neurons (blue) after the treatment. Lines connect group averages. *p < 0.05, t test. D, Experimental schematic. NRG1 treatment was initiated at P100 with concurrent monocular deprivation (3 d) after DE (from P90 to P100). Cumulative distributions of ODI are shown. NRG1 treatment inhibited ocular dominance plasticity in DE adults (red). *p < 0.001, Kruskal–Wallis test, H(2) = 15.77.
NRG1 reverses reactivation of critical period by dark exposure
The reduction in the excitability of RS neurons of the visual cortex over development contribute to the decline in ocular dominance plasticity with age (Takesian and Hensch, 2013). However, we have previously shown that robust ocular dominance plasticity can be reactivated in the adult mouse visual cortex after complete visual deprivation through dark exposure (He et al., 2007; Montey and Quinlan, 2011; Eaton et al., 2016). Single unit recordings from layer 2.3 of the primary visual cortex were used to ask if dark exposure regulates the strength of excitation onto FS INs. Ten days of dark exposure initiated in adulthood (P90) significantly decreased the excitability of FS INs and increased the excitability of RS neurons (spontaneous firing rates, FS: DE, 1.74 ± 0.07, n = 7; RS: DE, 1.22 ± 0.04, n = 7; Fig. 4C). To ask whether NRG1 could reverse the effects of DE, NRG1 treatment was initiated at P97, coincident with the last 3 d of dark exposure. NRG1 significantly increased the excitability of FS INs (+NRG1, 2.11 ± 0.07; Fig. 4C, red) and decreased excitability of RS neurons (+NRG1, 0.48 ± 0.04; Fig. 4C, blue) in DE visual cortex. To ask how the increase in FS IN excitability impacted the reactivation of ocular dominance plasticity by dark exposure, dark-exposed adults received NRG1 treatment with concurrent brief (3 d) monocular deprivation. NRG1 prevented the induction of ocular dominance plasticity in dark-exposed adults [blue, MD; red, DE+MD; green, DE+NRG1+MD; *H(2) = 15.77, p < 0.001, Kruskal–Wallis test, n = 8 per condition; Fig. 4D]. Together, this suggests that dark exposure in adults may enable ocular dominance plasticity by decreasing the strength of excitation onto FS INs, and reversing this effect with NRG1 is sufficient to disable this plasticity.
Inhibition of ErbB4 reactivates the critical period in adults
To examine the contribution of excitation onto FS INs in controlling the expression of ocular dominance plasticity in adults, we inhibited signaling through erbBs, the receptor tyrosine kinase that transduces the effects of endogenous NRGs. Adult wild-type mice were treated with the small erbB receptor tyrosine kinase inhibitor PD158780 (330.18 Da; for erbB4, IC50 = 52 nm; 10 mg/kg, i.p., two times per day for 3 d starting at P90). Whole-cell recordings of pharmacologically isolated mEPSCs in slices of visual cortex prepared from G42 mice revealed that treatment with the erbB inhibitor significantly decreased the amplitudes, but not the frequencies, of mEPSCs onto layer 2/3 PV INs (Fig. 5).

ErbB inhibition reduces excitation onto PV+ interneurons. A, Representative examples of 2 s current traces of pharmacologically isolated mEPSCs recorded from PV interneurons from layer 2/3 of visual cortex slices after vehicle (black) or erbB inhibitor (PD158780; 10 ng/kg, 2 times per day for 3 d; red). B, The treatment reduced the amplitude (left; *p = 0.029, t test) but not the frequency (Right, p = 0.971, t test) of the mEPSCs. The numbers of mice, neurons are indicated within the columns. C, Average of all mEPSCs recorded in both conditions. Isolated mEPSCs were first averaged by neuron and then by condition. D, Significant difference in the cumulative distribution of mEPSC amplitudes in vehicle-treated (black) versus erbB inhibitor-treated (red; *p < 0.001, Kolmogorov–Smirnov test) mice. Vh, Vehicle.
To determine whether erbB inhibition could be used to control excitability of FS INs in vivo, PD158780 was delivered to adult wild-type mice. A single dose of the erbB inhibitor (10 mg/kg, i.p.) induced a 25.1 ± 2.2% decrease in spontaneous firing frequency of FS INs within 15 min of delivery (n = 3; data not shown). Longer-duration treatment with PD158780 (10 mg/kg, i.p., two times per day for 3 d starting at P90) did not impact waveform characteristic of the clustering of single units acquired from layer 2/3 based on waveform parameters (Fig. 6A–C; RS, n = 34,8; +inh, n = 32, 8; FS, n = 20, 8; +inh, n = 18, 8). However, inhibition of erbB significantly decreased the excitability of FS INs (average ± SEM, evoked, 2.38 ± 0.20; +inh, 3.65 ± 0.11; spontaneous, 0.39 ± 0.07; +inh, 1.31 ± 0.06; Fig. 6D,F) and increased the excitability of RS neurons (evoked, 7.52 ± 0.34; +inh, 6.05 ± 0.13; spontaneous, 2.34 ± 0.06; +inh, 1.79 ± 0.07; Fig. 6E,G). To determine how decreasing the excitability of FS INs in adults impacted ocular dominance plasticity, we examined the effect of erbB inhibition on the response to concurrent brief (3 d) monocular deprivation initiated at P90 (Fig. 7A,B). Treatment with the erbB inhibitor enabled a shift in VEP ocular dominance in response to brief MD, which is not seen at this age in the absence of PD158780 [*H(2) = 15.24, p < 0.001, Kruskal–Wallis test; Fig. 7C]. Thus, inhibition of erbB reactivated the critical period for ocular dominance plasticity.

erbB inhibition decreases excitability of FS INs in vivo. A, Experimental schematic. Treatment with the ErbB inhibitor (erb inh.) was initiated on P90. B, C, The treatment does not change the characteristics of single-unit waveforms (B) or the sorting into two clusters based on waveform characteristics (C; red, FS; blue, RS). D, E, Average ± SEM poststimulus time histograms for fast-spiking (red) and regular-spiking (blue) neurons in binocular visual cortex before and after treatment with the erbB inhibitor. Activity was evoked with 100% contrast, 0.05 cycles/° gratings reversing at 1 Hz. F, G, Significant difference in average visually evoked (F) and spontaneous (G) spike rates in FS (red) and RS (blue) neurons before and after treatment. Activity was averaged over 500 ms. Lines connect group averages. *p < 0.05, t test.

erbB inhibition reactivates ocular dominance plasticity in adults. A, Experimental schematic. Treatment with the ErbB inhibitor (erb inh) with concurrent monocular deprivation (3 d) was initiated at P90. B, Average VEP waveforms in response to stimulation of contralateral (Contra) and ipsilateral (Ipsi) eyes in binocular controls (top), after monocular deprivation (middle), and after erbB inhibition with concurrent monocular deprivation (bottom). C, Cumulative distributions of ODI. The erbB inhibition enabled a shift in ocular dominance in response to MD (green). *p < 0.001, Kruskal–Wallis test, *H(2) = 15.24.
ErbB inhibition enables recovery from chronic monocular deprivation in adults
The developmental reduction in ocular dominance plasticity is thought to limit the ability of adults to recover from the amblyopia. To determine whether erbB inhibition could be used to reverse the effects of long-term monocular deprivation, subjects received PD158780 (10 mg/kg, i.p., two times per day for 3 d starting at P90) after monocular deprivation from P14 to P90 (Fig. 8A). Single-unit activity from layer 2/3 of binocular visual cortex revealed that ErbB inhibition decreased the excitability of FS INs (red; average ± SEM evoked: cMD, 6.61 ± 0.09, n = 18, 8; +inh, 4.93 ± 0.17, n = 16, 8; spontaneous: cMD, 1.85 ± 0.06; +inh,1.25 ± 0.06; Fig. 8B,D) and increased the excitability of RS neurons (evoked: cMD, 1.62 ± 0.09, n = 38, 8; +inh, 3.21 ± 0.21, n = 32, 8; spontaneous: cMD, 0.20 ± 0.02; +inh, 1.09 ± 0.08; Fig. 8C,E). To ask whether decreasing the excitability of FS INs enabled recovery from chronic MD, treatment with erbB inhibitor was initiated at eye opening at P90 (open, deprived eye; filled, nondeprived eye; Fig. 8F–H). Eye opening alone did not produce spontaneous recovery of ocular dominance; however, recovery of normal ocular dominance was enabled by treatment with PD158780 (10 mg/kg, i.p., two times per day for 3 d starting at P90) initiated at eye opening [*H(2) = 15.37, p < 0.001; Kruskal–Wallis test; Fig. 8G,H]. Thus, a reduction in excitability of FS INs was sufficient to reactivate plasticity in the adult visual cortex and enable recovery from chronic monocular deprivation.
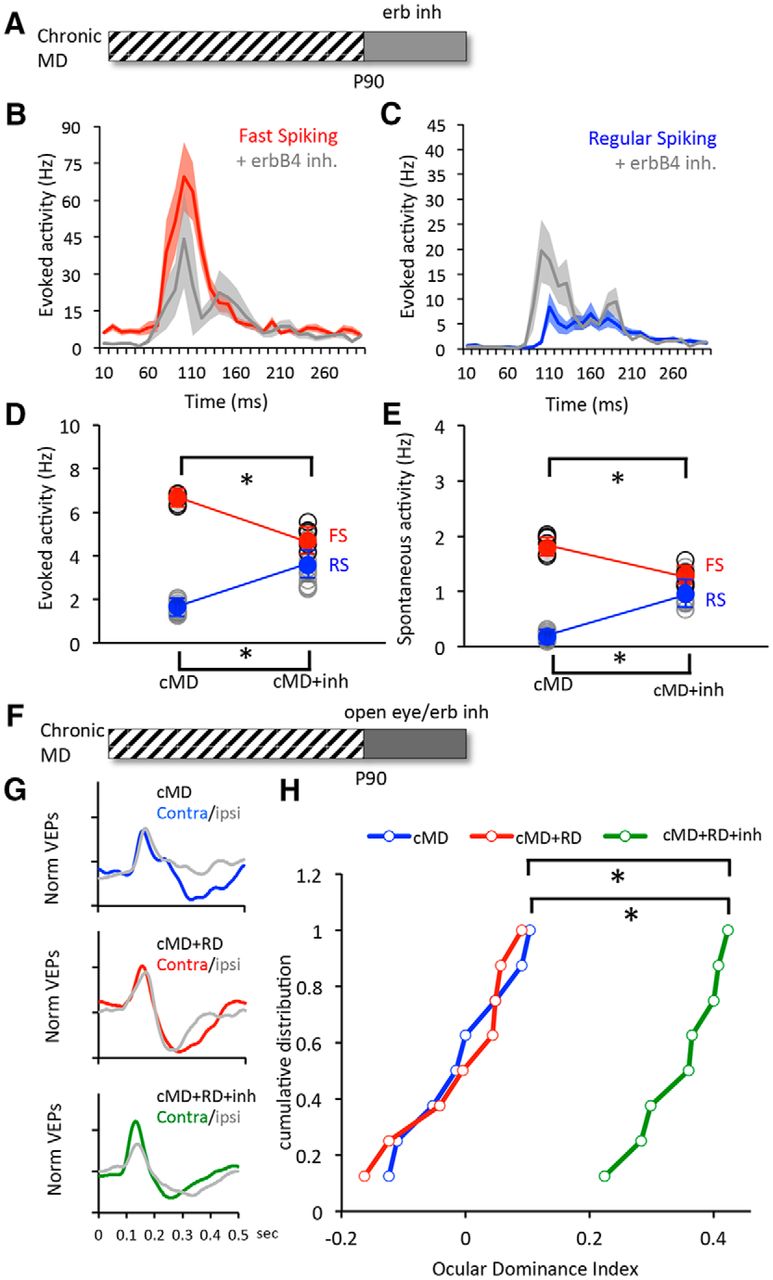
ErbB inhibition allows for recovery from amblyopia in adults. A, Experimental schematic. Subjects received monocular deprivation from eye opening (∼P14) until adulthood (P90), followed by treatment with ErbB inhibitor (3 d). B, C, Average ± SEM poststimulus time histograms for fast-spiking (B, red) and regular-spiking (C, blue) neurons in deprived visual cortex before and after treatment with erbB inhibitor. Activity was evoked with 100% contrast, 0.05 cycles/° gratings reversing at 1 Hz. D, E, Significant difference in average visually evoked (D) and spontaneous (E) spike rates in FS (red) and RS (blue) neurons before and after treatment. Activity was averaged over 500 ms. Lines connect group averages. *p < 0.05, t test. F, Experimental schematic. Subjects received monocular deprivation from eye opening until adulthood, followed by treatment with the ErbB inhibitor and concurrent reverse deprivation (open, the closed eye; filled, the open eye, 3 d). G, Average VEP waveforms in response to stimulation of contralateral (Contra) and ipsilateral (Ipsi) eye after chronic MD (top), chronic MD followed by reverse deprivation (middle), and chronic MD followed by reverse deprivation with erbB inhibition (bottom). H, Cumulative distributions of ODIs. The erbB inhibition (green) enabled a significant recovery from chronic monocular deprivation (green). *p < 0.001; Kruskal–Wallis test, H(2) = 15.37.
Discussion
Fast-spiking interneurons in the visual cortex exert powerful control over pyramidal neuron excitability and plasticity. Here we show that the NRG/erbB signaling pathway controls the strength of excitation onto FS INs, and can be manipulated to reversibly regulate ocular dominance plasticity. Treatment with the neurotrophin NRG1 increases the excitability of FS INs; however, the impact of this increase depends on the initial state of excitatory input. During the critical period, when excitation onto FS INs is predicted to be optimal, NRG1 treatment induced a precocious termination of the critical period. In contrast, in NARP−/− mice, in which reduced excitatory drive onto FS INs prevents the initiation of the critical period, NRG1 treatment rescues FS IN excitability and ocular dominance plasticity. Importantly, the reactivation of the critical period in adults by dark exposure was reversed by NRG1 treatment, suggesting that dark exposure reduces excitation onto FS INs into a range permissible for ocular dominance plasticity. In addition, NRG/erbB4-dependent maintenance of excitation onto FS INs plays a fundamental role in the active constraint of plasticity in the adult visual cortex. Conditional deletion of erbB4 in PV+ interneurons reduces the excitability of FS INs and renders the visual cortex insensitive to regulation by NRG1. Inhibition of endogenous NRG signaling in adults, via inhibition of erbB, decreases the excitability of FS INs and reactivates the critical period for ocular dominance plasticity. The decrease in FS IN excitability mediated by inhibition of erbB is sufficient for recovery from long-term monocular deprivation, reinforcing the idea that the developmental loss of plasticity limits the recovery from amblyopia in adulthood. This work demonstrates that the strength of excitation onto FS INs is an important locus for bidirectional control of critical period plasticity and identifies NRG/erbB4 signaling as a target for therapeutic enhancement of plasticity in adults.
Role of excitation onto FS INs in the response to MD
A decrease in the amplitude and frequency of mEPSCs in FS INs is an early response to monocular deprivation in young mice at the peak of the critical period for ocular dominance plasticity (Kuhlman et al. 2013). The decrease in FS IN excitability would enable the enhancement of excitability of downstream RS neurons necessary to engage spike-timing-dependent mechanisms of synaptic plasticity. A developmental slowing of plasticity at excitatory synapses onto FS INs may explain the requirement for longer durations of monocular deprivation with age (Sawtell et al., 2003; Pham et al., 2004; Lehmann and Löwel, 2008; Huang et al., 2010; Kameyama et al., 2010). Indeed, the rapid decrease in excitation onto FS INs induced by monocular deprivation is lost relatively early in development, at ages where ocular dominance plasticity can be engaged by longer durations of MD. The dependence on erbB4 for the maintenance of excitatory synapses onto FS INs appears to also increase with age, as mEPSCs recorded from PV+ FS INs in the prefrontal cortex of erbB4−/− mice are normal at P21 but are reduced later in development (Yang et al., 2013). Direct reduction of the strength of excitation onto FS INs via erbB inhibitions in adults reactivates plasticity and may compensate for the developmental loss of plasticity at these synapses
Parallel pathways for the regulation of excitatory synaptic strength on FS INs
A subset of signaling pathways that regulate synapse strength and number are highly enriched at excitatory synapses onto FS INs. The two pathways examined here, NRG/erbB4 and NARP, have both been shown to regulate the stability of AMPARs at excitatory synapses onto FS INs, albeit by different mechanisms. erbB4 and NARP are also targets for signaling pathways that enable rapid plasticity at these synapses (Cho et al., 2008; Chang et al., 2010; Tamura et al., 2012; Vullhorst et al., 2015). NARP [also known as NP2 (neural pentraxin 2)] forms an AMPAR-binding complex with NP1 (neuronal pentraxin 1) and NPR (neuronal pentraxin receptor; O'Brien et al., 1999; Xu et al., 2003), enabling coregulation of GluR4-containing AMPARs and surface NARP expression (Chang et al., 2010; Pelkey et al., 2015). In contrast, NRG is thought to induce a tyrosine kinase-dependent stabilization of PSD-95 and subsequent recruitment or maintenance of synaptic AMPARs (Li et al., 2007; Ting et al., 2011). In addition to the relatively rapid increase in mEPSC amplitudes onto FS INs observed by us and others in response to NRG1 treatment in cortex [Abe et al., 2011; but see Fazzari et al. (2010) regarding hippocampus], a slower latency increase in excitatory synapse number has been observed in response to manipulations of NRG/erb signaling. Accordingly, long-term treatment with NRG1 results in a delayed increase in the frequencies of mEPSCs in FS INs, and decreased mEPSC frequencies by erbB4 deletion emerges late in development (Abe et al., 2011; Yang et al., 2013).
However, a single dose of NRG1 has also been shown to reduce the excitability and increase the action potential threshold in erbB4+ hippocampal neurons (Janssen et al., 2012). Activation of erbB4 also induces the internalization of α1-containing GABAA receptors and GluN2b-containing NMDARs in hippocampal interneurons identified by live labeling with erbB4 antibody (Mitchell et al., 2013; Vullhorst et al., 2015), which would increase the excitability of erbB4+ interneurons and enhance the stability of excitatory synapses. Our demonstration that NRG1 treatment can rescue the deficits of the NARP−/− mice, including a reversal of FS IN hypoexcitability and a recovery of ocular dominance plasticity, supports the independence of these signaling pathways. Interestingly, NRG/erbB4 and NARP/NPR complexes both interact with molecules of perineuronal nets, dense specializations of the extracellular matrix that surround FS INs, suggesting a possible locus for coregulation of these pathways.
Distribution of erbB4 in mammalian cortex
Our demonstration of the enrichment of erbB4 receptors in parvalbumin+ interneurons in the mouse visual cortex confirms and extends previous reports of the selectivity of erbB4 expression (Fazzari et al., 2010; Neddens and Buonanno, 2010; Bean et al., 2014; Lu et al., 2014). Although we cannot rule out the possibility of indirect effects, our data strongly support that peripheral delivery of NRG1 directly activates cortical erbB4 receptors to increase FS IN excitability. Indeed, a single low dose of the small (71 amino acid) NRG1 peptide significantly increased the phosphorylation of cortical erbB4 receptors, a traditional measure of NRG1 influx into the brain (Rösler et al., 2011). Previous work has demonstrated that radiolabeled NRG1 delivered peripherally can be detected in brain within 15 min of a single injection, with no changes in Tmax detected from 20 min to 4 h after injection (Rösler et al., 2011; Li Y et al., 2012). The influx of radiolabeled NRG1 into brain is saturable (reduced by inclusion of nonradiolabeled NRG1-1 peptide) and blocked by antibodies to NRG1 receptors (Kastin et al., 2004), suggesting that NRG1 crosses the blood–brain barrier through receptor-mediated transport. Importantly, the effects of repeated injections of low-dose NRG1 that we observe, including the increase in mEPSC amplitudes recorded in FS INs and the increase in FS IN excitability recorded in vivo, were absent in the visual cortex of PV-erbB4−/− mice. Similar treatments with the small NRG1 peptide protects central neurons from toxic insults (Carlsson et al., 2011; Li KX et al., 2012; Depboylu et al., 2015). In the absence of erbB4, the baseline excitability of FS INs was significantly reduced, reinforcing the idea that the NRG1/erbB4 signaling pathway plays an important role in the maintenance of excitatory drive onto this class of interneuron.
Reactivation of the critical period in adulthood
Our demonstration that inhibition of erbBs reactivates the critical period for ocular dominance plasticity supports a model in which plasticity in adults is actively constrained by maintenance of strong excitation onto FS INs. Accordingly, dark exposure, which reactivates robust ocular dominance plasticity in adult mice, reduces the excitability of FS INs. Indeed, a reduction in the excitability of FS INs may be a mechanism common to several manipulations that reactivate the critical period in adulthood. For example, environmental enrichment, shown to reactivate robust ocular dominance plasticity in adults, reduces the expression of parvalbumin and GAD67 in inhibitory neurons of the visual cortex, indicative of a reduction in excitability (Sale et al., 2007; Donato et al., 2013). Similarly, treatment with the antidepressant fluoxetine, which reactivates rapid ocular dominance plasticity in adults, decreases the expression of PV in the visual cortex and decreases the number of perineuronal nets that colocalize with PV+ interneurons (Maya Vetencourt et al., 2008; Guirado et al., 2014). Fluoxetine also accelerates morphological plasticity of inhibitory dendrites induced by monocular deprivation (Chen et al., 2011). Thus, the strength of excitation onto FS INs is a key site for the regulation of the critical period, and NARP and NRG signaling complexes are emerging as powerful and reversible regulators of the strength of these synapses.
Footnotes
This work was supported by NIH Grants R01EY016431 (E.M.Q.) and R01EY025922 (E.M.Q. and A.K.).
The authors declare no competing financial interests.
- Correspondence should be addressed to either of the following: Elizabeth M. Quinlan at the above address. equinlan{at}umd.edu; or Alfredo Kirkwood at the above address. kirkwood{at}jhu.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}