

Abstract
Drosophila photoreceptors respond to oscillating light of high frequency (∼100 Hz), while the detected maximal frequency is modulated by the light rearing conditions, thus enabling high sensitivity to light and high temporal resolution. However, the molecular basis for this adaptive process is unclear. Here, we report that dephosphorylation of the light-activated transient receptor potential (TRP) ion channel at S936 is a fast, graded, light-dependent, and Ca2+-dependent process that is partially modulated by the rhodopsin phosphatase retinal degeneration C (RDGC). Electroretinogram measurements of the frequency response to oscillating lights in vivo revealed that dark-reared flies expressing wild-type TRP exhibited a detection limit of oscillating light at relatively low frequencies, which was shifted to higher frequencies upon light adaptation. Strikingly, preventing phosphorylation of the S936-TRP site by alanine substitution in transgenic Drosophila (trpS936A) abolished the difference in frequency response between dark-adapted and light-adapted flies, resulting in high-frequency response also in dark-adapted flies. In contrast, inserting a phosphomimetic mutation by substituting the S936-TRP site to aspartic acid (trpS936D) set the frequency response of light-adapted flies to low frequencies typical of dark-adapted flies. Light-adapted rdgC mutant flies showed relatively high S936-TRP phosphorylation levels and light–dark phosphorylation dynamics. These findings suggest that RDGC is one but not the only phosphatase involved in pS936-TRP dephosphorylation. Together, this study indicates that TRP channel dephosphorylation is a regulatory process that affects the detection limit of oscillating light according to the light rearing condition, thus adjusting dynamic processing of visual information under varying light conditions.
SIGNIFICANCE STATEMENT Drosophila photoreceptors exhibit high temporal resolution as manifested in frequency response to oscillating light of high frequency (≤∼100 Hz). Light rearing conditions modulate the maximal frequency detected by photoreceptors, thus enabling them to maintain high sensitivity to light and high temporal resolution. However, the precise mechanisms for this process are not fully understood. Here, we show by combination of biochemistry and in vivo electrophysiology that transient receptor potential (TRP) channel dephosphorylation at a specific site is a fast, light-activated and Ca2+-dependent regulatory process. TRP dephosphorylation affects the detection limit of oscillating light according to the adaptation state of the photoreceptor cells by shifting the detection limit to higher frequencies upon light adaptation. This novel mechanism thus adjusts dynamic processing of visual information under varying light conditions.
Introduction
As they respond to light, fly photoreceptors exhibit an unusually high temporal resolution. The detection limit of the fly's response to oscillating light intensities undergoes substantial changes during dark and light adaptation (Wu and Wong, 1977). This adaptive processing of visual information allows small flying animals to cope with rapidly occurring natural contrast changes. Although the phenomenology of these processes is well defined, the underlying molecular mechanisms are largely unknown (Song et al., 2012). The waveform and kinetics of the light response are determined downstream of phospholipase C (PLC) in the inositol lipid phototransduction cascade, via Ca2+-dependent effects on the transient receptor potential (TRP) channels. However, the underlying molecular mechanism is still unclear (Gu et al., 2005; Katz and Minke, 2012).
TRP channels are crucial components in a myriad of biological processes, especially of sensory and neuronal functions (for review, see Minke and Cook, 2002; Clapham, 2003; Montell, 2005; Myers et al., 2008; Bohlen et al., 2011). Many of the TRP channels undergo phosphorylation and dephosphorylation. The role of TRP phosphorylation has been investigated mainly in TRP vanilloid receptor 1 (TRPV1). These studies examined the effects of mutating serine/threonine residues, which constitute primary targets of protein kinase Cε (Cesare et al., 1999; Bhave et al., 2003), protein kinase CβII (Li et al., 2014), protein kinase A (Bhave et al., 2002), and Ca2+/calmodulin-dependent kinase II (Jung et al., 2004). Mutating these sites caused sensitization or desensitization of TRPV1 toward activation by heat, capsaicin, protons, G-protein-coupled receptors (Kumar et al., 2017), or a variety of chemicals. However, the physiological roles of these phosphorylations and their control of TRPV1 activity in vivo remain largely unknown (Yao et al., 2005; Winter et al., 2013).
Recently, we identified light-regulated phosphorylation sites of the Drosophila TRP channel using quantitative mass spectrometry (Voolstra et al., 2010, 2013). Many (15 of 28) of the identified phosphorylation sites displayed enhanced phosphorylation in the light, while a single phosphorylation site in the middle of the C-terminal domain, S936, was predominantly phosphorylated in the dark and became dephosphorylated upon illumination (Voolstra et al., 2010, 2013). The physiological roles of pS936-TRP dephosphorylation remain unknown.
A prospective pS936-TRP phosphatase is retinal degeneration C (RDGC), which is a Ca2+-dependent rhodopsin phosphatase originally found in Drosophila photoreceptors. The rdgC306-null mutant exhibits light-dependent retinal degeneration (Steele and O'Tousa, 1990; Steele et al., 1992). RDGC is a member of a divergent family of protein serine/threonine phosphatases that binds Ca2+/calmodulin and contains 2 Ca2+-binding EF-hand motifs (Steele et al., 1992; Sherman et al., 1997; Lee and Montell, 2001; Ramulu and Nathans, 2001; Ramulu et al., 2001). Rhodopsin dephosphorylation in Drosophila eye homogenates and in vivo was abolished in the rdgC306 mutant (Byk et al., 1993), indicating that RDGC promotes rhodopsin dephosphorylation (Steele and O'Tousa, 1990; Steele et al., 1992; Byk et al., 1993; Vinós et al., 1997; Lee and Montell, 2001). Two homologues of RDGC expressed in the mammalian retina were found, but their targets are still unknown (Sherman et al., 1997; Ramulu and Nathans, 2001; Ramulu et al., 2001).
Here, we show by an in vivo combination of biochemistry and electrophysiology that TRP channel dephosphorylation at a specific site is a fast, light-activated and Ca2+-dependent regulatory process. We further found that this process, by shifting the detection limit of flies toward higher frequencies upon light adaptation, affects the detection limit of high-frequency oscillating light according to the adaptation state of the photoreceptor cells. The data indicates that wild-type (WT) flies differ in their response to oscillating light of high frequency depending on whether they are light or dark adapted. The data obtained from trpS936A and trpS936D transgenic flies indicates that this difference is due to the phosphorylation state of S936-TRP in WT flies. This novel mechanism thus adjusts dynamic processing of visual information under changing light conditions.
Materials and Methods
Fly stocks and illumination conditions.
The following strains and mutants of Drosophila melanogaster were used: yw, w Oregon R (here referred to as WT), trp14 (Wang et al., 2005), w;;p[rh1-trpD621G],trpP343 (Liu et al., 2007), rdgC306 (Steele and O'Tousa, 1990; Steele et al., 1992), yw;;trpP343, yw;inaD1 (Tsunoda et al., 1997), w,norpAP24 (Bloomquist et al., 1988), w;inaCP209 (Smith et al., 1991), yw;P[rh1-trp-egfp];trpP343 (Katz et al., 2013), P[rh1-rh1Δ356];rdgC306,ninaEI17 (Vinós et al., 1997). yw;p[rh1-trpS936A];trpP343, yw;p[rh1-trpS936D];trpP343, and yw;p[rh1-trpWT],trpP343 (see Site-directed mutagenesis and generation of transgenic flies). Experiments were conducted with flies of either sex.
Flies were illuminated with an 18 W fluorescent lamp, 2000 lux, unless noted otherwise. To investigate TRP phosphorylation at S936, flies were dark-adapted for 12–18 h and were then illuminated for 1 h or vice versa. To analyze kinetics of TRP dephosphorylation, WT flies were dark-adapted overnight and illuminated for different periods using a bright white light-emitting diode (50,000 lux at the level of the fly). After illumination, flies were immediately shock-frozen in liquid nitrogen and heads were subjected to Western blot analysis. To assess S936 phosphorylation in different light intensities, flies were dark-adapted overnight and then subjected to different light intensities for 15 min using a KL1500LCD cold light source (Schott). Unattenuated light intensity was 1500 lux and attenuation was achieved using neutral density filters. Eye cups were prepared from dark-adapted WT flies under dim red illumination (Schott RG630 filter and Schott KL1500LCD cold light source) in Ringer's solution [125 mm NaCl, 5 mm KCl, 10 mm N-tris(hydroxymethyl)methyl-2-aminoethane-sulphonic acid (TES), 20 mm glucose, 8 mm MgSO4] using a scalpel. Eye cups were then transferred to standard bath (120 mm NaCl, 5 mm KCl, 10 mm TES, 4 mm MgCl2, 1.5 mm CaCl2, 25 mm proline, 5 mm alanine, 1 mm glucose, 1% FCS, 0.1% DMSO) or to Ca2+-free bath (120 mm NaCl, 5 mm KCl, 10 mm TES, 4 mm MgCl2, 1.6 mm EGTA, 25 mm proline, 5 mm alanine, 1 mm glucose, 1% FCS, 0.1% DMSO), and were illuminated or kept in the dark for 15 min before immunoblot analysis.
Site-directed mutagenesis and generation of transgenic flies.
A 3′ trp fragment was amplified from WT head cDNA using the primers forward, 5′-TATTAGGTACCCATCTCGGAGCGAGCCGAC-3′ and reverse, 5′-CTCACCGGGCTTGCTGATAGTGCCAGCT-3′. The PCR product was digested with KpnI and SacI and cloned into a pBluescript II SK (+) vector (Stratagene). Site-directed mutagenesis was performed using the QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene) following the manufacturer's instructions with the primer 5′ GGCTGCCGACGAGGTTGCCTTGGCCG-3′, which exchanges a codon for Ser with a codon for Ala (GCC) and the primer 5′-GCTGCCGACGAGGTTGACTTGGCCGATGACGA-3′ that exchanges a codon for Ser with a codon for Asp (GAC). After digestion with DraIII and SacI, the mutagenized fragment was cloned into trp full-length cDNA in the pENTR1A vector (Invitrogen). From there, the trp cDNA was recombined into a modified Yellow C4 vector containing the ninaE minimal promoter (base pairs −833 to +67) and 3′ untranslated region of ninaE along with a DEST cassette (Richter et al., 2011) using the Gateway cloning system (Invitrogen). P-element-mediated transformation of Drosophila was performed as described previously (O'Tousa, 1992). The constructs were injected in yw embryos and crossed into the trpP343 mutant background. The transgenic fly expressing WT TRP was generated likewise without the mutagenesis.
Antibodies.
For immunoprecipitation, a polyclonal α-TRP antibody (Bähner et al., 2000) and a monoclonal α-GFP antibody (Roche) were used. For Western blot analyses, the following antibodies were used: monoclonal α-TRP antibody (MAb83F6, Developmental Studies Hybridoma Bank), polyclonal α-TRP antibody (Bähner et al., 2000), polyclonal monoclonal α-β-tubulin antibody (E7, Developmental Studies Hybridoma Bank), polyclonal α-PLC antibody (Huber et al., 2000), polyclonal α-TRPL antibody (Xue et al., 2011), polyclonal α-Gqα antibody (Elia et al., 2005). To generate polyclonal α-INAD and α-PKC antibodies, fragments spanning the last 300 aa (Tsunoda et al., 1997) or amino acids 232–369, respectively, were heterologously expressed in bacteria with a His tag. The polypeptides were cleaned up using Ni-NTA agarose (Qiagen) according to the manufacturer's protocol. Immunization of rabbits was done by Pineda Antibody Service. Antisera were cleaned up using a HiTrap protein A HP column (GE Healthcare Life Sciences) according to the manufacturer's protocol. For immunocytochemistry, the monoclonal α-TRP antibody (MAb83F6) was used.
Immunoprecipitation.
For immunoprecipitation, 50 fly heads were homogenized in 0.2 ml of extraction buffer supplemented with protease and phosphatase inhibitors [50 mm Tris-HCl, pH 8.0; 150 mm NaCl; 1% Triton X-100; 50 μm (4-amidinophenyl)-methanesulfonyl fluoride hydrochloride monohydrate; 10 μg/ml aprotinin, leupeptin, and pepstatin A; 10 mm benzamide; 10 mm sodium fluoride; 1 mm orthovanadate; 10 mm β-glycerophosphate; 500 nm cantharidine; 10 mm sodium pyrophosphate) using a micropestle (Carl Roth). Head homogenates were extracted on ice for 30 min and centrifuged for 10 min at 16,000 × g. Ten microliters of the supernatant were saved as input control. The remaining supernatant was incubated with 50 μl of protein G or protein A agarose beads (Pierce) and 3 μg of α-GFP or α-TRP antibody (see Antibodies), respectively, for 1 h at 4°C. After four washes with wash buffer (same as extraction buffer but with 0.1% Triton X-100), precipitated proteins were eluted with 25 μl of SDS sample buffer [200 mm Tris, pH 6.8, 1.92% (w/v) SDS, 2% (v/v) 2-mercaptoethanol, 0.004% (w/v) bromphenol blue] at 80°C for 10 min. Fifteen microliters of the eluate were subjected to SDS-PAGE.
Dot blot and Western blot analysis.
Dot blot analysis was performed as described previously (Voolstra et al., 2013). Briefly, 2.5 μg of each peptide were applied onto a nitrocellulose membrane. After blocking, the membrane was incubated with α-pS936-TRP antibody. Western blotting was performed as described previously (Voolstra et al., 2013) using polyvinylidene difluoride membranes (Bio-Rad). Chemiluminescent signals were recorded using an XRS+ documentation system (Bio-Rad).
To assess signals with another antibody, membranes were either stripped or a parallel blot was performed. Quantification of Western blot bands was accomplished using the Image Lab 4.0 software (Bio-Rad). Bands were selected manually with background subtraction (disk size, 10 mm) enabled. The volume (Int) value that reflects band area and intensity was used. Signals obtained from the phosphospecific α-pS936 antibody were normalized to signals obtained with the pan-specific monoclonal α-TRP antibody. For quantification of transgenic trp expression, signals obtained with the monoclonal α-TRP antibody were normalized to α-β-tubulin antibody signals.
Candidate screen for kinases and phosphatases of S936.
The screen of fly strains with alterations in kinases and phosphatases expressed in the eye was performed as described previously (Voolstra et al., 2013) except that the α-pS936 antibody was used.
Immunocytochemistry of fly eyes.
Immunocytochemistry of fly eyes was performed as described previously (Voolstra et al., 2010). α-TRP (MAb83F6, Developmental Studies Hybridoma Bank) was used as primary antibody. Secondary antibody was α-mouse Cy5 (Dianova). AlexaFluor 546-coupled phalloidin (Life Technologies) was used to label the actin cytoskeleton of the rhabdomeres.
Electroretinogram.
Electroretinograms (ERGs) were recorded from immobilized flies at the age of 0–3 d, as described previously (Peretz et al., 1994). To prevent possible degeneration of the rdgC mutant and to reduce variability among responses of individual flies of the various Drosophila strains, flies of all strains were raised at 19°C. Then, 48 h before the experiment, they were raised in total darkness. Extracellular ERG light responses were measured at 20°C with standard glass micropipettes filled with Ringer's solution (in mm: 130 NaCl, 2 KCl, 5 MgCl2, 2 CaCl2, 10 mm HEPES, pH 7.0) introduced through the cornea into the extracellular matrix of the eye under dim red light (Schott 630 nm edge filter). Flies were grounded via a reference electrode placed on the thorax in a drop of electrode gel. Light from a xenon high-pressure lamp (Photon Technology International, LPS 220, operating at 50 W) passed via red (Schott 630 nm edge filter) or green (Balzers K-55 broad-band green filter) filters was delivered to the compound eye via a light guide. The green light, which is absorbed by both states of fly Rh1, rhodopsin and metarhodopsin, ensures strong activation of Rh1-rhodopsin and hence robust light excitation. At the same time, the green light ensures an efficient metarhodopsin-to-rhodopsin photoconversion. Accordingly, the use of green light prevented the induction of the prolonged depolarizing afterpotential (for review, see Minke, 2012), which constitutes a saturated response that extends in the dark for many minutes. Signals were amplified using a homemade amplifier. The maximal luminous intensity at the eye surface was ∼3.5 logarithmic intensity units above the intensity for a half-maximal response.
Oscillating light stimulation and data analysis.
In vivo stimulation with oscillating light was obtained by using a custom-made electrical circuit driving a 527 nm light-emitting diode. The duty cycle of the square light pulses could be controlled with an accuracy of 1 Hz. A custom-made phototransistor was used to monitor the oscillating light.
To determine the effective intensity of the oscillating light, the peak ERG amplitudes in response to increasing intensities of the green (Balzers K55) light pulses were measured up to saturated amplitude. Then, after 3 min of dark adaptation, the same fly was exposed to the 527 nm oscillating light while its intensity was adjusted to elicit nearly saturating negative ERG amplitude (see Fig. 4C). This adjusted intensity of the oscillating light was determined once each day of the experiment. To determine the time required for the oscillating light at a specific frequency to elicit a significant oscillating response at this frequency (frequency-locked response), a Fourier transform was calculated from time segments of 200 ms after oscillating light onset. When the Fourier transform of a specific time segment reached a significant amplitude (in mV2/Hz), which was larger than the noise, at the frequency of the light stimulus (see Fig. 6B–F), the time from light onset to the specific time segment was plotted in the histograms of Figure 5.
Results
Dephosphorylation of TRP at S936 depends on elevation of cellular Ca2+ levels
Previously, using quantitative mass spectrometry, we identified S936 as a phosphorylation site of the TRP channel that was predominantly phosphorylated in the dark and became dephosphorylated upon illumination (Voolstra et al., 2010, 2013). We generated a phosphospecific antibody that detects TRP phosphorylated at S936 (Voolstra et al., 2010). This antibody will hereafter be referred to as α-pS936-TRP. To verify the specificity of α-pS936-TRP, we performed a dot blot analysis using phosphopeptides and dephosphopeptides (Fig. 1A). As a result, the antibody strongly detected solely the phosphopeptide against which it was raised. To examine whether the α-pS936-TRP can be used for a quantitative study, we examined the relation between the amount of TRP protein loaded and pS936-TRP signal intensity. To this end, we mixed head extracts from dark-adapted WT flies and from trpP343-null mutants in varying proportions and performed Western blot analyses. Membranes were probed with α-TRP and, after stripping, with α-pS936-TRP. The signal intensities of the bound antibodies linearly increased with increasing amounts of TRP protein (data not shown). The results thus showed that the method and detection system we used allowed proper quantification within a certain range of protein amounts loaded on the gel.

Dephosphorylation of TRP at S936 depends on elevation of cellular Ca2+ levels. Immunoblot analyses of S936-TRP phosphorylation and total TRP amounts using a phosphospecific α-pS936-TRP and a pan-specific α-TRP antibody. A, Dot blot analysis testing the specificity of the α-pS936-TRP antibody. TRP phosphopeptides and dephosphopeptides (2.5 μg each) harboring the phosphorylation sites T849, T864, and S936 were spotted onto a nitrocellulose membrane. B, Flies expressing defective TRP channels and control flies were light (white bars) or dark (black bars) adapted for 1 h before Western blot analysis of fly head extracts. The figure shows a representative immunoblot of three independent experiments from which the graph was obtained. Error bars show SEM (N = 3). P values were calculated using Welch's t test and are presented above the bars. C, Eye cups from WT flies were prepared under dim red light and incubated in buffer containing Ca2+ (+Ca2+) or in buffer lacking Ca2+ and containing 1.6 mm EGTA (−Ca2+) for 15 min. During incubation, eye cups were illuminated (white bars) or kept in the dark (black bars). Intact flies were illuminated or kept in the dark for 15 min before heads were subjected to immunoblot analysis (heads). The figure shows a representative immunoblot. The graph was obtained from 5–7 independent experiments for each condition. Error bars show SEM. Using two-way ANOVA, the initial P values for the comparison of light and dark, as well as the comparison +Ca2+ eye cups, −Ca2+ eye cups, and heads were <0.0001. To compare specific datasets, we did a Bonferroni post hoc test. The P values from this test are presented above the bars. Molecular mass markers (in kDa) are indicated to the left of the blots.
To characterize the process of light-dependent dephosphorylation of pS936-TRP, we analyzed the Ca2+ dependence of this process using Drosophila mutants with reduced light-induced Ca2+ influx (Fig. 1B). Due to a mutation in the TRP box domain, the trp14 mutants show a transient response to prolonged light and ∼60% TRP expression level relative to WT flies (Wang et al., 2005). Western blot analysis of trp14 fly heads using the α-pS936-TRP revealed elevated TRP phosphorylation at S936 regardless of the light conditions. This indicates that activation of the phototransduction cascade with transient Na+ and Ca2+ influx into the photoreceptor cell is insufficient to promote pS936 dephosphorylation. Similarly, we examined a transgenic fly expressing a mutant TRP channel, in which Asp 621 was exchanged by Gly in the pore region (trpD621G). This mutation drastically reduces the Ca2+ permeation of TRP and thus strongly decreases the Ca2+ influx during sustained illumination (Liu et al., 2007). We observed strong TRP phosphorylation at S936 in both light-adapted and dark-adapted trpD621G transgenic flies. This result indicates that transient light response and thus transient influx of Ca2+ and Na+ is not sufficient to trigger dephosphorylation at pS936-TRP when the Ca2+ influx is attenuated.
To provide additional, genetically independent, evidence for the role of Ca2+ influx in dephosphorylation of TRP at S936, we performed Western blot analyses of eye cup preparations using the α-pS936-TRP (Fig. 1C). Eye cups from WT flies (see Materials and Methods) were incubated for 15 min in Ringer's solution containing Ca2+ (Fig. 1C, +Ca2+) or in Ringer's solution lacking Ca2+ and containing 1.6 mm EGTA (Fig. 1C, −Ca2+). During incubation, the eye cups were illuminated or kept in the dark. As a control, intact flies were illuminated or kept in the dark for 15 min before heads were subjected to Western blot analysis (Fig. 1C, heads). The results showed that the phosphorylation level of S936-TRP was significantly decreased by Ca2+ addition, independent of the light–dark regime (Fig. 1C). The results also showed that the phosphorylation level of S936-TRP was significantly decreased upon illumination in a Ca2+-independent manner (according to ANOVA analysis). However, in the absence of Ca2+, the effect of light on S936-TRP phosphorylation was small and not significant (according to t test analysis; Fig. 1C). Together, the use of mutants with reduced light-induced Ca2+ influx indicated that S936-TRP phosphorylation is Ca2+-dependent. This conclusion was supported by modulation of extracellular Ca2+ in the eye cup preparation.
pS936-TRP dephosphorylation in the light is rapid and occurs gradually as a function of light intensity
To investigate the kinetics of light-induced dephosphorylation of pS936-TRP, we used flies that were kept in the dark and were then illuminated with intense white light for different periods of time before they were shock-frozen in liquid nitrogen. pS936-TRP dephosphorylation was determined by using the phosphospecific α-pS936-TRP and total amounts of TRP were monitored with a pan-specific α-TRP (Fig. 2A). The results show that 45% of the phosphorylated TRP channels became dephosphorylated at S936 within 10 s of illumination. After 60 s of illumination, 82% of the phosphorylated TRP channels were dephosphorylated. To investigate the dependence of the dephosphorylation reaction on light intensity, we dark-adapted flies for 12–18 h and exposed them to different light intensities for 15 min. Fly heads were then subjected to Western blot analyses using α-pS936-TRP and α-TRP (Fig. 2B). We observed a gradual reduction of phosphorylated pS936-TRP with increasing illumination intensity. Since phosphorylation at a single site is a binary event, gradual dephosphorylation must result from dephosphorylation of different numbers of TRP molecules. The duration of 15 min was chosen because previous and new data showed that the dephosphorylation reaction of S936 at intense background illumination was completed at this time point (Fig. 2A; Voolstra et al., 2010).

Light-dependent dephosphorylation of pS936-TRP is rapid and graded as a function of light intensity. A, Time course of dephosphorylation of pS936-TRP. WT flies were kept in the dark overnight and were then illuminated with bright white light for the indicated periods. Immediately after illumination, flies were shock-frozen in liquid nitrogen to stop the dephosphorylation reaction. A representative blot and a graph obtained from three independent experiments are shown. Error bars show SEM. B, Dark-adapted WT flies were exposed to different light intensities or darkness for 15 min before Western blot analysis of head extracts. The graph was derived from three independent experiments. C, D, rdgC-null mutant (rdgC306) and WT flies were light (white bars) or dark (black bars) adapted for 1 h (C) or overnight (D) before Western blot analyses of heads. The figures show representative blots from three independent experiments from which the graphs were obtained. Error bars show SEM. P values were calculated by Welch's t test and are presented above the bars. Molecular mass markers (in kDa) are indicated to the left of the blots.
Together, these results show that phosphorylated TRP channels at S936 in the dark undergo relatively rapid (time scale of seconds) dephosphorylation to a level that inversely correlates with light intensity.
The RDGC phosphatase dephosphorylates the TRP channel at S936
An equilibrium between protein kinase and phosphatase activities determines the fraction of phosphorylated TRP channels at S936 upon specific illumination conditions. To search for kinases and phosphatases that phosphorylate and dephosphorylate S936-TRP, we performed a candidate screen using the α-pS936-TRP as described previously for the TRP phosphorylation sites T849 and T864 (Voolstra et al., 2013). We analyzed 83 fly strains with impairments in kinases and phosphatases expressed in the eye. Of the 83 fly strains tested, only the rdgC-null mutant, rdgC306 (Steele et al., 1992; Sherman et al., 1997; Ramulu and Nathans, 2001; Ramulu et al., 2001), exhibited a significantly altered phosphorylation/dephosphorylation pattern of S936-TRP (Fig. 2C,D). We found elevated phosphorylation of S936-TRP in light-adapted (Fig. 2C,D) and in prolonged dark-adapted rdgC306 flies (Fig. 2D). The photoreceptors of the rdgC306 mutant degenerate due to constant phosphorylation of Rh1 (Vinóset al., 1997). Therefore, as a control, we also used a transgenic rdgC mutant that expresses truncated Rh1 and cannot undergo Rh1 phosphorylation (P[rh1-rh1Δ356];rdgC306,ninaEI17) which means it does not degenerate. This transgenic fly showed a level of S936-TRP phosphorylation after 1 h of dark adaptation similar to that of the rdgC mutant and WT flies (data not shown).
Interestingly, the S936-TRP phosphorylation level in 12 h dark-adapted WT flies was smaller (63.4 ± 6.7%) than the phosphorylation level of 12 h dark-adapted rdgC306 flies, thus showing hyperphosphorylation of S936-TRP in rdgC306 relative to WT flies (Fig. 2D). Assuming that this hyperphosphorylation represents maximal S936-TRP phosphorylation, it allowed estimating the fraction of S936-TRP channel subunits, which undergo light-dependent dynamic changes of their phosphorylation state in WT flies. Since ≤100% of the TRP molecules can be phosphorylated in the 12 h dark-adapted rdgC mutant, ≤63.4% of TRP molecules are phosphorylated at S936 in 12 h dark-adapted WT flies. The 12 h dark-adaptation regime corresponds to that used to measure the time course of pS936-TRP dephosphorylation, in which flies were dark-adapted overnight (Fig. 2A). After 10 s of illumination, 55% (±2.6%) of the previously phosphorylated TRP channels remained phosphorylated (Fig. 2D). It can be thus inferred that ≤34.9% (0.634 × 0.55 = 0.349) of TRP channels undergo dephosphorylation during 10 s of illumination. We therefore propose that during alternating dark/light, in a time scale of seconds, only a fraction of TRP channel subunits undergo dephosphorylation at pS936-TRP (see Discussion).
The hyperphosphorylation of S936-TRP in rdgC306 flies in prolonged dark adaptation (Fig. 2D) is consistent with similar observations of Rh1 hyperphosphorylation in this mutant (Vinós et al., 1997). Thus, the reduced S936 phosphorylation in 12 h light-adapted rdgC306 flies compared with 12 h dark-adapted rdgC306 flies (Fig. 2D) suggests that additional light-dependent protein phosphatases, other than RDGC, dephosphorylate pS936-TRP. Meanwhile, the increased S936-TRP phosphorylation of light-adapted rdgC306 flies suggests participation of light-dependent protein kinase in S936-TRP phosphorylation. Together, the data suggest a complex regulation of S936-TRP phosphorylation with participation of RDGC in this process (see Discussion).
Generation of transgenic flies that express TRP channels with altered S936-TRP phosphorylation
To investigate the physiological roles of light-dependent pS936 dephosphorylation in the most direct manner, we generated a transgenic fly strain that expresses a TRP channel in which Ser 936 was exchanged to Ala (trpS936A), thus preventing phosphorylation permanently. We also generated a transgenic fly that expresses a TRP channel in which Ser 936 was exchanged with Asp (trpS936D) mimicking permanent phosphorylation. In addition, we generated a fly transgenically expressing an unmodified TRP channel (trpWT) as a control with a similar genetic background. All transgenes were expressed under control of the rh1 (rhodopsin 1) promoter on a trpP343 (trp-null mutant) background (all transgenic flies will be designated hereafter as trpS936A, trpS936D, and trpWT). To rule out possible effects of the transgenic expression of TRP per se, we performed several control experiments. In principle, an abnormal light response due to impaired function of TRP channels may occur via strongly reduced channel protein level (Wang et al., 2005), failure to interact with the INAD (inactivation no afterpotential D) scaffold protein (Tsunoda et al., 1997), failure to form homomultimers, or mislocalization of the TRP channel in the photoreceptors (Katz et al., 2013). Therefore, using Western blot, coimmunoprecipitation (co-IP), and immunocytochemical analyses, we examined whether the TRPS936A and TRPS936D mutant channels are expressed at a level comparable to WT TRP, are associated with the INAD scaffold protein, form multimers with WT TRP, and are localized to the rhabdomeres. Protein levels of TRPS936A and TRPS936D in the transgenic flies were 84.0 ± 7.4% and 120.7 ± 23.0%, respectively, of WT TRP (Fig. 3A, wild type). trpWT Flies exhibited 103.1 ± 19.5% of WT TRP levels. The ∼16% reduction of TRP levels in the trpS936A transgenic fly relative to WT flies is unlikely to account for a significant abnormality in a physiological phenotype because of the large abundance of TRP in the photoreceptors (∼7.5 × 105 TRP molecules in a single cell; Henderson et al., 2000; Katz and Minke, 2009). Indeed, it was shown that drastically reduced expression of TRP still resulted in normal light response with little if any physiological consequences (Wang et al., 2005). Using co-IP, we investigated the capability of TRPS936A and TRPS936D to interact with the INAD scaffold protein (Fig. 3B; Huber et al., 1996; Chevesich et al., 1997; Tsunoda et al., 1997) and to form multimers with an eGFP-tagged WT TRP (Katz et al., 2013; Fig. 3C). Both properties of the WT TRP (i.e., interaction with INAD and multimer formation) were not affected by the exchange of S936 to A or D (Fig. 3B,C). Other signaling proteins of the phototransduction cascade were present in comparable amounts to their WT levels (Fig. 3D). We also examined the rhabdomeric localization of the TRPS936A and TRPS936D proteins using immunocytochemistry and found that it was normally localized to the rhabdomeres as was WT TRP (Fig. 3E). Together, all the control studies indicated that the trpS936A and trpS936D transgenes were suitable for examining the effect of TRP permanent dephosphorylation, or the effect of a phosphomimetic mutation at the S936 site, on the physiological response to light.

Generation of transgenic flies that express TRP channels with altered S936-TRP phosphorylation. A, Heads of flies were subjected to Western blot analyses assessing TRP and tubulin. For quantification, the integrated density values of the TRP bands were divided by the integrated density values of the tubulin bands. These values were normalized to the WT value, which was set to 100%. The graph was obtained from four independent experiments. Error bars show SEM. The trpS936A, trpS936D, and trpWT transgenes were expressed in a trpP343-null mutant background. Flies of the same genotype were dark adapted for 1 h and subjected to Western blot analysis using the α-pS936-TRP antibody (bottom). As expected, α-pS936-TRP did not bind to the mutated TRP channels in trpS936A and trpS936D flies. B, TRPS936A or TRPS936D binds to INAD: Co-IP of TRPS936A or TRPS936D with the INAD scaffold protein. Protein extracts from fly heads of the indicated fly strains were coimmunoprecipitated using a polyclonal α-TRP antibody. Extracts (input) and coimmonoprecipitates (IP) were separated by SDS-PAGE, blotted to PVDF membranes, and probed with α-INAD and monoclonal α-TRP antibodies. C, Co-IP of TRPS936A or TRPS936D with eGFP-tagged WT TRP. Protein extracts from fly heads were coimmunoprecipitated using a α-GFP antibody. Extracts (input) and coimmonoprecipitates (IP) were separated by SDS-PAGE, blotted to PVDF membranes, and probed with a polyclonal α-TRP antibody. The upper band in the panels represents eGFP-tagged TRP. The lower band represents WT TRP, TRPS936A, or TRPS936D, respectively. D, Western blot analyses assessing expression of major signaling proteins in various fly strains as indicated. Molecular mass markers are indicated to the left of the blots. E, Immunocytochemical localization of TRP in photoreceptors. Cross sections through the eyes of WT, trpS936A, and trpS936D transgenic flies were probed with an α-TRP (green) and with phalloidin (red), which labels the rhabdomeres. Overlay of both colors in the merged panels appears yellow. Scale bar, 10 μm.
Preventing phosphorylation of S936-TRP abolishes the difference in frequency response between dark-adapted and light-adapted flies
To study the effect of the trpS936A and trpS936D mutations on the physiological response to light, we used the ERG signal. The ERG is an extracellular voltage recording that reflects the summed electrical activity of the eye in response to light stimulation (Minke, 1982). The main components of the ERG light response are as follows: (1) the extracellularly recorded photoreceptor potential; (2) the “on” and “off” transients, at the beginning and the end of the light pulse, arising from the second order lamina neurons; and (3) the slow response of the pigment (glia) cells (Minke, 1982). The photoreceptor potential is the physiological response to light arising from the light-induced openings of the TRP and TRPL channels. The depolarization of the photoreceptor cell triggers the induction of the corneal positive “on” transient, via sign-inverting synapse between the photoreceptor axon and the large monopolar neurons of the lamina. These postsynaptic transients disappear during oscillating light frequencies of >10 Hz (Fig. 4C). The response of the pigment cells appears as a corneal-negative slow rise and slow decay of the ERG after light on and off, respectively. These slow components arise from an increase in extracellular K+, resulting from K+ efflux from the photoreceptor cells via the TRP and TRPL channels, which depolarize the pigment cells surrounding the photoreceptor cells (Minke, 1982). These components cannot follow light frequencies of >10 Hz and constitute the steady corneal-negative response to oscillating light, on which the oscillating response of the photoreceptors is superimposed (Fig. 4C).

Frequency response to oscillating light of light adapted flies. A, B, Graphs showing the frequency response to intense oscillating green (527 nm) light with an intensity that elicited near-saturating ERG amplitude. The graphs plot the log of the peak-to-peak oscillating ERG amplitude as a function of log stimulus frequency. The graphs were measured in light-adapted fly strains including trpWT, trpS936A, and trpS936D transgenic flies (Fig. 4A) and WT, rdgC306 mutant, and rh1Δ356;ninaEI17, rdgC306 transgenic flies (Fig. 4B), as indicated. All ERG responses were measured after the oscillating light elicited stable steady-state oscillating responses. The differences between the various fly strains at low frequencies are not significant (p > 0.05). The frequency response of trpS936D transgenic flies was significantly smaller than that of the other fly strains at high frequencies. Error bars show SEM; N = 4–15 for each fly strain. P values were calculated by Welch's t test and are presented for trpS936D near the averaged values at the inset. Insets, The frequency response of A and B at high frequencies in expanded linear frequency scale. The P values comparing trpS936D and WT are as follows: 50 Hz, not significant, p = 0.0621; 55 Hz, not significant, p = 0.0616; 60 Hz, *p = 0.0263; 65 Hz, *p = 0.0195; 70 Hz, *p = 0.0109; 75 Hz, *p = 0.0116; 80 Hz, **p = 0.004; 85 Hz, **p = 0.0044; 90 Hz, **p = 0.0023. C, A representative example of an ERG response to oscillating light of 10 Hz recorded from a light-adapted WT fly. The initial positive “spike” is the “on” transient coming from the lamina neurons. The upper red trace depicts the light stimulus measured simultaneously by a phototransistor.
The trpl302 mutant (lacking the TRPL channel) occasionally displays oscillating voltage responses arising from the synaptic terminal of R1–R6 photoreceptors due to a secondary mutation in the inebriated gene. These oscillating light responses appear in the ERG or intracellular measurements of the light response to a constant light pulse (Leung et al., 2000) but they do not appear in whole-cell recordings from isolated ommatidia, in which the synaptic terminals are removed. These oscillatory responses interfere with measuring the in vivo frequency response to oscillating light in trpl mutant background. Therefore, ERG recordings were performed in trpWT, trpS936A, and trpS936D on single null trpP343 background. None of these flies displayed oscillating voltage responses upon constant light stimulation.
A physiological manifestation of the light response kinetics in vivo is the ability of the light response to follow oscillating light of increasing frequencies up to the flicker fusion frequency at which the light response no longer follows the oscillating light. To measure the effect of the Drosophila TRP channel dephosphorylation at S936 on the frequency response in vivo, we measured the ERG response to intense (near saturation) oscillating green (527 nm) light. The response to the oscillating light was measured during increasing stimulus frequencies from 10 to 100 Hz in light-adapted WT (and trpWT), rdgC306 (and P[rh1-rh1Δ356];rdgC306,ninaEI17), trpS936A, and trpS936D transgenic flies (Fig. 4A,B). In response to oscillating light of gradually increased frequency, the frequency responses of the tested light-adapted fly strains were similar. This was illustrated by plotting the log of the peak-to-peak oscillating ERG amplitude as a function of log stimulus frequency (Fig. 4A,B). Strikingly, the frequency response of light-adapted trpS936D flies at high frequencies was different from that of the other fly strains and was confined to lower frequencies (Fig. 4A; see below).
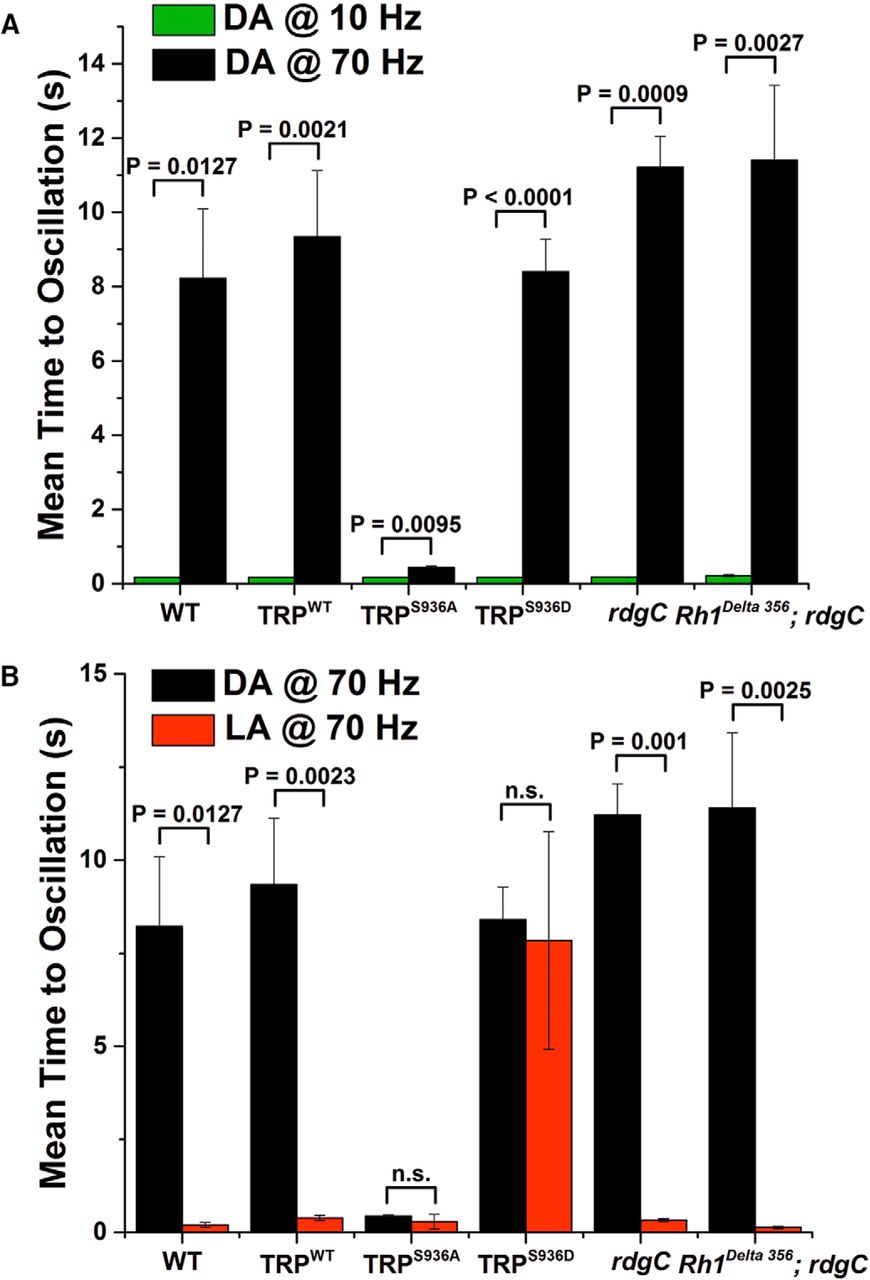
Under dark-adapted conditions, all tested fly strains revealed a quick onset (<0.3 s) of frequency-locked response to oscillating light of low frequencies (e.g., 10 Hz; Fig. 5A, green columns). In contrast, at higher stimulus frequencies (>70 Hz), dark-adapted fly strains did not reveal frequency-locked response to the oscillating light, even 6 s after light onset (Fig. 6). Illumination time >8 s of intense oscillating light (>70 Hz) was required for the fly strains to show significant oscillatory-locked response (Fig. 5A, black columns). The trpS936A fly constituted a pronounced exception: it revealed a quick onset (<0.3 s) of frequency-locked response to oscillating light of high frequency under dark-adapted conditions (Fig. 5A, black column, TRPS936A). In contrast to the fly strains under dark-adapted conditions, fly strains under light-adapted conditions revealed a quick (<0.3 s) onset of frequency-locked response to oscillating light of high frequency (>70 Hz; Fig. 5B, red columns). However, the trpS936D flies constituted a pronounced exception relative to all other light-adapted fly strains: light-adapted trpS936D flies failed to reveal frequency-locked response to oscillating light of high frequency even 6 s after light onset (Fig. 6). Illumination time of ∼8 s intense oscillating light was required for this light-adapted fly strain to show significant frequency-locked response to oscillating light of high frequency (Fig. 5B, red column, TRPS936D). Surprisingly, rdgC mutant flies (and P[rh1-rh1Δ356];rdgC306,ninaEI17, serving as a control that does not undergo retinal degeneration) did not reveal any phenotype compared with WT flies in response to high stimulus frequencies (Fig. 5B; see Discussion).

Preventing phosphorylation of S936-TRP in transgenic Drosophila abolished the difference in frequency response between dark-adapted and light-adapted flies. A, Histogram plotting the time required for the ERG response of dark-adapted fly strains to follow the oscillating 527 nm light at 10 (green) and 70 Hz (black). A marked difference is observed between the frequency response of dark-adapted flies at 10 and 70 Hz light stimulus frequencies. At 10 Hz stimulus frequency, dark-adapted WT flies could follow the stimulus frequency shortly after stimulus onset, in a manner similar to that of light-adapted flies (B). However, at 70 Hz stimulus frequency, dark-adapted WT flies could not follow the stimulus frequency immediately after stimulus onset and required ∼8 s to follow this stimulus frequency. This behavior was found in WT and in transgenic flies expressing WT TRP (trpWT), in rdgC306 flies (and rh1Δ356;rdgC306 serving as a control), and in light-adapted trpS936D transgenic flies. Strikingly, dark-adapted trpS936A transgenic flies, which cannot undergo phosphorylation of TRP at S936, exhibited a light response to the oscillating light shortly after light onset at both 10 and 70 Hz. Error bars are SEM; N = 4–11 for each fly strain. P values were calculated by Welch's t test and are presented above the error bars. The differences among the black bars of the various fly strains (except for the trpS936A) are not significant (p > 0.05). B, Histograms showing the time required for the ERG response to follow 70 Hz oscillating light in dark-adapted (black) and light-adapted (red) fly strains. A marked difference is observed between the frequency response of dark-adapted and light-adapted flies at 70 Hz in most but not all fly strains. Accordingly, light-adapted WT flies could follow the high stimulus frequency shortly after stimulus onset. However, dark-adapted WT flies required ∼8 s to follow the high stimulus frequency. This behavior was found in WT and in transgenic flies expressing WT TRP (trpWT), rdgC mutant flies (and rh1Δ356;ninaEI17, rdgC306). Strikingly, trpS936A transgenic flies, which cannot undergo phosphorylation at the S936-TRP site, revealed oscillatory-locked response to the oscillating light shortly after light onset at both dark-adapted and light-adapted flies, while trpS936D revealed oscillatory-locked response to the oscillating light long after light onset at both dark-adapted and light-adapted flies. Error bars are SEM; N = 5–9 for each fly strain. P values were calculated by Welch's t test and are presented above the error bars. The differences among the black bars of the various fly strains (except for the trpS936A) are not significant (p > 0.05). The relatively large SEM of light-adapted trpS936D arise from three of eight flies that showed time to oscillatory-locked response similar to that of WT flies. This result suggests that the phosphomimetic effect is weaker than actual phosphorylation. The frequency response of few trpS936D flies was below the noise level at 70 Hz and therefore the time to oscillatory-locked response was measured at 60 Hz, which represents a lower limit for 70 Hz that could not be measured accurately.

Frequency response amplitude to oscillating light in dark-adapted and light-adapted flies measured 6 s after light onset. A, C, E, Representative short segments of ERG responses to intense (near saturation) oscillating 527 nm light of 70 Hz, measured 6 s after light onset in dark-adapted (DA) and light-adapted (LA) fly strains: WT (A); trpS936A (C), and trpS936D (E) transgenic flies. B, D, F, The amplitude of the Fourier transform of the ERG responses to oscillating light of 70 Hz, measured 6 s after light onset in dark-adapted (DA) and light-adapted (LA) flies. The Fourier transform was calculated from time segments of 200 ms, 6 s after oscillating light onset. The graphs plot the Fourier transform of ERG responses as a function of frequency obtained from light-adapted and dark-adapted fly strains as indicated. A prominent peak at 70 Hz is observed in light-adapted WT and trpS936A fly strains but not in trpS936D transgenic flies, while in dark-adapted flies, only the trpS936A fly showed a pronounced peak at 70 Hz.
To summarize, the data indicates that WT flies differ in their frequency-locked response to oscillating light of high frequency depending on whether they are light or dark adapted. The data obtained from trpS936A and trpS936D flies indicate that this difference is due to the S936-TRP phosphorylation state.
Discussion
The speed of converting light-intensity changes into neuronal activity is critical for the behavior of small flying organisms, such as flies. A large body of information already exists on Drosophila phototransduction, which consists of sequential activation and termination of a cascade of enzymatic reactions with feedforward and feedback loops (Katz and Minke, 2009; Montell, 2012; Hardie and Juusola, 2015). The speed of this signaling cascade is adjusted according to the adaptation state of the photoreceptors, as light-adapted flies exhibit a higher temporal resolution (and frequency response) of the visual system than dark-adapted flies (Wu and Wong, 1977; Song et al., 2012). The higher temporal resolution of light-adapted flies is reflected in the ability of the visual system to follow higher frequencies of oscillating light, but the underlying molecular mechanism is not clear. Interestingly, the studies of Hardie and colleagues suggested that the TRP channel is a critical protein in Ca2+-mediated light adaptation (Gu et al., 2005).
The results of the present study show that in dark-adapted WT flies, when most TRP molecules are phosphorylated at the S936 site, the ability to follow oscillating light is initially limited to relatively low frequencies (i.e., <70 Hz). However, after ∼10 s of illumination with intense oscillating light, the photoreceptors begin showing frequency-locked response to oscillating light of higher frequencies. Light-adapted flies are able to follow high-frequency oscillating light almost immediately (within 0.3 s). We suggest that this change in temporal resolution of the light response depends on dephosphorylation of pS936-TRP. Accordingly, when this site is phosphorylated, the maximally detected stimulus frequency is relatively low, while upon light adaptation, Ca2+-dependent dephosphorylation of S936-TRP sites shifts the maximally detected frequency of the light stimulus to higher frequencies. Support for this mechanism came from our biochemical experiments showing similar temporal dynamics of the dephosphorylation of pS936-TRP sites and the frequency-locked physiological response. The above notion was further supported by the experiments in which S936-TRP could not undergo phosphorylation and remained constantly dephosphorylated in the trpS936A transgenic flies. The relatively fast onset of frequency-locked response to oscillating lights of high frequency, regardless of the dark/light adaptation regime, fits well with the constant dephosphorylation state of its S936 site. Furthermore, the shift of the frequency response of the phosphomimetic trpS936D transgenic flies to lower frequencies in light-adapted flies and the relatively long time required to show frequency-locked response to oscillating light of high frequency in both dark-adapted and light-adapted flies also support the above notion. Interestingly, the trpS936A transgenic flies revealed characteristics of light-adapted WT flies under dark-adapted conditions, while the phosphomimetic trpS936D transgenic flies revealed characteristics of dark-adapted WT flies under light-adapted conditions. These results strongly support the notion that the difference in the frequency response at high frequencies between dark-adapted and light-adapted WT flies depends on the phosphorylation state of TRP at the S936 site.
The observation of frequency-locked response at high frequency in light-adapted rdgC mutant flies, in spite of the phosphorylation of a significant fraction of its TRP molecules at the S936 site, is apparently not compatible with the above notion. A possible reconciliation may arise from the assumption that the RDGC phosphatase is not the only Ca2+-dependent phosphatase of the photoreceptor cells. Thus, in hyperphosphorylated dark-adapted rdgC306, a sufficient number of pS936-TRP molecules capable of affecting the frequency response undergoes dephosphorylation locally at the sites of photon absorption by other, still unidentified, light-dependent and Ca2+-dependent phosphatase(s). Experimental support for this hypothesis came from the observation that the phosphorylation level of dark-adapted pS936-TRP in rdgC306 flies was largely reduced upon light adaptation (Fig. 2D). Additionally, analyses of the time course of pS936-TRP dephosphorylation revealed that only a fraction of TRP channels undergoes light-induced dephosphorylation during a time scale of ∼10 s (Fig. 2A). Note that this is approximately the time required for dark-adapted WT flies to show frequency-locked response to oscillating lights of >70 Hz. It is also important to note that TRP molecules form tetramers to yield functional channels. Dephosphorylation of a single-channel subunit might be sufficient to affect the kinetics of the whole tetramer. Thus, a high-frequency-locked response could be obtained by a relatively small fraction of pS936-TRP molecules that undergo light-dependent dephosphorylation.
Evidence showing that activation of the TRP channels constitutes the limiting factor that determines the speed of phototransduction came from a recent study. In this study, the lipid composition of the photoreceptor membranes was modulated (Randall et al., 2015). Accordingly, dietary-induced replacement of polyunsaturated fatty acids (PUFAs) led to replacement of PUFAs by saturated fatty acids in the plasma membrane, leading to a large shift of both the frequency response to light toward low frequencies and the quantum bump latency distribution toward longer latencies. Importantly, all these effects stemmed from effects on the TRP channels (Randall et al., 2015). The authors proposed that the above changes in membrane lipid composition changed the stiffness of the plasma membrane, leading to changes in the threshold of TRP channel activation (Katz and Minke, 2012; Kohn et al., 2015; Randall et al., 2015).
Due to the similarity in the effect of PUFA reduction and S936-TRP phosphorylation on the frequency response of Drosophila photoreceptors, we suggest that both TRP modulations may exert their effect via changes in the activation threshold of TRP channels.
Footnotes
This work was supported by grants from the National Eye Institute (R01 EY 03529), the Israel Science Foundation, and the Deutsch-lsraelische Projektkooperation to B.M., and by grants from the Deutsche Forschungsgemeinschaft (Vo 1741/1-1, Hu 839/2-6, Hu 839/7-1). Dr. Ben Katz was a postdoctoral fellow of the Teva National Network of Excellence and the Edmond and Lilly Safra Center. We thank Drs. Moshe Parnas, Shahar Frechter, and Shirley Weiss for useful comments on the manuscript. We also thank Mr. Anatoly Shapochnikov for the construction of the stimulating light source of variable frequencies and Katherina Beck for help with generating transgenic flies.
The authors declare no competing financial interests.
- Correspondence should be addressed to Baruch Minke, Department of Medical Neurobiology, Faculty of Medicine, Hebrew University, Jerusalem 91120, Israel. baruchm{at}ekmd.huji.ac.il





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}