

Abstract
Spinocerebellar ataxia 17 (SCA17) is caused by polyglutamine (polyQ) repeat expansion in the TATA-binding protein (TBP) and is among a family of neurodegenerative diseases in which polyQ expansion leads to preferential neuronal loss in the brain. Although previous studies have demonstrated that expression of polyQ-expanded proteins in glial cells can cause neuronal injury via noncell-autonomous mechanisms, these studies investigated animal models that overexpress transgenic mutant proteins. Since glial cells are particularly reactive to overexpressed mutant proteins, it is important to investigate the in vivo role of glial dysfunction in neurodegeneration when mutant polyQ proteins are endogenously expressed. In the current study, we generated two conditional TBP-105Q knock-in mouse models that specifically express mutant TBP at the endogenous level in neurons or in astrocytes. We found that mutant TBP expression in neuronal cells or astrocytes alone only caused mild neurodegeneration, whereas severe neuronal toxicity requires the expression of mutant TBP in both neuronal and glial cells. Coculture of neurons and astrocytes further validated that mutant TBP in astrocytes promoted neuronal injury. We identified activated inflammatory signaling pathways in mutant TBP-expressing astrocytes, and blocking nuclear factor κB (NF-κB) signaling in astrocytes ameliorated neurodegeneration. Our results indicate that the synergistic toxicity of mutant TBP in neuronal and glial cells plays a critical role in SCA17 pathogenesis and that targeting glial inflammation could be a potential therapeutic approach for SCA17 treatment.
SIGNIFICANCE STATEMENT Mutant TBP with polyglutamine expansion preferentially affects neuronal viability in SCA17 patients. Whether glia, the cells that support and protect neurons, contribute to neurodegeneration in SCA17 remains mostly unexplored. In this study, we provide both in vivo and in vitro evidence arguing that endogenous expression of mutant TBP in neurons and glia synergistically impacts neuronal survival. Hyperactivated inflammatory signaling pathways, particularly the NF-κB pathway, underlie glia-mediated neurotoxicity. Moreover, blocking NF-κB activity with small chemical inhibitors alleviated such neurotoxicity. Our study establishes glial dysfunction as an important component of SCA17 pathogenesis and suggests targeting glial inflammation as a potential therapeutic approach for SCA17 treatment.
Introduction
Polyglutamine (polyQ) diseases are a family of neurodegenerative disorders caused by CAG trinucleotide repeat expansions, which are translated into a large polyQ tract that makes the disease proteins prone to misfold and aggregate (Orr and Zoghbi, 2007). Currently, the polyQ disease family consists of nine members, including Huntington's disease, spinobulbar muscular atrophy, dentatorubral-pallidoluysian atrophy, and spinocerebellar ataxia (SCA) types 1, 2, 3, 6, 7, and 17. Among these diseases, SCA17 is caused by >42 CAG repeats in the TATA box-binding protein (TBP) gene (Koide et al., 1999; Nakamura et al., 2001), which encodes a general transcription factor that plays critical roles in transcriptional initiation (Nikolov and Burley, 1994). SCA17 patients show late-onset symptoms that include ataxia, dystonia, parkinsonism, and psychiatric abnormalities, accompanied by progressive neurodegeneration in the cerebellum (Koide et al., 1999; Nakamura et al., 2001; Rolfs et al., 2003; Bauer et al., 2004; Bruni et al., 2004; Toyoshima et al., 2004). Although the exact pathological mechanisms of SCA17 remain elusive, emerging evidence indicates that transcriptional dysregulation caused by mutant TBP represents a major form of toxicity (Yang et al., 2016). Indeed, the generation of several SCA17 rodent models has enabled the identification of transcriptional defects that account for the neuronal toxicity of SCA17 (Friedman et al., 2007; Chang et al., 2011; Huang et al., 2011; Kelp et al., 2013; Yang et al., 2014; S. Huang et al., 2015; Cui et al., 2016).
As neuronal survival is preferentially compromised in the brains of polyQ disease patients, most efforts have been devoted to researching the toxic mechanisms caused by mutant polyQ proteins in neurons. Nonetheless, in recent years, research has revealed that glia, the most abundant cells in the CNS and which provide support and protection for neurons, are also actively involved in the pathogenesis of polyQ diseases. For example, in Huntington's disease, the expression of mutant huntingtin (htt) in glial cells can lead to the development of Huntington's disease phenotypes in mice (Shin et al., 2005; Bradford et al., 2009; Tong et al., 2014; B. Huang et al., 2015; Oliveira et al., 2016). These previous studies, however, investigated the glial toxicity of polyQ proteins when these proteins are overexpressed via a transgenic approach. Since overexpression of transgene can confound glial functions and phenotypes (Kagawa et al., 1994; Stefanova et al., 2001), how mutant polyQ proteins in glial cells contribute to neurodegeneration when they are expressed at the endogenous level remains unknown.
In the present study, we generated two lines of conditional TBP-105Q knock-in (KI) mice, which specifically express mutant TBP in neurons or astrocytes at the endogenous level. We found that mutant TBP expression in astrocytes was important for the development of severe neurotoxicity when neuronal cells also express mutant TBP in SCA17 mice. Coculture of neuronal and glial cells also showed that mutant TBP in astrocytes promotes neuronal injury. Inflammatory pathways, including the nuclear factor κB (NF-κB) signaling pathway, were excessively activated in astrocytes expressing mutant TBP, and chemical blocking of this activation in astrocytes ameliorated neurotoxicity in a noncell-autonomous manner. These findings demonstrate that, although severe neuronal degeneration requires synergistic effects of mutant proteins in both neuronal and glial cells, targeting inflammation in astrocytes could be a potentially effective approach for treating SCA17 and other polyQ diseases.
Materials and Methods
Mice.
All the mice (C57BL/6) were bred and maintained in the animal facility at Emory University under specific pathogen-free conditions. All procedures were performed in accordance with the NIH and United States Public Health Service Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee at Emory University, which is accredited by the American Association for Accreditation of Laboratory Care.
Floxed TBP-105Q KI or germline-KI mice were generated as described previously (Huang et al., 2011; B. Huang et al., 2015). CaMKIIα-Cre and GFAP-Cre mice were purchased from The Jackson Laboratory. To generate CaMKIIα-KI or GFAP-KI mice, floxed TBP-105Q KI mice were crossed with CaMKIIα-Cre or GFAP-Cre mice. Genotyping primers used were as follows: for TBP, forward: 5′-CCA CAG CCT ATT CAG AAC ACC-3′, reverse: 5′-AGA AGC TGG TGT GGC AGG AGT GAT-3′; for Cre, forward 5′-GCG GTC TGG CAG TAA AAA CTA TC-3′, reverse: 5′-TGT TTC ACT ATC CAG GTT ACG G-3′.
Antibodies and drugs.
Antibodies were obtained commercially for the following antigens: expanded polyglutamine (1C2, Millipore/Millipore Bioscience Research Reagents), N-terminal 1–20 amino acids of TBP (1TBP18, Thermo Fisher Scientific), vinculin (V9131, Sigma), calbindin-D-28k (C8666, Sigma), NeuN (ABN78, Millipore), GFAP (GA5, Cell Signaling Technology), Cre (C7988, Sigma) MAP-2 (H300, Santa Cruz Biotechnology), NF-κB (p65; D14E12, Cell Signaling Technology), and p-NF-κB (phospho-p65; 93H1, Cell Signaling Technology). Hoechst 33258 (Invitrogen) was used for nuclear labeling. Secondary antibodies were purchased from Jackson ImmunoResearch Laboratories. Sigmafast 3,3′-diaminobenzidine tablets and cytosine β-d-arabinofuranoside were purchased from Sigma. QNZ and BAY 11-7082 were purchased from Enzo Life Sciences.
Mouse behavioral studies.
Mouse body weight was measured monthly, and survival was monitored regularly.
For the rotarod test (Rotamex, Columbus Instruments), mice were trained on the rotarod at 5 rpm for 10 min on 3 consecutive days. For the actual test, the rotarod was set to accelerate to 40 rpm over a 5 min period. Latency to fall was recorded automatically by the instrument, and the performance of each mouse was measured by averaging three trials.
For the balance beam test, a 0.6-cm-thick, 0.8-m-long wooden stick was suspended from a platform on both sides by metal grips. A bright light source at the starting point and a dark box at the end point were used to entice the mice to walk through the stick. Each mouse was trained for 3 consecutive days with three runs per day. For data collection, a stopwatch was used to record the time each mouse took to walk through the stick. The result was the average of three trials.
In the startle response test, prepulse inhibition of mice was detected in a SR-LAB (San Diego Instruments) test station. For testing, mice were placed into the startle apparatus and acclimated for a 5 min period, during which a background noise was administered. Four different intensities of acoustic prepulse stimuli were applied (2, 4, 8, and 16 dB lower than pulse stimuli), and the prepulse stimuli were always 100 milliseconds before the onset of pulse stimuli.
The novel object recognition test was performed as described previously (Leger et al., 2013). Mice at 4–18 months of age were habituated for 5 min on 3 consecutive days in the empty open-field box. Twenty-four hours after the last day of habituation, mice were familiarized for 10 min for one time in the open-field box in which two identical objects were placed. Testing commenced 24 h after familiarization in the open-field box, in which one of the identical objects was replaced by a novel object. Mice were subjected to one test session, each with a maximum duration of 10 min.
For the forced swim test, the mice were placed individually into a round, opaque plastic cylinder (18 cm in height, 15 cm in diameter) filled with water (25°C) at a depth of 12 cm. Immobility time, defined as floating or the absence of active behaviors, such as swimming or struggling to escape, was measured. Slight movements of the feet and tail necessary to keep the head above water were excluded as mobility. Each mouse was measured for 6 min by a trained observer who was kept blind to the genotypes of the mice.
For the tail suspension test, mice were suspended by taping the tail (∼1 cm from tip of tail) to a horizontal bar at a height of 40 cm from the table surface for 6 min. The trial was conducted for a duration of 6 min, and the immobility time was recorded manually via stopwatch by a trained observer who was blind to the genotypes of the mice. Mice were considered immobile when they hung passively and motionlessly without escape-oriented behaviors.
Mouse stereotaxic injection.
AAV-Synapsin-Cre and AAV-GFAP-Cre viruses were purchased from Vector Biolabs. The method for stereotaxic injection was adopted from a previous study (Yang et al., 2017). To inject the viruses into the cerebellum, mice were anesthetized with 1.5% isoflurane inhalation and stabilized in a stereotaxic instrument (David Kopf Instruments). The location of the injected site was determined according to the distance from bregma: anteroposterior, −6.3 mm; mediolateral, ±1 mm; dorsoventral, −1.5 mm. Small holes were drilled on the skull, and a 30 gauge Hamilton microsyringe was used to deliver the virus at a speed of 200 nl per minute (bilateral injection, 2 μl in each side). Meloxicam was given as analgesics, and after surgery the mice were placed on a heated blanket to recover from anesthetic.
Western blotting.
Mouse brain tissues were lysed in ice-cold RIPA buffer containing a protease inhibitor mixture. The lysates were incubated on ice for 30 min, sonicated, and centrifuged at top speed for 10 min. The supernatants were collected and subjected to SDS-PAGE. The proteins in the gel were transferred to a nitrocellulose membrane. The blot was blocked with 5% milk/PBS for 1 h at room temperature and incubated with a primary antibody in 3% BSA/PBS overnight at 4°C. After three washes in PBS, the blot was incubated with HRP-conjugated secondary antibodies in 5% milk/PBS for 1 h at room temperature. The blot was then washed three times in PBS and developed using ECL Prime (GE Healthcare).
Primary neuron and astrocyte coculture.
Enriched glial cultures were prepared following the protocols described previously (Shin et al., 2005). For neuron/astrocyte coculture, astrocytes were plated on coverslips. In rescue experiments, cultured astrocytes were treated with 20–40 nm QNZ for 30 min to 4 h (20 or 40 nm for 30 min, 20 or 40 nm for 2 h, 20 or 40 nm for 4 h) or 5–20 μm BAY 11-7082 for 1–24 h (20 μm for 1 h; 5 μm for 3, 6, 12, or 24 h) to suppress the expression of phospho-NF-κB p65. Neuronal cultures were prepared from the cerebral cortex of mouse pups at postnatal day 0. Viable cells were plated at 1 × 106 cells/ml on poly-d-lysine-coated plastic culture 12-well plates (Corning Costar) in B27-supplemented Neurobasal-A medium (Invitrogen). To reduce glial proliferation, we added cytosine arabinoside to the cultures 1 d after plating (5 × 10−6 m final concentration). At 7–8 DIV, most cells were mature neurons with elongated processes, and they were used for coculture with astrocytes. After washing, the astrocyte-plated coverslips were placed over the cultured neurons in B27-supplemented Neurobasal-A medium for >2 d before being examined. Astrocytes attached to coverslips were unable to grow because serum-free medium and cytosine arabinoside suppressed their growth. Neurons were cocultured with drug-treated or untreated astrocytes for >2 d, astrocytes on the coverslips were removed, and the medium was changed to fresh B27-supplemented Neurobasal-A medium. Neurons were cultured for 24 h before immunofluorescent examination.
Immunofluorescent staining and Immunohistochemistry.
The method was adopted from our previous study (Xiang et al., 2017). For immunofluorescent staining of neurons, neurons were fixed with 4% paraformaldehyde (PFA) for 10 min. Fixed cells were washed three times with PBS and blocked with 3% BSA plus 5% normal donkey serum/PBST (0.2% Triton X-100 in PBS) for 1 h at room temperature. Primary antibodies were diluted in blocking buffer and incubated with the cells at 4°C overnight, followed by three washes with PBS and incubation with fluorescent secondary antibodies and nuclear dye. After three washes, the cells were ready for examination using a Zeiss (Axiovert 200M) microscope with a digital camera (Orca-100, Hamamatsu Photonics) and OpenLAB software (Improvision).
To prepare brain sections, mice were deeply anesthetized and perfused with saline, followed by fixation. Brains were postfixed overnight in 4% PFA and switched to 30% sucrose at 4°C. After being completely sunk, brains were sectioned at 40 μm with a cryostat at −20°C. For immunofluorescent staining, the brain sections were subjected to the same method described above for cultured cells. For immunohistochemistry, brain sections were subjected to primary antibodies, biotinylated secondary antibodies (Vector Laboratories), and the Vectastain ABC HRP kit (Vector Laboratories). When the 1C2 antibody was used for immunohistochemistry, fixed brain sections were treated with 44% formic acid for 1 min and then with 88% formic acid for 10 min for antigen retrieval before incubation with the 1C2 antibody. The brain sections were then incubated in 3,3′-diaminobenzidine solution and examined under a Zeiss AXIO light microscope system.
To measure immunostaining intensity, ImageJ software was used. Colored images were first converted to 8-bit black-and-white images. The “Threshold” function was used to adjust the background to highlight specific staining. The same threshold was applied to all images analyzed. Finally, the “Measure” function was used to quantify staining intensity in each image.
Microarray assay.
Microarray assay was performed as described previously (Friedman et al., 2007). Total cortical astrocyte RNA was extracted with TRIzol reagent (Invitrogen) and further purified with RNeasy columns (Qiagen) following the manufacturer's instructions. RNA was isolated from postnatal 0–3 d mouse cortex of both genotypes (WT and TBP-105Q KI littermates). The murine 430 2.0 genome array (Affymetrix) was used to perform the independent hybridizations (five for TBP-105Q KI vs four for WT) of synthesized cRNA. GCOS software (Affymetrix) was subjected to background adjustment and normalized with RMA Express, through which raw data were obtained. RMA-normalized values were averaged across hybridizations. The number of probe sets that met the threshold p value (<0.05) was ascertained. Chips were not used in the final results if they showed signs of RNA degradation, contamination, poor amplification, or abnormal hybridization for chip quality control. Real-time-PCR was performed for a subset of interested genes identified to confirm the expression changes between WT and TBP-105Q KI astrocytes.
Experimental design and statistical analysis.
For mouse behavioral analysis, each group consisted of at least 10 sex-matched animals. For Western blotting analysis and immunostaining, data were generated from three or more experiments, and the results were expressed as mean ± SEM. The data were statistically analyzed via GraphPad Prism 5 software, and statistical significances were calculated based on Student's t test, one-way ANOVA, or two-way ANOVA. A p value <0.05 was considered significant.
Results
Generation and characterization of neuronal TBP-105Q KI (CaMKIIα-KI) mice
We first crossed floxed TBP-105Q KI mice with CaMKIIα-Cre transgenic mice to generate a KI mouse model that specifically expresses mutant TBP in the neurons of the forebrain (CaMKIIα-KI). Via Western blotting analysis using primary cultured cortical neurons and astrocytes, we confirmed that mutant TBP was only expressed in the neurons of CaMKIIα-KI mice, but not in the astrocytes. In contrast, germline-KI mice express mutant TBP in both neurons and astrocytes (Fig. 1A). We also compared mutant TBP expression in dissected brain tissues. Among all the brain regions examined (cortex, hippocampus, and striatum), CaMKIIα-KI mice showed lower levels of mutant TBP than germline-KI mice (Fig. 1B; cortex: t = 7.857, df = 4, p = 0.0014; hippocampus: t = 3.494, df = 4, p = 0.0250; striatum: t = 3.843, df = 4, p = 0.0184; Student's t test), which is in agreement with the fact that mutant TBP is only expressed in the neurons of CaMKIIα-KI mice.

Characterization of germline-KI and CaMKIIα-KI mice. A, Western blotting analysis of mutant TBP expression in primary cultures of cortical neurons and astrocytes from WT, CaMKIIα-KI, and germline-KI mice using 1C2 antibody. Vinculin was used as loading controls. B, Western blotting and quantitative analysis of mutant TBP expression in the cortex, hippocampus, and striatum of WT, CaMKIIα-KI, and germline-KI mice (Student's t test, n = 3). 1C2 antibody was used to detect soluble mutant TBP, and 1TBP18 was used to detect aggregated mutant TBP. C, D, Body weight and survival of male and female germline-KI (C) and CaMKIIα-KI (D) mice, which were measured monthly (two-way ANOVA, n = 12). Data are represented as mean ± SEM. *p < 0.05; **p < 0.01.
We systematically compared SCA17 phenotypes in germline-KI and CaMKIIα-KI mice. Germline-KI mice exhibited body weight loss and a shortened life span (Fig. 1C; body weight male: F = 432.6, p < 0.0001; body weight female: F = 400.0, p < 0.0001; survival male, p < 0.0001; survival female, p < 0.0001; two-way ANOVA), consistent with our previous report (S. Huang et al., 2015). Nonetheless, CaMKIIα-KI mice displayed normal body growth and no early death throughout their entire lifetimes compared with their wild-type littermates (Fig. 1D).
One hallmark of SCA17 is impaired motor function, which has been routinely evaluated in SCA17 mouse models using the rotarod and balance beam tests (Yang et al., 2014; S. Huang et al., 2015). In germline-KI mice, significant deficits in rotarod and balance beam performance could be found as early as 3 and 6 months of age, respectively (Fig. 2A; rotarod male: F = 54.53, p < 0.0001; rotarod female: F = 46.17, p < 0.0001; balance beam male: F = 18.30, p = 0.0079; balance beam female: F = 9.656, p = 0.0145; two-way ANOVA). However, we were unable to detect such deficits during the life span of CaMKIIα-KI mice (Fig. 2B).

Neurological symptoms of germline-KI and CaMKIIα-KI mice. A, B, Rotarod and balance beam performance of male and female germline-KI (A) and female CaMKIIα-KI (B) mice were measured at different ages (two-way ANOVA, n = 12). C, Prepulse inhibition of 3- to 4-month-old germline-KI mice was tested by acoustic startle, using prepulses of 16, 8, 4, and 2 dB lower than the pulse (100 dB; two-way ANOVA, n = 12). D, Prepulse inhibition of CaMKIIα-KI mice was tested by acoustic startle every 4 months (two-way ANOVA, n = 12). E, Novel object recognition test of 4-month-old germline-KI and 18-month-old CaMKIIα-KI mice, forced swim test of 2-month-old germline-KI and 3-month-old CaMKIIα-KI mice, and tail suspension test of 3-month-old germline-KI and CaMKIIα-KI mice (Student's t test, n = 12). Data are represented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
SCA17 patients also display nonmotor symptoms, such as dementia and psychiatric abnormalities; accordingly, a global cerebral atrophy is seen in SCA17 patient brains (Koide et al., 1999; Nakamura et al., 2001; Rolfs et al., 2003; Manto, 2005). To check whether such nonmotor deficits could be recapitulated in our mouse models, we performed a battery of behavioral tests. Prepulse inhibition of startle response measures sensorimotor gating (Braff and Geyer, 1990), and such a defect has been observed in a SCA17 transgenic rat model (Amato et al., 2016). A significant decrease in prepulse inhibition was found in 3-month-old germline-KI mice (Fig. 2C; F = 34, p < 0.0001; two-way ANOVA), but not in CaMKIIα-KI mice at any ages tested (Fig. 2D). Novel object recognition is a classic test for evaluating recognition memory (Antunes and Biala, 2012). We found that 4-month-old germline-KI mice spent significantly less time exploring the novel object, indicating memory defects. In contrast, no such defect was found in 18-month-old CaMKIIα-KI mice (Fig. 2E; germline-KI: t = 7.002, p = 0.0001; CaMKIIα-KI: t = 0.4339, p = 0.6758; Student's t test). We also performed the forced swim and tail suspension tests, both of which are used to assess depressive-like behaviors in mice (Xiang et al., 2015). Germline-KI mice, but not CaMKIIα-KI mice, showed significant deficits in both tests (Fig. 2E; germline-KI forced swim: t = 3.998, p = 0.0479; CaMKIIα-KI forced swim: t = 0.6099, p = 0.5524; germline-KI tail suspension: t = 3.744, p = 0.0025; CaMKIIα-KI tail suspension: t = 0.9520, p = 0.3585; Student's t test). In summary, germline-KI mice exhibited defective performance in all the behavioral tests, including both motor and nonmotor functions, whereas CaMKIIα-KI mice showed no obvious defects performing these tests.
Characterization of SCA17 neuropathology in SCA17 KI mouse models
The severe neurological symptoms of germline-KI mice could be attributable to the expression of mutant TBP in non-neuronal cells and peripheral tissues, such as muscle (S. Huang et al., 2015), making it important to compare neuropathology in the brains of germline-KI and CaMKIIα-KI mice. Thus, we collected tissues from different brain regions and used Western blotting to compare neuropathology in germline-KI and CaMKIIα-KI mice. Neuronal loss and astrocyte reactivity are two hallmarks of neurodegenerative diseases, which can be assessed by the neuronal marker NeuN and astrocyte marker GFAP (Yu et al., 2003; Serrano-Pozo et al., 2011; Ben Haim et al., 2015). In the cortex, mutant TBP was detected in both germline-KI and CaMKIIα-KI mice. Nonetheless, germline-KI mice showed the lowest level of NeuN and highest level of GFAP compared with wild-type and CaMKIIα-KI mice (Fig. 3A; cortex NeuN: F = 6.086, p = 0.0360; WT vs CaMKIIα-KI, q = 0.5009; WT vs germline-KI, q = 4.5015; CaMKIIα-KI vs germline-KI, q = 4.2006; cortex GFAP: F = 42.48, p = 0.0003; WT vs CaMKIIα-KI, q = 7.2367; WT vs germline-KI, q = 13.0074; CaMKIIα-KI vs germline-KI, q = 5.7707; one-way ANOVA with Tukey's post hoc tests), indicating that germline-KI mice have more severe neurodegeneration in the cortex than CaMKIIα-KI mice. CaMKIIα promoter is not active in the cerebellum, and no obvious expression of mutant TBP was seen in the cerebellum of CaMKIIα-KI mice. On the other hand, mutant TBP was present in the cerebellum of germline-KI mice, and so was neurodegeneration, as evidenced by a loss of the Purkinje cell marker calbindin, and increased GFAP expression (Fig. 3B; cerebellum calbindin: F = 6.970, p = 0.0272; WT vs CaMKIIα-KI, q = 0.1188; WT vs germline-KI, q = 4.6310; CaMKIIα-KI vs germline-KI, q = 4.5121; cerebellum GFAP: F = 8.419, p = 0.0178; WT vs CaMKIIα-KI, q = 0.5056; WT vs germline-KI, q = 5.2810; CaMKIIα-KI vs germline-KI, q = 4.7754; one-way ANOVA with Tukey's post hoc tests).

Neuronal loss and reactive astrocytes in germline-KI and CaMKIIα-KI mice. A, B, Western blotting analysis of NeuN and GFAP levels in the cortex (A) and calbindin and GFAP levels in the cerebellum (B) of 3-month-old germline-KI and CaMKIIα-KI mice. 1C2 was used to indicate the expression of mutant TBP. Vinculin was used as loading controls. The ratios of NeuN, calbindin, or GFAP to vinculin on Western blots are presented beneath the blots (one-way ANOVA, n = 3). Arrows indicate mutant TBP. C, Immunohistochemistry staining of NeuN and GFAP in the cortex of germline-KI and CaMKIIα-KI mice. 1C2 staining was used to show the expression of mutant TBP (scale bars, 20 μm). D, Immunohistochemistry staining of calbindin and GFAP in the cerebellum of germline-KI and CaMKIIα-KI mice (scale bars: for calbindin, 50 μm; for GFAP and 1C2, 20 μm). E, Quantitative analysis of NeuN, calbindin, and GFAP staining intensity in D (one way ANOVA, n = 5). F, G, Nissl staining (F) and quantification (G) of neurons in the cortex of WT, CaMKIIα-KI, and germline-KI mice (scale bar, 20 μm; one-way ANOVA, n = 5). Data are represented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
The Western blotting results were further supported by immunohistochemical studies. In the cortex of germline-KI mice, we saw a significant loss of NeuN staining and an increase of GFAP staining. Such changes were less dramatic in the cortex of CaMKIIα-KI mice, which also expressed mutant TBP, as evidenced by immunostaining with 1C2 antibody that selectively labels polyQ-expanded proteins (Fig. 3C). In the cerebellum, severe degeneration of Purkinje cells and robust reactive astrocytes were observed in germline-KI mice, but not in CaMKIIα-KI mice, whose cerebella showed much weaker 1C2 staining for mutant TBP (Fig. 3D). Quantification of NeuN staining in the cortex, calbindin staining in the cerebellum, and GFAP staining in both the cortex and cerebellum also confirmed that germline-KI mice had more severe neurodegeneration than CaMKIIα-KI mice (Fig. 3E; NeuN: F = 27.58, p = 0.0009; WT vs CaMKIIα-KI, q = 2.9896; WT vs germline-KI, q = 10.2146; CaMKIIα-KI vs germline-KI, q = 7.2250; calbindin: F = 105.2, p < 0.0001; WT vs CaMKIIα-KI, q = 1.9868; WT vs germline-KI, q = 16.6891; CaMKIIα-KI vs germline-KI, q = 18.6759; cortex GFAP: F = 8.647, p = 0.0171; WT vs CaMKIIα-KI, q = 0.8402; WT vs germline-KI, q = 5.4611; CaMKIIα-KI vs germline-KI, q = 4.6209; cerebellum GFAP: F = 12.17, p = 0.0077; WT vs CaMKIIα-KI, q = 0.7071; WT vs germline-KI, q = 6.3640; CaMKIIα-KI vs germline-KI, q = 5.6569; one-way ANOVA with Tukey's post hoc tests). This result was further corroborated by traditional Nissl staining (Fig. 3F,G; F = 57.14, p < 0.0001; WT vs CaMKIIα-KI, q = 4.164; WT vs germline-KI, q = 14.67; CaMKIIα-KI vs germline-KI, q = 10.05; one-way ANOVA with Tukey's post hoc tests).
Mutant TBP expression in astrocytes causes mild SCA17 phenotypes
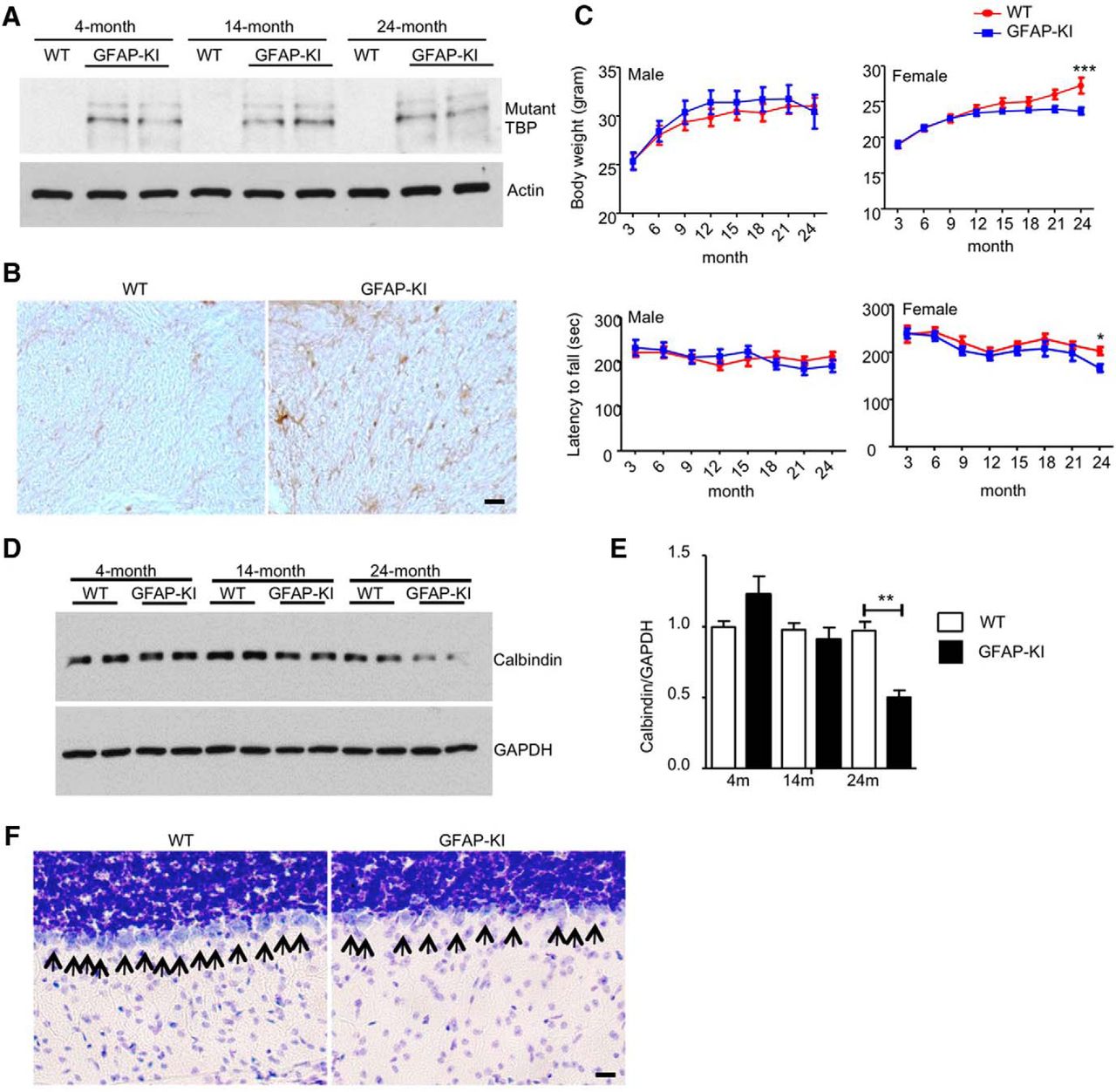
We also generated a KI mouse model that specifically expresses mutant TBP in the astrocytes (GFAP-KI) by crossing floxed TBP-105Q KI mice with GFAP-Cre transgenic mice. Using Western blotting to analyze the corpus callosum tissues isolated from these KI mice, we confirmed that mutant TBP was expressed in the glial cell-enriched corpus callosum (Fig. 4A). Immunohistochemical staining also revealed abundant staining of mutant TBP in the white matter of the cerebellum (Fig. 4B). However, GFAP-KI mice lived normally as wild-type mice and showed no obvious symptoms until they were >20 months old. We found that old female GFAP-KI mice at 21 months of age stopped gaining weight, and the body weight difference between wild-type and GFAP-KI mice became significant at 24 months (Fig. 4C; male: F = 1.634, p = 0.2033; female: F = 13.93, p = 0.0003; two-way ANOVA). Also, GFAP-KI mice at 24 months of age had a decline in rotarod performance (Fig. 4C; male: F = 0.0002185, p = 0.9882; female: F = 6.735, p = 0.0105; two-way ANOVA). Despite their mild neurological phenotypes, GFAP-KI mice displayed an age-related decrease of calbindin in the cerebellum, which was revealed by Western blotting (Fig. 4D). Quantitation of the ratio of calbindin to GAPDH on Western blot also demonstrated a significant decrease in calbindin in the cerebellum of 24-month-old GFAP-KI mice (Fig. 4E; t = 10.14, p = 0.002; Student's t test), indicating a loss of Purkinje cells. By Nissl staining, we found a significantly reduced number of Purkinje cells in the cerebellum of 24-month-old GFAP-KI mice (Fig. 4F; WT, 13.6 ± 0.6782; GFAP-KI, 9.4 ± 0.600; t = 4.638, df = 8, p = 0.0017; Student's t test). Although such pathological changes occur at a much later stage than in germline-KI mice, these results suggest that mutant TBP expression in the astrocytes is sufficient to cause late-onset SCA17 phenotypes and neuropathology in mice.

Pathological phenotypes of GFAP-KI mice. A, Western blotting analysis of mutant TBP expression in the corpus callosum of different ages of GFAP-KI mice using 1C2 antibody. Actin was used as loading controls. B, Immunohistochemistry staining of mutant TBP in the white matter of cerebellum of GFAP-KI mice using 1C2 antibody (scale bar, 50 μm). C, Top, Body weight of male and female GFAP-KI mice was measured every 3 months (two-way ANOVA, n = 10). Bottom, Rotarod performance of male and female GFAP-KI mice was measured every 3 months (two-way ANOVA, n = 10). D, Western blotting analysis of calbindin levels in the cerebellum of different ages of GFAP-KI mice. GAPDH was used as loading controls. E, Quantitative analysis of Western blotting results in D (Student's t test, n = 3). F, Nissl staining of Purkinje neurons in the cerebellum of WT and GFAP-KI mice (scale bar, 20 μm). Data are represented as mean ± SEM. *p < 0.05; **p < 0.01; ***p < 0.001.
Synergistic expression of mutant TBP caused Purkinje cell degeneration
To precisely control mutant TBP expression in neurons and astrocytes, we used adeno-associated viruses encoding Cre recombinase under the control of neuronal-specific Synapsin promoter (AAV-Synapsin-Cre) or astrocyte-specific GFAP promoter (AAV-GFAP-Cre). We injected AAV-Synapsin-Cre or AAV-GFAP-Cre, or both viruses, into the cerebellum of 3-month-old floxed TBP-105Q KI mice to induce mutant TBP expression in different types of cells (Fig. 5A). One month after viral injection, we confirmed the expression of Cre recombinase and mutant TBP (Fig. 5B,C). Importantly, only a combination of AAV-Synapsin-Cre and AAV-GFAP-Cre, but not AAV-Synapsin-Cre or AAV-GFAP-Cre alone, led to a significant reduction of the calbindin level (Fig. 5C,D; mutant TBP: F = 31.29, p = 0.0007; Synapsin vs GFAP, q = 0.9704; Synapsin vs Synapsin/GFAP, q = 10.14; GFAP vs Synapsin/GFAP, q = 9.167; calbindin: F = 5.658, p = 0.0223; uninjected vs Synapsin/GFAP, q = 5.595; one-way ANOVA with Tukey's post hoc tests). By performing immunohistochemistry and Nissl staining, we found damaged Purkinje cell morphology and reduced Purkinje cell number in mice given injections of both AAV-Synapsin-Cre and AAV-GFAP-Cre (Fig. 5E–G; F = 12.30, p = 0.0002; uninjected vs Synapsin/GFAP, q = 7.614; Synapsin vs Synapsin/GFAP, q = 6.621; GFAP vs Synapsin/GFAP, q = 6.621; one-way ANOVA with Tukey's post hoc tests). These results demonstrate that synergistic expression of mutant TBP in neurons and astrocytes could expedite and exacerbate Purkinje cell degeneration.

Synergistic toxicity of mutant TBP. A, Schematic representation of stereotaxic injection strategy using AAV-Synaspin-Cre and AAV-GFAP-Cre to express Cre in neuronal cells and astrocytes, respectively. B, Double immunostaining confirmed the expression of Cre recombinase in calbindin-positive Purkinje cells or GFAP-positive astrocytes in mice given injections of AAV-Synaspin-Cre or AAV-GFAP-Cre (scale bar, 20 μm). C, Western blotting analysis of Cre, mutant TBP, and calbindin in the cerebellum of floxed TBP-105Q KI mice given injections of AAV-Synaspin-Cre (Synapsin), AAV-GFAP-Cre (GFAP), or both (Synapsin/GFAP). Vinculin was used as loading controls. D, Quantitative analysis of Western blotting results in C (one-way ANOVA, n = 3). E, Immunohistochemistry staining of calbindin in the cerebellum of floxed TBP-105Q KI mice given injections of different viruses (scale bar, 20 μm). F, G, Nissl staining (F) and quantification (G) of Purkinje neurons in the cerebellum of floxed TBP-105Q KI mice given injections of different viruses (scale bar, 20 μm; one-way ANOVA, n = 5). Data are represented as mean ± SEM. **p < 0.01; ***p < 0.001.
Mutant TBP-expressing astrocytes lead to neurotoxicity via enhanced inflammation
Glia, specifically astrocytes, support and protect neurons by providing essential nutrients and maintaining a homeostatic environment (Kimelberg and Nedergaard, 2010; Sofroniew and Vinters, 2010). In CaMKIIα-KI mice, expression of mutant TBP in neuronal cells alone did not yield overt neurotoxicity, whereas expression of mutant TBP in astrocytes in GFAP-KI mice had late-onset neurodegeneration, suggesting that mutant TBP may produce severe neurotoxicity when it is expressed in both neuronal and glial cells. Indeed, germline-KI mice exhibited the most severe SCA17 phenotypes, and injection of both AAV-Synapsin-Cre and AAV-GFAP-Cre exacerbated Purkinje cell degeneration, which led us to propose that neurodegeneration is caused by the synergistic effects of mutant TBP in both neuronal and glial cells. To directly explore this hypothesis, we generated different combinations of neuron/astrocyte cocultures using primary neurons and astrocytes prepared from wild-type and germline-KI pups (Fig. 6A). In this coculture system, the survival and morphology of neuronal cells depend primarily on support from cultured astrocytes, whereas in the brain, neuronal survival is determined by the function of multiple types of neuronal and non-neuronal cells. Thus, this coculture system provides a sensitive assay to assess the protective capacities of astrocytes by examining the morphology of cocultured neurons (Shin et al., 2005).

Neuronal morphology in primary neuron/astrocyte cocultures. A, Schematic representation of the primary neuron/astrocyte coculture system. B–E, Immunostaining of the morphology of neurons collected from WT or germline-KI (KI) mice cocultured with WT or KI astrocytes using MAP2 antibody. DAPI was used to stain the nucleus (scale bars: for 20X, 20 μm; for 63X, 10 μm). F, Quantitative analysis of MAP2 staining intensity in B–E (Student's t test, n = 5). **p < 0.01. Data are represented as mean ± SEM.
By immunostaining using an antibody against the neuronal process marker MAP2, we observed normal morphology of wild-type neurons cultured with wild-type astrocytes (Fig. 6B). Interestingly, wild-type neurons cultured with mutant TBP-expressing astrocytes displayed dystrophic neurites (Fig. 6C), whereas the structure of mutant TBP-expressing neurons cultured with wild-type astrocytes remained mostly intact (Fig. 6D). As expected, the most severe neurodegeneration was found in mutant TBP-expressing neurons cultured with mutant TBP-expressing astrocytes (Fig. 6E), which was verified by quantifying MAP2 immunostaining in the different cocultures (Fig. 6F; WT neuron: t = 5.916, p = 0.0041; KI neuron: t = 7.341, p = 0.0018; Student's t tests). These results provide strong evidence that mutant TBP expression could impair the protective functions of astrocytes, which renders the neurons more vulnerable to mutant TBP toxicity.
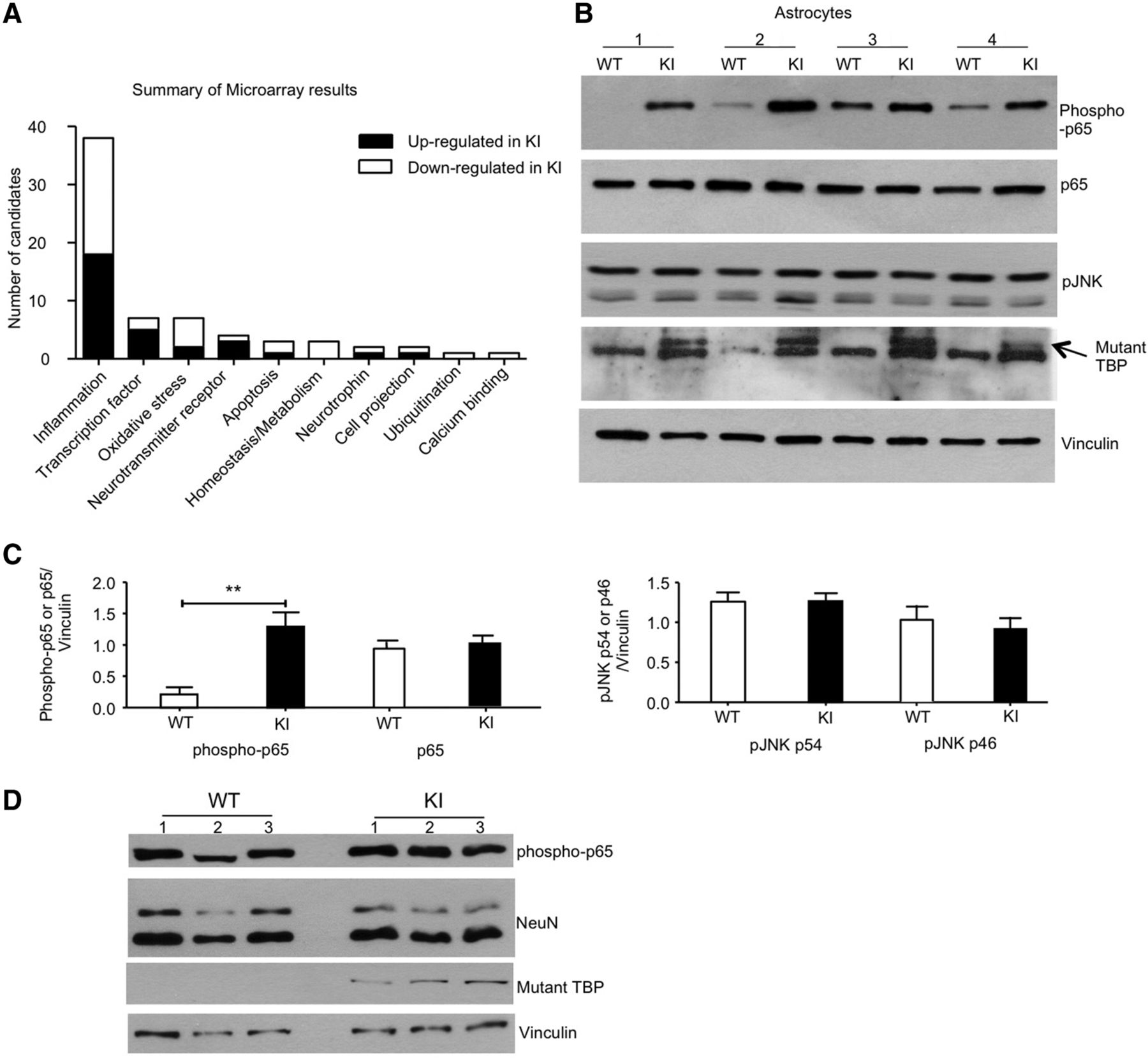
To systematically evaluate the astrocytic dysfunction caused by mutant TBP, we performed microarray analysis using primary astrocyte cultures. Indeed, mutant TBP expression disrupted a wide spectrum of cellular processes in astrocytes, but most of the altered transcripts were found to be involved in the inflammatory signaling pathway (Fig. 7A). To confirm the microarray result, we focused on NF-κB and JNK, two well established inflammatory signaling pathways (Ip and Davis, 1998; Lawrence, 2009). Western blotting revealed a significant increase in the phosphorylation level on NF-κB subunit p65 in germline-KI astrocytes (Fig. 7B,C; phospho-p65: t = 4.645, p = 0.0017; Student's t tests), indicating that NF-κB signaling is hyperactivated in the astrocytes when mutant TBP is present. In contrast, the phosphorylation level of JNK did not change much between wild-type and germline-KI astrocytes (Fig. 7B,C; p54: t = 0.0044, p = 0.9966; p46: t = 0.4169, p = 0.6913; Student's t tests). To examine whether this increase in phosphorylated p65 is specific to astrocytes, we used cultured primary neurons for Western blotting and found comparable levels of phosphorylated p65 in primary cultured neurons isolated from wild-type and germline-KI mice (Fig. 7D), suggesting that mutant TBP-caused NF-κB activation specifically occurs in the astrocytes.

Abnormal activation of inflammatory signaling pathways in germline-KI astrocytes. A, Summary of microarray result showing transcriptional dysregulation in primary cultured germline-KI astrocytes. B, Western blotting analysis of phospho-p65, p65, and pJNK levels in primary WT and germline-KI astrocytes. The arrow indicates mutant TBP. Vinculin was used as loading controls. C, Quantitative analysis of the ratios of phosphorylated proteins (p65, pJNK54, pJNK64) to the loading control vinculin on Western blot results in Figure 6B (Student's t test, n = 3). D, Western blotting analysis of phospho-p65 levels in primary cultured WT and KI neurons. **p < 0.01. Data are represented as mean ± SEM.
Suppressing NF-κB signaling ameliorated neurotoxicity caused by mutant TBP-expressing astrocytes
Finally, we tested whether suppressing NF-κB signaling in astrocytes could be used as a potential therapy for SCA17 treatment. Because it remains unknown whether NF-κB inhibitors can pass through the blood–brain barrier and because glia–neuron cocultures enable us to uncover the direct protective effects of astrocytes, we treated mutant TBP-expressing astrocytes with QNZ or BAY 11-7082, two potent inhibitors of NF-κB activation (Pierce et al., 1997; Tobe et al., 2003). Primary neurons isolated from germline-KI mice were cocultured with astrocytes treated with the inhibitors, and MAP2 immunostaining was performed to examine neuronal morphology. Remarkably, neurons cocultured with mutant TBP-expressing astrocytes treated with either QNZ or BAY 11-7082 showed significantly improved morphology compared with untreated controls (Fig. 8A,B; F = 70.14, p < 0.0001; KI no treatment vs KI plus QNZ, q = 18.3648; KI no treatment vs KI plus BAY 11-7082, q = 16.4425; one-way ANOVA with Tukey's post hoc tests). The treatment did not alter wild-type neuron morphology, indicating that NF-κB inhibitors do not generally improve neuronal outgrowth and branching, but rather protect mutant TBP-mediated neurotoxicity. To further verify that these inhibitors could reduce phosphorylation of p65 in the cocultures, we performed Western blotting and obtained strong evidence that both inhibitors could rapidly and significantly decrease phosphorylated p65 in dose- and time-dependent manners (Fig. 8C,D; BAY 11-7082: F = 211.9, p < 0.0001; 1 h/20 μm vs 0, q = 25.0934; 3 h/5 μm vs 0, q = 35.0236; 6 h/5 μm vs 0, q = 35.3531; 12 h/5 μm vs 0, q = 36.9517; 24 h/5 μm vs 0, q = 37.1538; QNZ: F = 101.8, p < 0.0001; 0.5 h/20 nm vs 0, q = 2.0886; 0.5 h/40 nm vs 0, q = 16.9130; 2 h/20 nm vs 0, q = 19.7328; 2 h/40 nm vs 0, q = 22.9402; 4 h/20 nm vs 0, q = 23.0709; 4 h/40 nm vs 0, q = 23.1247; one-way ANOVA with Tukey's post hoc tests).

Blocking NF-κB signaling in astrocytes alleviated neurotoxicity in primary neuron/astrocyte cocultures. A, Immunostaining of neuronal morphology in cocultures with primary astrocytes treated with QNZ or BAY 11-7082. DAPI was used to stain the nucleus (scale bar, 50 μm). B, Quantitative analysis of MAP2 staining intensity in A (one way ANOVA, n = 5). C, D, Western blotting and quantitative analysis of phospho-p65 levels in primary astrocytes treated with BAY 11-7082 (C) or QNZ (D) at different concentrations for different lengths of time (one way ANOVA, n = 3). Data are represented as mean ± SEM. ***p < 0.001.
Discussion
In summary, our present study suggests that mutant TBP expression in neuronal and glial cells exerts synergistic toxic effects that lead to neurodegeneration in SCA17 mice. The noncell-autonomous mechanism underlying neurotoxicity is derived from excessive inflammatory activity in the astrocytes. Blocking NF-κB-mediated inflammatory activity by chemical inhibitors restored the protective capacities of astrocytes impaired by mutant TBP expression, thereby serving as a potential therapy to alleviate SCA17 neurotoxicity.
Glia are well known to provide support and protection to neurons in the CNS via various mechanisms (Fields and Stevens, 2000; Maragakis and Rothstein, 2001; Bazargani and Attwell, 2016; von Bernhardi et al., 2016). Therefore, it is not surprising that glial dysfunction could contribute to neurodegenerative diseases (Lobsiger and Cleveland, 2007). For example, glial mutant polyQ protein aggregates and loss of glia are found in several polyQ diseases, including Huntington's disease, SCA1, and SCA2 (Probst-Cousin et al., 2004; Shin et al., 2005; Shiwaku et al., 2010). In addition, glia-specific expression of mutant huntingtin or ataxin-7 leads to neurodegeneration and disease phenotypes in mouse models (Custer et al., 2006; Bradford et al., 2009; B. Huang et al., 2015), providing direct evidence supporting the causative role of glial dysfunction in polyQ disease pathogenesis. However, it is noteworthy that many previous studies used transgenic models, which overexpress mutant polyQ proteins, to investigate the toxicity of mutant protein in neurons and glial cells. It was reported that overexpression of mutant TBP in Purkinje cells driven by the Purkinje-specific promoter (Pcp2/L7) could lead to early neurological symptoms and Purkinje cell degeneration in transgenic mice (Chang et al., 2011). Given that overexpression of polyQ proteins, even in the wild-type forms, could cause disease-like phenotypes (Fernandez-Funez et al., 2000; Monks et al., 2007; Nedelsky et al., 2010), whether expressing mutant polyQ proteins at an endogenous level in neuron or glia could lead to neurodegeneration remained to be tested.
We addressed this important issue using conditional TBP-105Q KI mice and provided several lines of evidence indicating that mutant TBP expression in glia under physiologically relevant conditions is still an integral component of SCA17 pathogenesis. We found that neuron-specific TBP-105Q KI (CaMKIIα-KI) or astrocyte-specific TBP-105Q KI (GFAP-KI) mice showed much more delayed and milder phenotypes, as well as less severe neurodegeneration, than germline-KI mice that express mutant TBP in both neurons and glia. A potential caveat is that in the brain of germline-KI mice, mutant TBP is also expressed in other types of cells, which could confound the synergistic toxicity between neurons and astrocytes. Therefore, we performed additional experiments by injecting AAV-Synapsin-Cre or AAV-GFAP-Cre into the cerebellum of floxed TBP-105Q KI mice to induce mutant TBP expression exclusively in neurons or astrocytes. Our result indicates that synergistic expression of mutant TBP in neurons and astrocytes, rather than in each cell type alone, could facilitate and exacerbate Purkinje cell degeneration, which is consistent with our findings from germline-KI mice. Additionally, wild-type neurons cocultured with mutant astrocytes showed degenerative phenotypes, whereas mutant neurons cocultured with wild-type astrocytes were primarily protected from degeneration. Given the important role of glia in maintaining neuronal survival, it is possible that restoring glial functions could be used as a therapeutic approach to delay or treat the pathogenesis of neurodegenerative diseases. In support of this idea, a recent publication reported improved phenotypes and extended survival of Huntington's disease transgenic mice after striatal transplantation of normal glial progenitor cells (Benraiss et al., 2016).
We should note that the severe phenotypes of germline TBP-105Q mice are also influenced by peripheral mutant TBP in muscle (S. Huang et al., 2015). Nevertheless, germline-KI mice and the mutant neurons cocultured with mutant astrocytes showed more severe neuronal degeneration, suggesting that the neurotoxicity of mutant TBP results from its synergistic effects in both neurons and glia. Additionally, neither CaMKIIα-KI nor GFAP-KI mice had any overt signs of neurodegeneration until very late stages of life, whereas in germline-KI mice, significant neuronal loss was seen as early as 3 months of age (S. Huang et al., 2015). Therefore, mutant TBP expression in both neurons and glia could synergistically expedite disease onset and progression, which also highlights the importance of ubiquitous expression to faithfully recapitulate the pathogenesis of SCA17 and other neurodegenerative diseases.
Inflammatory activation in the CNS has been documented in several polyQ diseases, such as Huntington's disease and SCA3 (Evert et al., 2001, 2006; Sapp et al., 2001; Dalrymple et al., 2007; Crotti et al., 2014); however, whether the immune response is the cause or effect of neurodegeneration remains to be confirmed. By performing transcriptome analysis on primary astrocyte cultures, we identified activated inflammatory pathways in the presence of mutant TBP. More importantly, blocking the proinflammatory NF-κB pathway completely rescued the impaired neuronal morphology in neuron/astrocyte cocultures. Interestingly, activated NF-κB signaling is also found in the astrocytes of a Huntington's disease transgenic mouse model (Hsiao et al., 2013), suggesting that inflammatory activation in astrocytes could be a common pathological event in polyQ diseases. Furthermore, astrocytes are known to produce multiple proinflammatory molecules (Sofroniew, 2015). Given all the evidence, our present study establishes inflammatory activation in the astrocytes as a major contributor to the pathogenesis of SCA17. In addition, targeting the NF-κB pathway in astrocytes could serve as a potential therapy for the treatment of SCA17, and perhaps other polyQ diseases as well.
Footnotes
This work was supported by NIH Grants AG19206 and NS101701 (X.-J.L.) and AG031153 and NS045016 (S.L.). We thank Xiangya Hospital for supporting Y.Y.'s study at Emory University and Cheryl Strauss for critical reading of this manuscript.
The authors declare no competing financial interests.
- Correspondence should be addressed to either Xiao-Jiang Li or Shihua Li, Department of Human Genetics, Emory University School of Medicine, 615 Michael Street, Atlanta, GA 30322. xli2{at}emory.edu or sli{at}emory.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}