

Abstract
Motoneurons establish a critical link between the CNS and muscles. If motoneurons do not develop correctly, they cannot form the required connections, resulting in movement defects or paralysis. Compromised development can also lead to degeneration because the motoneuron is not set up to function properly. Little is known, however, regarding the mechanisms that control vertebrate motoneuron development, particularly the later stages of axon branch and dendrite formation. The motoneuron disease spinal muscular atrophy (SMA) is caused by low levels of the survival motor neuron (SMN) protein leading to defects in vertebrate motoneuron development and synapse formation. Here we show using zebrafish as a model system that SMN interacts with the RNA binding protein (RBP) HuD in motoneurons in vivo during formation of axonal branches and dendrites. To determine the function of HuD in motoneurons, we generated zebrafish HuD mutants and found that they exhibited decreased motor axon branches, dramatically fewer dendrites, and movement defects. These same phenotypes are present in animals expressing low levels of SMN, indicating that both proteins function in motoneuron development. HuD binds and transports mRNAs and one of its target mRNAs, Gap43, is involved in axonal outgrowth. We found that Gap43 was decreased in both HuD and SMN mutants. Importantly, transgenic expression of HuD in motoneurons of SMN mutants rescued the motoneuron defects, the movement defects, and Gap43 mRNA levels. These data support that the interaction between SMN and HuD is critical for motoneuron development and point to a role for RBPs in SMA.
SIGNIFICANCE STATEMENT In zebrafish models of the motoneuron disease spinal muscular atrophy (SMA), motor axons fail to form the normal extent of axonal branches and dendrites leading to decreased motor function. SMA is caused by low levels of the survival motor neuron (SMN) protein. We show in motoneurons in vivo that SMN interacts with the RNA binding protein, HuD. Novel mutants reveal that HuD is also necessary for motor axonal branch and dendrite formation. Data also revealed that both SMN and HuD affect levels of an mRNA involved in axonal growth. Moreover, expressing HuD in SMN-deficient motoneurons can rescue the motoneuron development and motor defects caused by low levels of SMN. These data support that SMN:HuD complexes are essential for normal motoneuron development and indicate that mRNA handling is a critical component of SMA.
Introduction
Neuronal development is controlled at many levels. This includes transcriptional programs that dictate the type of neuron, RNA transport for localized RNA translation, and downstream pathways important for the formation of neuronal structures necessary for their function, such as axons, dendrites, and synapses. The disruption of any of these steps results in a motoneuron that either does not develop at all or one that develops, but does not function properly. For motoneurons, the initial developmental programs that drive differentiation are well understood. However, much less is known about the later steps in motoneuron development that encompass axon branch and dendrite formation.
We have previously shown that the survival motor neuron (SMN) protein is required for these later stages of motoneuron development. Using a genetic zebrafish model expressing no zebrafish SMN and only low levels of human SMN (hereafter referred to as mz-smn mutants), we found that motoneurons are born and take on the correct fates but fail to form robust axons with the normal level of branching, and their dendrites are reduced in number and length (Hao le et al., 2013). We and others have shown that neuromuscular junction (NMJ) formation is also affected under conditions of low SMN (Kariya et al., 2008; Boon et al., 2009; Kong et al., 2009; Ling et al., 2012). In the context of the disease spinal muscular atrophy (SMA), which is caused by low levels of SMN, we have hypothesized that this abnormal development generates a motoneuron that will eventually fail to function. Indeed, zebrafish with low levels of SMN and deficient motoneuron development have significant movement defects supporting that they are functionally compromised (Hao le et al., 2013). Our goal is to elucidate how SMN functions in motoneuron development as a way to better understand normal development and the motoneuron defects in SMA.
One indication of how SMN may be affecting motoneuron development has come from studies revealing that SMN interacts biochemically with a number of RNA binding proteins (RBPs) (Fallini et al., 2012). One of these RBPs, HuD, a member of the ELAV family of RBPs is expressed in the nervous system soon after motoneurons are born and is thought to play a role in their development. RBPs have been shown to function in transporting mRNAs to axons and dendrites during development and regeneration after injury (Holt and Schuman, 2013; Hörnberg and Holt, 2013). For example, limiting the amount of the RBP ZBP1 decreases the levels of axonal mRNAs and negatively affects regeneration after injury (Donnelly et al., 2013), and mouse knockdown of the RBP IMP2 resulted in disruption of commissural axon pathfinding in vivo (Preitner et al., 2016). Locally translated RNAs affect both axonal branching and elongation of regenerating axons, supporting a critical role for RBPs and their mRNA cargos in regeneration and function (Donnelly et al., 2013). Recently, analysis in cultured primary mouse motoneurons showed that SMN and HuD affect local translation in growth cones (Fallini et al., 2016; Donlin-Asp et al., 2017). These studies indicate that SMN may facilitate HuD binding to its target mRNAs, ultimately affecting mRNA transport into axons. This has not, however, been tested in vivo in developing motoneurons.
To elucidate the function of SMN and HuD in vivo in motoneuron development, we have taken a direct approach to analyze SMN:HuD complexes in vivo. By performing motoneuron-specific biochemistry, we find that SMN and HuD interact in motoneurons, but only during phases of robust motoneuron development. We generated HuD mutants and find that they exhibit defects similar to animals expressing low levels of SMN supporting a role for these proteins in motoneuron development. We also analyzed growth-associated protein 43 (Gap43), a protein involved in axonal outgrowth during development and regeneration whose mRNA is a target of HuD. We found that Gap43 mRNA levels were decreased in both HuD and mz-smn mutants. Expressing HuD in motoneurons of mz-smn mutants, rescued all of the motoneuron defects and the Gap43 levels. Together, these data support a key role for SMN, HuD, and mRNA handling in motoneuron development and suggest that disrupting the interaction between these two proteins can cause SMA phenotypes.
Materials and Methods
Zebrafish maintenance.
Zebrafish used in this study were on the *AB/TL (Tupfel long fin) background. smnY262stop mutants (smn1fh229) (Boon et al., 2009) and the generation of mz-smn mutants have been described previously (Hao le et al., 2013). Adult zebrafish and embryos were kept at temperatures between 27°C and 29°C and maintained by standard protocols (Westerfield, 1995).
Generation of HuD mutant fish using CRISPR/Cas9.
Two different guide RNAs were designed and used to target zebrafish HuD exon 3, using protocols based on Talbot and Amacher (2014); for mutagenesis, guide RNAs were coinjected with nuclear localized Cas9 (Jao et al., 2013)
HuD CRISPR target 1 is GGGGCAGGTAGTTGACGATG at the end of HuD exon3. We designed a guide oligo sequence, including a Cas9 binding scaffold, CRISPR target, and T7 promoter as follows: AAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAACCATCGTCAACTACCTGCCCCTATAGTGAGTCGTATTACGC.
HuD CRISPR target 2 is GGAGGGTCCGTTGGCTGTGG at the beginning of HuD exon3. A guide oligo sequence, including a Cas9 binding scaffold, CRISPR target, and T7 promoter, was designed as follows: AAAGCACCGACTCGGTGCCACTTTTTCAAGTTGATAACGGACTAGCCTTATTTTAACTTGCTATTTCTAGCTCTAAAACCCACAGCCAACGGACCCTCCTATAGTGAGTCGTATTACGC. Both of these synthetic single DNA strands were amplified using oligos GCGTAATACGACTCACTATAG and AAAGCACCGACTCGGTGCCAC and the program: 95°C for 1 min (95°C 15 s, 60°C 30 s, 72°C 20 s) for 40 times, 72°C for 5 min. The 120 bp PCR product was purified on 3% agarose gel. PCR product was used to synthesize this gRNA using Maxiscript T7 kit (Invitrogen).
For CRISPR mutagenesis, Cas9 mRNA was synthesized from pCS2-nCas9n (Jao et al., 2013) using mMessage mMachine Sp6 kit (Invitrogen). The gRNA (40 ng/μl) and cas9 mRNA (80 ng/μl) were mixed together, and 1 nl was injected into 1 cell stage embryos. Injected embryos were raised for 3 months and outcrossed to wild-type fish for screening. Embryos were screened using High Resolution Melt Analysis (HRMA) (Dahlem et al., 2012) on CFX machine (Bio-Rad). HRMA primers for screening and genotyping HuD target 1 were HuD_HRM_E3_F: TCACCCATGCAGACC and HuD_HRM_E3_R: GACTCGATCTCACCAATG. HRMA primers for screening and genotyping HuD target 2 were HuD_HRM_E3_F2: TAATCAGCAACATGGAGCCTCAG and HuD_HRM_E3_R2: TGAGGTTGGTCTTGCTGTCATC. Samples were run using 95°C 3 min (95°C 15 s, 58°C 20 s, 70°C 20 s) 45×, 65°C 30 s, 95°C 15 s. Amplification and melt curves were analyzed using Precision Melt Analysis software (Bio-Rad). Heterozygote samples with deflections were selected for sequencing. Primers for sequencing HuD target 1 were HuD_Seq_E3_F: CACGTCCTTCAACTCGTCTTTTG and HuD_Seq_E3_R: CCTGTGATTTTGTCTCGAACCAG. Primers for sequencing HuD target 2 were HuD_Seq_E3_F2: TTACGGTGGGTGTGCAAGAATC and HuD_Seq_E3_R2: TCTCACCAATGCTGCCAAAGAG. Injected F0 fish carrying mutations were crossed to wild-type fish to raise lines. F1 fish were raised and sequenced to confirm the mutation before generating F2 mutant lines. Subsequent generations of both heterozygous and homozygous animals were genotyped by HRMA. This was possible in the homozygotes because the 2 bp lesion created a distinctive melt curve that clearly differentiated it from heterozygotes or wild-types.
HuD homozygous mutants were viable, but not very good breeders. After verification of the motoneuron phenotype in all of the mutant lines, we used a 2 bp insertion allele (hudos55), generated at CRISPR target 2, for all other experiments in the paper.
Scoring embryos based on motor axon defects at 26 hours post fertilization (hpf).
Embryos were fixed at ∼26 hpf. If not expressing Tg(mnx1:GFP), they were processed for znp1 (synaptotagmin) antibody labeling as described below. Embryo trunks were mounted laterally between two coverslips. Ventrally projecting CaP motor axons from hemisegments 6–15 (10 axons, one per hemisegment) were analyzed. Based on motor axon defects, embryos were placed in one of four categories: severe, moderate, mild, or no defects as previously described (Carrel et al., 2006). Embryos with at least four severe motor axon defects (truncations or excessively branched at the truncation) or greater than four moderate mutations (excessively branched, bifurcating, innervating neighboring myotome) were categorized as “severe.” Embryos with at least four moderate motor axon defects, two severe defects, or greater than four mild defects (some ectopic branches but normal length and morphology, axons lacking perfect morphology but no excessive branches) were categorized as “moderate.” Embryos with at least two mild defects or one moderate defect were categorized as “mild,” and embryos categorized as “no defects” had motor axons indistinguishable from wild-types. For each experiment ∼25 embryos were scored from three separate clutches. Mean ± SEM of the 3 experiments were plotted and statistics performed using the Mann–Whitney nonparametric rank test.
Generation of Tg(Mnx1:Gal4).
To generate transgenic lines expressing Gal4 in motoneurons, the plasmid mnx1-3 × 125bp:Gal4-VP16 plasmid (Zelenchuk and Brusés, 2011) was coinjected with Tol2 mRNA (final concentration 5 ng/μl DNA and 100 ng/μl mRNA) into 1 cell stage embryos. Injected fish were raised to adulthood, outcrossed to wild-types, and F1s screened for the transgene using PCR. PCR primers: GGAAGCTTATGAAGCTACTGTCTTCTATC and AAGGATCCACATATCCAGAGCGCCGTAGG. F0s that resulted in positive F1s were used to generate lines. Positive F1 transgenic fish were confirmed by PCR as adults (fin clip) and crossed into HuD and mz-smn mutants. Line designation, Tg(mnx1-3X125:Gal4-VP16)os57.
Generation of Tg(UAS:mCherry-HuD) on HuD−/− and mz-smn−/− backgrounds.
5XUAS from 5XUAS:GFP plasmid (Biswas et al., 2014) was PCR amplified using primers ATAGAGCTCTGCAGGTCGGAGTACTGTCC and ATGCGGCCGCGCTAGCCAATTCCCTATTC. The PCR product was digested with SacI and NotI and cloned into a pBlueScript with two SceI sites. mCherry-HuD from clontech C1 vector (Fallini et al., 2011) was PCR amplified using ATGCGGCCGCATGGTGAGCAAGGGCGAGG and CGCTGCGGCCGCTTAAGATACATTGATGAG primers. The resulting PCR product was digested with NotI and cloned into the pBlueScript with 5XUAS to generate pBlueScript: 5XUAS:mCherry-HuD plasmid. This plasmid was coinjected with SceI enzyme into Tg(mnx1:Gal4):mz-smn−/− and Tg(mnx1:Gal4):HuD−/− embryos at the 1 cell stage. Injected embryos that had mCherry expression in motoneurons at 24 hpf were raised to adulthood. F1 transgenics from these F0 fish were used for rescue and behavior experiments. Genotype was confirmed by PCR or HRMA. Line designation, Tg(5XUAS:mCherry-HuD)os58.
Generation of transgenic Tg(mnx1:mCherry-HuD).
mCherry-HuD from clontech C1 vector (Fallini et al., 2011) was PCR amplified using ATGCGGCCGCATGGTGAGCAAGGGCGAGG and CGCTGCGGCCGCTTAAGATACATTGATGAG primers. This PCR product was digested with NotI and cloned into the pBlueScript containing the zebrafish mnx1 promoter (Hao le et al., 2015). Injected embryos that had mCherry expression in motoneurons at 24 hpf were raised to adulthood. F1 transgenics from these F0 fish were crossed to mz-smn mutants for rescue experiments. Line designation, Tg(mnx1:mCherry-HuD)os59.
Generation of Tg(mnx1:RFP-SMNE134K) and Tg(mnx1:RFP-SMNQ282A) transgenics.
Site-directed mutations E134K and Q282A were introduced into human SMN full-length construct mnx1:hs:RFP-SMN using QuikChange II Site-Directed Mutagenesis Kit (Agilent, catalog #200523). Mnx1:hs:RFP-SMN construct plasmid was previously described (Hao le et al., 2015). Primers for E134K mutagenesis were GTGGTTTACACTGGATATGGAAATAGAAAGGAGCAAAATCTG and CAGATTTTGCTCCTTTCTATTTCCATATCCAGTGTAAACCAC. Primers for Q282A mutagenesis were CTGGCTATTATATGGGTTTCAGAGCAAATCAAAAAGAAGG and CCTTCTTTTTGATTTGCTCTGAAACCCATATAATAGCCAG. DNA plasmids were coinjected with SceI into 1 cell stage embryos as previously described (Hao le et al., 2015). Line designation, Tg(mnx1:1.5hsp70:RFP-SMNE134K)os60 and Tg(mnx1:1.5hsp70:RFP-SMNQ282A)os61.
Immunoprecipitation.
A total of 300 2 d post fertilization (dpf) embryos were collected and placed in a Petri dish containing 100 ml ice-cold PBS. Embryos were pipetted up and down several times to remove yolks and heads. The trunks were transferred to a 5 ml glass homogenizer. Embryos were homogenized in 1 ml lysis buffer (20 mm Tris, pH 7.5, 150 mm NaCl, 0.5% Triton X-100, 1 mm EDTA, 0.5 mm PMSF in isopropanol, and 1 tablet of Roche protease inhibitor per 10 ml). Samples were then centrifuged for 15 min at 12,000 × g at 4°C; 10 μl of supernatant was saved for Western blot analysis. The remaining supernatant was transferred to a clean tube containing 10 μl of Anti-RFP mAb-Magnetic Beads (MBL, catalog #M165-11). Controls were magnetic beads with no antibody attached. Samples were rotated overnight at 4°C. Samples were then placed onto a magnetic bar for 5 min. Supernatant was removed. Beads were washed 3× in wash buffer (20 mm Tris, pH 7.5, 150 mm NaCl, 0.5% Triton X-100). Beads were then boiled in 2× SDS buffer (100 mm Tris, pH 6.8, 4% SDS, 0.2% bromophenol blue, 20% glycerol, 200 mm DTT) to elute samples.
Whole-mount immunofluorescence staining.
This protocol is based on Hao le et al. (2013). Briefly, zebrafish embryos or larvae were anesthetized with tricaine (Sigma, A-5040) and then fixed in 4% PFA in PBS overnight at 4°C. The embryos were then washed in 1× PBS for 10 min, distilled H2O for 10 min followed by a 15 min incubation at room temperature with −20°C acetone. Embryos were then washed with distilled H2O for 20 min and then incubated overnight at 4°C with znp1 (synaptotagmin 2) (Developmental Studies Hybridoma Bank, University of Iowa) diluted 1/100 or acetylated tubulin (Invitrogen) diluted in 1/1000 in PBDT buffer (1% DMSO, 1% BSA, 0.5% Triton X-100) and 2.5% normal goat serum. Samples were washed 5 × 10 min with PBST at room temperature and incubated overnight at 4°C with AlexaFluor-488 goat-anti-mouse IgG (Invitrogen) diluted 1/400 in PBDT and 2.5% normal goat serum. Samples were washed for 5 × 10 min in PBST, mounted on a slide with ProLong Gold antifade reagent (Invitrogen), and images captured with a Leica TCS SL scanning confocal microscope system. Motor axons were scored as previously described (Carrel et al., 2006).
Western blot analysis.
This protocol is based on Hao le et al. (2011). Briefly, three zebrafish embryos were placed in 15 μl blending buffer (62.6 mm Tris, pH 6.8, 5 mm EDTA, and 10% SDS) and boiled for 10 min. Samples were then diluted with an equal volume of 2× SDS loading buffer (100 mm Tris, pH 6.8, 4% SDS, 0.2% bromophenol blue, 20% glycerol, and 200 mm DTT) and boiled for 2 min. One-third of each sample from three embryos (∼50 μg) was resolved on a 7% polyacrylamide gel. The gel was electrotransferred to Hybond-P PVDF membrane (GE Healthcare). Membranes were probed with SMN mouse monoclonal antibody 2E6 (from Dr. Glenn Morris), anti β-actin (1/5000; Santa Cruz Biotechnology) or anti HuD E-1 (1/500, Santa Cruz Biotechnology). Signal was detected with HRP-conjugated goat anti-mouse antibody (1/5000; Jackson ImmunoResearch Laboratories), ECL reagents, and Hyperfilm ECL (GE Healthcare Bioscience).
For the heat shock experiments, 10 or 24 hpf embryos were transferred to a 1 ml Eppendorf tube containing fish water (30 embryos/tube) and placed in a 37°C water bath for 1 h. Western blots were performed 36 h after heat shock.
Single-cell injection and tracing.
This protocol is based on Hao le et al. (2015). Briefly, DNA plasmid mnx1:0.6hsp70:GFP (Dalgin et al., 2011) was prepared (Plasmid Mini kit, QIAGEN) and diluted to 50 ng/μl in I-SceI buffer containing 10 mm Tris-HCl, 1 mm DTT, 10 mm MgCl2, pH 8.8, 5 units of SceI enzyme (New England Biolab), and 0.1% phenol red. DNA (50 ng/μl) was injected into embryos at the early 1 cell stage to 10% of the volume of the cell (∼1 nl). Injected embryos were transferred into fish water containing penicillin/streptomycin (Invitrogen) 1/100. Injected fish were screened for GFP-expressing CaP motor neurons at 26 hpf.
At 2 or 4 dpf, embryos were fixed in 4% PFA in PBS and 1% DMSO overnight at 4°C followed by storage in PBS. Images were captured with the Leica TCS SL scanning confocal microscope system. The motor axon branches at 2 dpf and motoneuron dendrites at 4 dpf were imaged using confocal microscopy (40× objective). Axons were measured at 2 dpf because the branches in wild-type larvae at 4 dpf become difficult to trace due to their increased complexity. Images were traced and measured using National Institutes of Health software ImageJ (Fiji). All images were set up as 512 × 512 pixels. The microscope calibration information (μm/pixel) was used to convert the ImageJ measurements (pixel) to microns. The actual size of the images for motor axon branches at 2 dpf was 288.4 × 288.4 μm (0.56 μm/pixel) and for motoneuron dendrites at 4 dpf was 93.75× 93.75 μm (0.18 μm/pixel). Each plot was from 100 to 400 individual dendrites or axon branches from 11 to 13 larvae and reported as mean ± SD.
Zebrafish motor behaviors.
Behavioral experiments were performed on 5 dpf larvae at 26°C-27°C. Larvae were preadapted to the testing arena for 20 min (4 × 4 grid) or 2 h (6 cm Petri dish). Larval behavior was captured with a MotionPro Y4 video camera (Integrated DNA Technologies) and analyzed with the FLOTE software package as previously described (Burgess and Granato, 2007a, b). Frame by frame, FLOTE tracks larval body position and conformation and then performs automated analysis of body curvature changes to extract kinematic details of swimming and turning movements. Based on defined kinematic parameters that distinguish discrete motor behaviors, FLOTE classifies each movement as a distinct form of a turn or swim. To evaluate nonevoked, gross movement over time, larvae were individually housed in a 4 × 4 grid (Wolman et al., 2011), and their position was recorded at 100 frames per second over a continuous 120 s period. The N per experimental group ranged from 30 to 61 larvae. To examine nonevoked swim and turn initiations and swimming performance, 20 larvae of identical genotype were grouped in a 6-cm-wide Petri dish, and 3 dishes were tested per genotype. For this analysis, we recorded at 1000 frames per second for 1 s at 5 s intervals to capture a total of 30 trials of 1 s recordings. For each recording, FLOTE determined whether each larva initiated a turn, swim, or remained stationary; only the first movement type in each 1 s recording was scored. The mean frequency in which a larva initiated a turn or swim within a 1 s recording period was plotted. To evaluate swimming performance, we analyzed kinematic parameters of slow “scoot” swims that included two or more swim half-cycles. A half swim cycle is defined by a single leftward or rightward tail undulation. All statistics for the behavioral data were analyzed by a one-way ANOVA with a Bonferroni post hoc test for significance and are noted in the figure legend.
Digital droplet PCR (ddPCR) for Gap43 mRNA analysis.
Wild-type, HuD−/−, mz-smn−/−, Tg(mnx1:Gal4);Tg(UAS:mCherry-HuD);mz-smn−/−, and Tg(mnx1:Gal4);Tg(UAS:mCherry-HuD);HuD−/− fish were set up into group crosses (3 sets of group crosses for each genotype). For each genotype, 30 embryos were collected combining embryos from the three different group crosses. At 2 dpf, embryos were dechorionated and transferred into Petri dishes containing 100 ml PBS and Tricaine (Sigma). Heads and yolks were removed using forceps, and the trunks were transferred to 1.5 ml Eppendorf tubes. RNA was isolated using Trizol (Invitrogen) and treated with RQ1 DNase (Promega). cDNA was made from 300 ng of RNA using iScript cDNA synthesis kit (Bio-Rad). The cDNA was diluted to an equivalent of 1, 5, and 10 ng RNA for the quantification of transcripts using ddPCR (Bio-Rad) (Iyer et al., 2014). ddPCR was performed in triplicates for each of 1, 5, and 10 ng RNA-equivalent cDNA sample to assay for GAP43 against the normalizing gene ubiquitin-conjugating enzyme E2A (Ube2a) (Xu et al., 2016). This was performed twice, resulting in 6 technical replicates. However, one data point for HuD−/− was 2.5 SDs from the mean of the other 5 data points and was thus removed as an outlier resulting in the HuD−/− dataset consisting of 5, not 6, data points. For ddPCR, ∼10,000–14,000 droplets were generated per sample with the necessary PCR reagents (Bio-Rad ddPCR Supermix for probes, no dUTP), primers (Integrated DNA Technologies) and probe (Integrated DNA Technologies), and the PCR was performed as described previously (Iyer et al., 2014). The fluorescence signal in the droplets was read and quantitated according to Poisson statistics using QuantaSoft software (Bio-Rad). The sequences of the primers and probes were as follows: GAP43 primers, 5′-CACCAAGATCCAGGCCAGCTTCC, 5′-CAGGGTCCGCTGCAGCAGTG and probe-FAM-5′-CCGACTGACGCCTCCACAGAAACACAG; and Ube2a primers, 5′-GCTGGAGTCCAACATATGACGTTTCC, 5′-CAGTCACGCCAGCTCTGTTCCAC and probe-HEX-5′-ATGAGCCGAACCCAAACAGCCCTGCC.
Experimental design and statistical analysis.
Zebrafish embryos and larvae cannot be designated as male or female. All experiments were performed on embryos or larvae from multiple group matings. The number of experiments and animals varied depending on the experiment and is therefore noted in the figure legends. The VasserStats statistical website (http://vassarstats.net/) or SigmaPlot was used to perform statistical analysis. The exact tests and parameters are noted in the text and figure legends.
Results
SMN interacts with HuD in motoneurons during development
Studies have shown that SMN and the neuron-specific RBP HuD interact biochemically in cultured primary cortical neurons (Akten et al., 2011), MN1-cells (Hubers et al., 2011), and in cultured mouse motoneurons and rat brain extract (Fallini et al., 2011). However, this has not been examined in motoneurons in vivo during development, which has implications for SMA. To this end, we performed motoneuron-specific coimmunoprecipitation experiments. To specifically analyze SMN from motoneurons in vivo, we took advantage of a transgenic line with motoneuron promoter mnx1 (also know as hb9) from zebrafish driving RFP-tagged human SMN (Fig. 1A) (Hao le et al., 2013), thus enabling us to express RFP-SMN in motoneurons. Using an anti-RFP antibody, we immunoprecipitated RFP-SMN from 2 dpf embryos, which is a time when motoneurons are forming extensive axon branches. We found that HuD did coimmunoprecipitate with SMN (Fig. 1B). Others had shown that SMN and HuD interact via the SMN tudor domain and that the human mutation SMNE134K disrupted the SMN:HuD interaction (Fallini et al., 2011; Hubers et al., 2011). Therefore, we generated a transgenic line with the mnx1 promoter driving RFP-SMNE134K (Fig. 1A). We found that SMNE134K in motoneurons was not able to precipitate HuD, indicating that this human mutation disrupted the interaction between SMN and HuD in vivo. We also generated a transgenic line expressing a c-terminal mutation, SMNQ82A (Carrel et al., 2006) (Fig. 1A). This mutation did not disrupt the SMN:HuD interaction supporting the specificity of this biochemical assay (Fig. 1B).

SMN and HuD coimmunoprecipitate from motoneurons, but only during the period of motor axon development. A, Schematic diagram of the transgenic constructs driving wild-type human (h) SMN, hSMNQ282A, and hSMNE134K in motoneurons. B, Immunoprecipitation using no antibody (No Ab) or anti-RFP (RFP Ab) and immunoblotting with SMN and HuD. Analysis was performed at 2 and 4 dpf. Total, Total protein.
We next asked whether this interaction between SMN and HuD was developmentally regulated. We had previously shown, using the Tg(mnx1:RFP-SMN) line, that SMN is present in motor axons when they are undergoing robust axon development and branching, which is essentially complete by 4 dpf (Hao le et al., 2015). We therefore asked whether SMN and HuD still coimmunoprecipitate at 4 dpf. We found that, at this time, they did not, suggesting that the interaction between SMN and HuD is developmentally regulated and occurs during the time period associated with axon branch development (Fig. 1B).
SMN is required for HuD expression, but only early in development
Because SMN and HuD form a complex in motoneurons, we wanted to address whether SMN had an effect on HuD expression. A number of studies have shown that SMN can regulate the levels of other proteins, such as some of the Gemin proteins (Feng et al., 2005) and Plastin3 (Bowerman et al., 2009; Hao le et al., 2012), and that SMN may function in the translational regulation of many proteins (Sanchez et al., 2013). Therefore, we analyzed whether SMN affected HuD protein levels. For these experiments, we used mz-smn mutants. In constructing this line, we crossed in the zebrafish hsp70 promoter driving RFP-SMN transgene (Tg(hsp70:RFP-SMN) onto mz-smn mutants. Under normal conditions, these fish express low levels of RFP-SMN due to the leaky hsp70 promoter and thus are a genetic model of SMA a motoneuron disease caused by low levels of SMN (Hao le et al., 2013, 2015). Using this line, we analyzed HuD protein levels and found that they were decreased (Fig. 2A). Because these fish carry the Tg(hsp70:RFP-SMN) transgene, we can heat shock them to increase SMN levels (Hao le et al., 2013). To address whether adding back SMN would lead to an increase in HuD, we heat-shocked animals at 10 and 24 hpf. Interestingly, we found that only when we heat-shocked animals and increased SMN at 10 h post fertilization (hpf), did we see an increase in HuD, but not when we heat-shocked and increased SMN at 24 hpf (Fig. 2A). Subjecting wild-type embryos to heat shock did not affect HuD levels (Fig. 2B). These data support that SMN has an effect on HuD levels that is developmentally regulated.

HuD levels are dependent on SMN. A, Western blot of wild-type (wt) and mz-smn mutants either not heat-shocked (−) or heat-shocked at 10 or 24 hpf. B, Western blot of wt embryos either not heat-shocked (−) or heat-shocked at 10 hpf. Protein was collected 36 h after heat shock. HuD, RFP-SMN, and zebrafish (zf) SMN were analyzed by Western blot. β-actin was used as a loading control.
HuD mutants have decreased axonal branches and dendrites
To better understand the function of HuD in motoneuron development, we generated zebrafish HuD mutants using CRISPR/Cas9 (Talbot and Amacher, 2014). Using two separate guide RNAs, we generated four unique HuD mutations that resulted in no evidence of HuD protein as revealed by Western blot supporting that these are null alleles (Fig. 3A). Unlike SMN (Boon et al., 2009; Hao le et al., 2013), we did not see evidence of any maternal HuD protein. To examine motoneuron development in HuD mutants, we examined motor axons at 1, 2, and 4 dpf and dendrites at 4 dpf. At 1 dpf, we found that motoneurons were present, but they did not extend axons normally (Fig. 3B–D; p < 0.0001 Mann–Whitney nonparametric rank test). At 2 and 4 dpf, we found that there were significant decreases in axonal branches (Fig. 3E–G; p < 0.0001, two-tailed t test of independent samples). At 4 dpf, HuD mutants exhibited a significant decrease in dendrites compared with wild-types (Fig. 4; p < 0.0001, two-tailed t test of independent samples). These phenotypes were very similar to those found in mz-smn mutants (Hao le et al., 2013), suggesting that decreasing either SMN or HuD has a negative effect on motoneuron development resulting in deficient maturation of axons and dendrites.

HuD mutants display motor axon defects similar to mz-smn mutant. A, Four HuD mutants were generated by CRISPR/Cas9 using two unique guide RNAs for HuD target 1 and HuD target 2. The size of the insertion (in) or deletion (del) is noted. Western blot analysis of protein from wild-type (wt) and the four HuD homozygous (−/−) mutants at 4 dpf. β-actin was used as a loading control. Lateral view of 26 hpf (B) wild-type (wt) and (C) HuD−/− os55 processed for znp1 (synaptotagmin) antibody labeling to visualize motor axons. Arrows indicate ventral motor axons. D, Quantification of wt and HuD−/− animals based on their motor axon defects at ∼26 hpf (for axon scoring rubric, see Materials and Methods). n = 73 (wt) and n = 60 (mutants) from three separate clutches of embryos. Data are mean ± SEM. ***p < 0.0001 (Mann–Whitney nonparametric rank test). E, Individual motoneurons were stochastically labeled by injecting mnx1:GFP into 1 cell stage embryos and imaged by confocal microscopy at 4 dpf. Arrows indicate axonal branches in the ventral muscle. F, Quantification of motor axon branching at 2 dpf using single axon labeling, axon tracing, and measurement of total axon and branch length (mean ± SD). ***p < 0.0001 (two-tailed t test of independent samples with equal variances and 26 df). n = 15 axons for wt, and n = 13 axons for mutants. Scale bars: B, C, 25 μm; E, F, 15 μm.

Motoneuron dendrites in HuD mutants are dramatically decreased in length. Individual GFP-labeled motoneurons in a wild-type (wt; A) and HuD (B) mutant at 4 dpf. C, Quantification of total dendrite length using dendrite tracing (see Materials and Methods). Data are mean ± SD. ***p < 0.0001 (two-tailed t test of independent samples with unequal variances and 13 df). n = 13 for wt, and n = 12 for mutants. Scale bar, 15 μm.
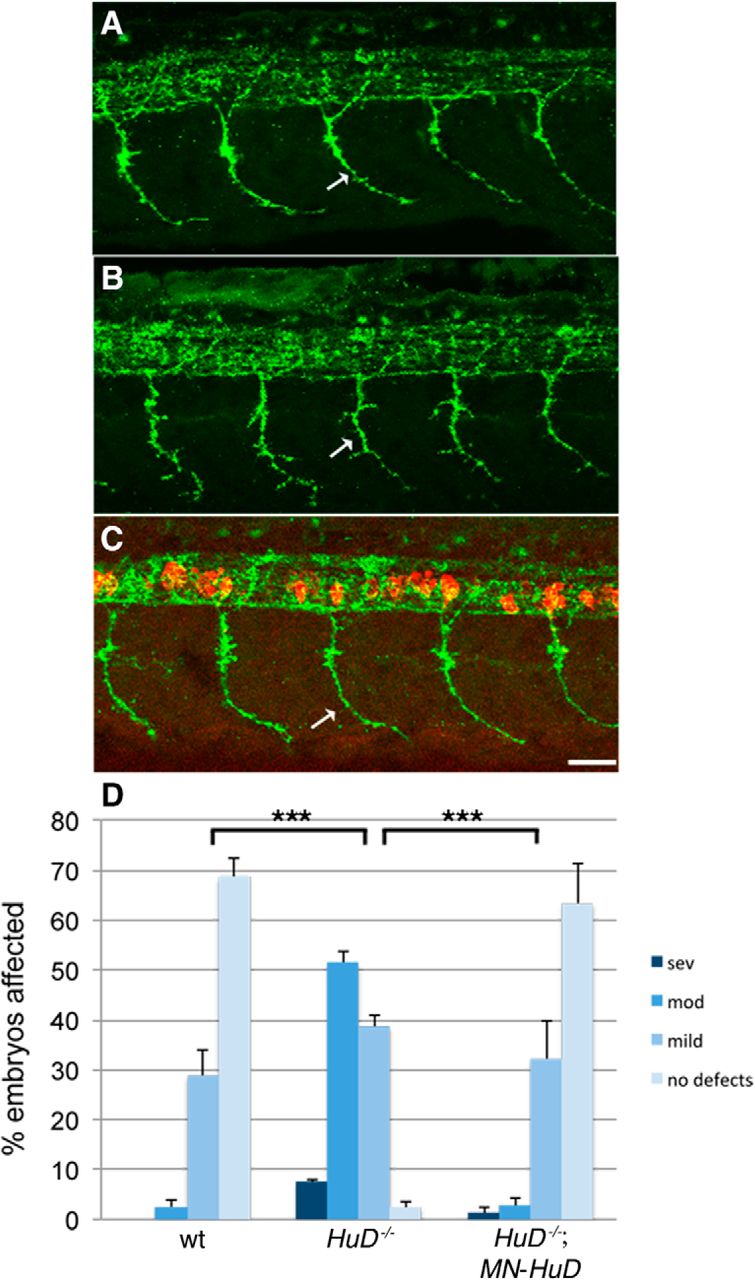
HuD is expressed in neurons throughout the central and peripheral nervous system (Marusich et al., 1994). Therefore, this effect on motoneuron development could be due to loss of HuD in cells other than motoneurons. To test this directly, we made a transgenic line expressing HuD in motoneurons using the Gal4:UAS system. For this, we generated Tg(mnx1:Gal4) and Tg(UAS:mCherry-HuD) and crossed these onto the HuD mutant line (for details, see Materials and Methods). We found that expressing HuD in motoneurons on a HuD mutant background rescued the motoneuron developmental defect at 26 hpf (Fig. 5; p < 0.0001, two-tailed Mann–Whitney nonparametric rank test; wt vs HuD−/− + MN-HuD, p = 0.61, two-tailed Mann–Whitney nonparametric rank test). We also analyzed motor axons at 2 dpf and dendrites at 4 dpf and found significant rescue when HuD was expressed just in motoneurons (Fig. 6; p < 0.0001, two-tailed t test of independent samples; wt vs HuD−/− + MN-HuD, p = 0.649 for axon branches and p = 0.23 for dendrites). These data support that HuD acts cell-autonomously in motoneurons and is essential for their normal development. We had previously shown that SMN also acts motoneuron cell-autonomously (Hao le et al., 2015), indicating that these two proteins are both integral in motoneurons for their development.

Motor axon defects in HuD mutants are rescued by expressing HuD in motoneurons. Lateral view of labeled motor axons from 26 hpf (A) wild-type, (B) HuD−/−, and (C) HuD−/−; MN-HuD. D, Quantification of animals based on their motor axon defects. n ranged from 75 to 80 for each experimental line from three separate clutches of embryos (mean ± SEM). p values were determined by Mann–Whitney nonparametric rank test (two-tailed). wt versus HuD−/−, ***p < 0.0001; HuD−/− versus HuD−/−; MN-HuD, ***p < 0.0001; wt versus HuD−/−; MN-HuD, p = 0.61. MN-HuD refers to Tg(mnx1:Gal4);Tg(UAS:mCherry-HuD). Scale bar, 25 μm.

Expressing HuD exclusively in motoneurons rescues the motor axon branching and dendrite defects in HuD mutants. Confocal image and tracing of individual axons in 2 dpf (A, A′) wild-type (n = 14), (B, B′) HuD−/− (n = 15), and (C, C′) HuD−/−; MN-HuD (n = 12) animals. D, Quantification of motor axons and branch length (mean ± SD). p values determined by a two-tailed t test of independent samples. wt versus HuD−/−: ***p < 0.0001 (equal variances, 27 df). HuD−/− versus HuD−/−; MN-HuD: ***p < 0.0001 (unequal variances, 15 df). wt versus HuD−/−; MN-HuD: p = 0.649 (equal variances, 24 df). Confocal images of dendrites in 4 dpf (E) wild-type (n = 14), (F) HuD−/− (n = 13), and (G) HuD−/−; MN-HuD (n = 12) animals. H, Quantification of total dendrite length (mean ± SD). p values determined by a two-tailed t test of independent samples. wt versus HuD−/−: ***p < 0.0001 (unequal variances, 15 df). HuD−/− versus HuD−/−; MN-HuD: ***p < 0.0001 (unequal variances, 15 df). wt versus HuD−/−; MN-HuD: p = 0.230 (unequal variances, 23 df). MN-HuD refers to Tg(mnx1:Gal4);Tg(UAS:mCherry-HuD). Scale bars: A–C, 50 μm; E–G, 15 μm.
HuD expressed in mz-smn motoneurons significantly rescues the motoneuron developmental defects and movement deficits
We showed above that HuD and mz-smn mutants cause a similar motoneuron phenotype and that decreasing SMN leads to decreased HuD levels. Therefore, we asked whether expressing HuD in mz-smn motoneurons could compensate and rescue the motoneuron developmental defects caused by low levels of SMN. We tested this in two ways. First, we used the Gal4:UAS system and generated Tg(mnx1:Gal4) and Tg(UAS:mCherry-HuD) lines on the mz-smn mutant background (for details, see Materials and Methods). We also generated another transgenic line expressing mCherry-HuD in motoneurons, Tg(mnx1:mCherry-HuD) and crossed these to the mz-smn mutants. Both transgenic lines expressed levels of mCherry-HuD comparable with the levels of endogenous HuD (data not shown). Using both approaches, we found that expressing HuD in mz-smn motoneurons significantly rescued the motor axon defects caused by low levels of SMN. We analyzed this at 1 dpf and found significant rescue based on motor axon defects (Fig. 7; p < 0.0001, two-tailed Mann–Whitney nonparametric rank test). We then analyzed individual motoneurons axons at 2 dpf and dendrites at 4 dpf using the Gal4:UAS system (Fig. 8). Analysis at the single-cell level revealed significant rescue for both axonal branches and dendrites (p < 0.0001 for both axon branches and dendrites, two-tailed t test of independent samples; for additional statistical analysis, see Fig. 8). These data reveal that expressing HuD in SMN-deficient motoneurons can rescue the developmental defects observed in axonal branching and dendrite formation. Although we found statistically significant rescue, wild-types were statistically better than mz-smn mutants carrying MN-HuD for these parameters, indicating that the rescue was not complete.

Expressing HuD in SMN-depleted motoneurons rescues the motor axon phenotype. Lateral view of 26 hpf (A) wild-type (wt), (B) mz-smn−/−, and (C) mz-smn−/−; MN-HuD1, processed for znp1/synaptotagmin antibody labeling to visualize motor axons. Arrows indicate ventral motor axons. D, Quantification of animals based on their motor axon defects as previously established. n = 71–81 for each experimental line from three separate clutches of embryos (mean ± SEM). p values were determined by Mann–Whitney nonparametric rank test (two-tailed). wt versus mz-smn−/−: ***p < 0.0001. mz-smn−/− versus mz-smn−/−; MN-HuD1: p < 0.0001. wt versus mz-smn−/−; MN-HuD1: ***p < 0.0001. E, Quantitation of animals based on their motor axon defects. n = 59–79 for each experimental line from three separate clutches of embryos (mean ± SEM). p values were determined by Mann–Whitney nonparametric rank test (two-tailed). wt versus mz-smn−/−: ***p < 0.0001; mz-smn−/− versus mz-smn−/−; MN-HuD2: p < 0.0001. wt versus mz-smn−/−; MN-HuD2: p < 0.0001. MN-HuD1 refers to Tg(mnx1:Gal4);Tg(UAS:mCherry-HuD). MN-HuD2 refers to Tg(mnx1:mCherry-HuD). Scale bar, 25 μm.

Expressing HuD in SMN-depleted motoneurons rescues the axon branching and dendrite defects. Confocal image and tracing of individual axons in 2 dpf (A, A′) wild-type (n = 12), (B, B′) mz-smn−/− (n = 13), and (C, C′) mz-smn−/−; MN-HuD (n = 14) animals. D, Quantification of motor axons and branch length (mean ± SD). p values determined by a two-tailed t test of independent samples. wt versus mz-smn−/−: ***p < 0.0001 (unequal variances, 15 df). mz-smn−/− versus mz-smn−/−; MN-HuD: ***p < 0.0001 (equal variances, 25 df). wt versus mz-smn−/−; MN-HuD: p = 0.042 (equal variances, 24 df). Confocal images of dendrites in 4 dpf (E) wild-type (n = 13), (F) mz-smn−/− (n = 12), and (G) mz-smn−/−; MN-HuD (n = 12) animals. H, Quantification of total dendrite length (mean ± SD). p values were determined by a two-tailed t test of independent samples. wt versus mz-smn−/−: ***p < 0.0001 (unequal variances, 15 df). mz-smn−/− versus mz-smn−/−; MN-HuD: ***p < 0.0001 (unequal variances, 15 df). wt versus mz-smn−/−; MN-HuD: ***p < 0.0001 (equal variances, 23 df). MN-HuD refers to Tg(mnx1:Gal4);Tg(UAS:mCherry-HuD). Scale bars: A–C, 50 μm; E–G, 15 μm.
We had previously shown that mz-smn mutant larvae were deficient in initiating bouts of spontaneous swims and turns, distance moved over time and per swim, and other kinematic parameters of swimming, together indicating that the motoneuron defects had a negative impact on motor function (Hao le et al., 2013). We asked, therefore, whether driving HuD in mz-smn−/− motoneurons not only rescued motoneuron development, but could also rescue motor behavior. To measure spontaneous movements, we used high-speed video capture and analysis on 5 dpf larvae. We first analyzed gross movement over a continuous 2 min period and found that both mz-smn and HuD mutants moved significantly less distance compared with wild-type larvae (Fig. 9A; p < 0.001, one-way ANOVA). Importantly, expressing HuD in motoneurons significantly rescued these movement defects in both mz-smn and HuD mutants (p < 0.01, one-way ANOVA). To determine whether the reduced overall movement by mz-smn and HuD mutants was due to reduced initiations of movement, we analyzed the frequency of swim and turn initiations. As with the gross movement, initiation frequency of both swims and turns was decreased in mz-smn and HuD mutants. These deficiencies were significantly rescued by driving HuD in motoneurons (Fig. 9B,C; see statistics). To look more closely at the rescue in mz-smn mutants, we analyzed kinematic parameters of nonevoked swimming events that included two or more swim half-cycles (a swim half-cycle involves a single, unilateral body bend). We found that swims in mz-smn mutants resulted in less distance moved compared with wild-types and that this deficit was rescued by expressing HuD in their motoneurons (Fig. 9D; p < 0.001, one-way ANOVA). In addition, both the mean body curvature change per swim half-cycle and the mean number of swim half-cycles per swim were also decreased in mz-smn mutants compared with wild-types, and these were rescued by expressing HuD in mz-smn−/− motoneurons (Fig. 9E,F; p < 0.001, one-way ANOVA). Wild-type and mz-smn−/−; MN-HuD fish were not statistically different (p > 0.05) for any of these tests, suggesting that these movements were fully rescued. These swim parameters were also decreased in HuD mutants compared with wild-type larvae and rescued by driving HuD in motoneurons (data not shown). Together, these results support that HuD can rescue the functional defects in motoneurons caused by low levels of SMN.

Motor behavior analysis. A, Gross movement over 120 s. Each dot represents individual larva. B, C, Mean frequency of swim or turn initiation in mz-smn−/− (B) and HuD−/− larvae (C). N = 3 groups of 20 larvae/group. D, Mean distance per swim. E, Mean body curvature change per half swim cycle. F, Mean number of half swim cycles per swim. D–F, N swims evaluated: wt = 155, mz-smn−/− = 40, mz-smn−/−; MN-HuD = 114. MN-HuD refers to Tg(mnx1:Gal4);Tg(UAS:mCherry-HuD). Data are mean ± SEM. *p < 0.05, **p < 0.01. ***p < 0.001 (ANOVA, Bonferroni post hoc analysis).
Gap43 RNA is decreased in mz-smn and HuD mutants
HuD binds to mRNAs affecting their stability and transport. If SMN and HuD are affecting mRNA, then we would expect to see an alteration in a HuD target mRNA. Gap43 is a neural-specific phosphoprotein, enriched in growth cones and involved in neural development, regeneration, and plasticity (Meiri et al., 1986; Benowitz and Routtenberg, 1987). Localized translation of Gap43 mRNA in axons is important for axonal growth (Donnelly et al., 2013). HuD binds a defined uridine-rich region of the Gap43 3′-UTR (Chung et al., 1997; Yoo et al., 2013) and decreasing HuD leads to decreased levels of Gap43 in PC12 cells and less stable Gap43 mRNA, suggesting the HuD binding to Gap43 stabilizes this message (Mobarak et al., 2000). Moreover, decreased levels of SMN in cultured neurons also leads to decreased axonal Gap43 (Fallini et al., 2016). We therefore asked whether Gap43 mRNA levels were altered in mz-smn and HuD mutants. We isolated trunks (spinal cord and muscle) from 2 dpf embryos and used digital droplet (dd) PCR to quantitate the levels of Gap43 mRNA compared with a control mRNA, ubiquitin-conjugating enzyme E2A (Ube2a). We found that, in both mutants, Gap43 was significantly decreased compared with control animals (Fig. 10; p < 0.001 wt vs mz-smn−/− and wt vs HuD−/−; ANOVA, Holm–Sidak post hoc analysis). To determine whether this decrease in Gap43 mRNA was due to RNA levels in motoneurons, we measured Gap43 mRNA in mz-smn and HuD mutants with HuD transgenically added back to motoneurons using the Gal4:UAS system. In these animals, Gap43 levels were not significantly different from levels in wild-type animals (Fig. 10; p = 0.84, wt versus mz-smn−/−; HuD MN; p = 0.78, wt versus HuD−/−; HuD MN). These data support that HuD effects Gap43 RNA levels in motoneurons and that SMN is critical for this process.

Decreased Gap43 mRNA levels in HuD and mz-smn mutants are rescued by HuD in motoneurons. Gap43 mRNA from 2 dpf embryos (30 animals/genotype) was analyzed by ddPCR and expressed as a ratio of control mRNA, Ube2a (mean ± SEM). MN-HuD refers to Tg(mnx1:Gal4);Tg(UAS:mCherry-HuD). ***p < 0.001 (ANOVA, Holm–Sidak post hoc analysis). ns, not significant.
Discussion
It remains unresolved how low levels of SMN directly lead to motoneuron defects, a hallmark of the childhood motoneuron disease SMA. It is clear that SMN helps facilitate the formation of RNA:protein complexes. This has been shown by extensive biochemistry for snRNP assembly (Pellizzoni et al., 2002; Pellizzoni, 2007) and more recently for mRNP assembly (Donlin-Asp et al., 2017). Because of the finding that low levels of SMN cause deficiencies in motoneuron development, SMN is present in motor axons during development, and in vitro has been shown to bind neuronal RBPs, we hypothesized that SMN:RBP complexes had a role in motoneuron development. Using a genetic model with low levels of SMN and analyzing these interactions directly in motoneurons, we demonstrate a physical protein–protein interaction between SMN and HuD in motoneurons during development. Moreover, we show that SMN levels dictate HuD protein levels. Generating HuD mutants revealed that they too had similar motoneuron developmental defects supporting that these proteins affect the same process. Expressing HuD in mz-smn mutant motoneurons rescued both the developmental defects at the cellular level, but also the motor deficits present in these mutants. Importantly, when SMN levels were low or when HuD was lacking, Gap43 mRNA, whose local translation affects axonal growth, was decreased. Together, these data support a role for SMN:HuD in motoneuron development and function. Mechanistically they implicate mRNA disruption as the cause of these defects.
RBPs bind mRNAs often keeping them translationally silent. In this way, mRNAs can be transported and then locally synthesized. When RBPs are missing, mRNAs are at risk of being degraded. Our in vivo data support this hypothesis as Gap43 mRNA was decreased when HuD was missing or when SMN levels were low. This is consistent with what was observed in cultured PC12 cells where Gap43 mRNA was less stable when HuD was decreased (Mobarak et al., 2000). Because HuD levels were also decreased in mz-smn mutants, low Gap43 levels could be due to low or absent HuD protein in both mutants. It is also possible that SMN helps facilitate HuD binding to Gap43; and when SMN levels are low, as in mz-smn mutants, this does not occur. In vitro data revealed that SMN facilitated the binding of the RBP IMP1 to β-actin RNA, thus supporting this mechanism (Donlin-Asp et al., 2017). Therefore, both mechanisms may be at play in vivo. RBPs bind mRNAs and transport them to axons and dendrites to be locally translated, and this is essential for neuronal development, including axonal branch formation (Holt and Schuman, 2013; Hörnberg and Holt, 2013; Wong et al., 2017). HuD binds GAP43 and β-actin, which are involved in axon growth and branch formation and need to be translated in the axon for these functions (Donnelly et al., 2013; Wong et al., 2017). Data from cultured PC12 cells also showed that both HuD and Gap43 were needed for neurite outgrowth (Mobarak et al., 2000). IMP2, an RBP expressed in commissural axons, binds RNAs highly enriched for functions related to axon development (Preitner et al., 2016). When IMP2 was depleted in chick embryos, commissural axon guidance was disrupted supporting a function for IMP2, like HuD, in axon development. This process is also dynamic as mRNAs in axons and growth cones change dramatically over development supporting that the requirement for particular mRNAs in distal axons changes over time (Zivraj et al., 2010; Shigeoka et al., 2016). Thus, we hypothesize that decreased HuD would result in less mRNA transport into axons, which would have a negative effect on axonal outgrowth and branch formation during development. It will be critical to identify what mRNAs are bound by the SMN:HuD complex in motoneurons and whether they are differentially localized during development.
HuD knock-out mice, like the zebrafish HuD mutants, are also viable and survive into adulthood (Akamatsu et al., 2005). Approximately 75% of young adult HuD−/− mice have a hindlimb clasping defect indicating the presence of motor-sensory deficits and have significant rotarod impairment supporting a motor defect. These mice also exhibit decreased dendritic branching in cortical neurons and CA3 hippocampal neurons (DeBoer et al., 2014), which is consistent with the dendritic defects we see in HuD mutant zebrafish motoneurons. A detailed analysis of motor nerves has not been published; however, these mice were less active specifically in walking and running and had increased stereotyped behaviors. This could be due to defects in both the cortical and motor systems. The motor behavior deficits observed in the zebrafish HuD mutants and mz-smn mutants were in initiating swims and turns and sustaining these movements, which together yielded reduced movement. Our observation of reduced body curvature/swim half-cycles in both mz-smn and HuD mutants reflects that the muscle is less innervated or activated. NMJs form on axonal branches; thus, this decreased movement is consistent with the decreased axonal branching observed in these mutants. This is also supported by the finding that partial motor nerve ablation causes a similar behavioral defect to what is seen in both mz-smn and HuD mutants (Rosenberg et al., 2012). Because the motoneuron dendrites are also reduced, motoneurons may receive less innervation contributing to the decreased activity. Because swimming involves alternating side muscle contractions caused by alternating activation of motoneurons, less active motoneurons would be expected to lead to a reduction in alternating muscle contractions and thus less distance moved. Indeed, both mutants were significantly worse than wild-types at these behaviors. Importantly, these defects were observed in both mz-smn and HuD mutants and were rescued by driving HuD in motoneurons indicating a cell-autonomous need for HuD in motoneuron development and function.
How could increasing HuD in SMN-deficient motoneurons lead to motoneuron rescue? We showed that SMN is required for normal HuD levels; thus, when SMN is depleted, so is HuD. It is intriguing that, only when SMN was increased by heat shock at 10, but not 24 hpf, did it increase HuD protein levels. This result suggests that SMN′s effect on HuD is temporally regulated having its effect before, but not after, motor axon outgrowth has been initiated. SMN is present in polysome fractions and affects protein levels acting to either activate or repress their expression (Sanchez et al., 2013). In this case, we do not know at what level SMN is affecting HuD, but previous work showed that SMN regulated Plastin 3 levels via a post-transcriptional mechanism (Hao le et al., 2012). Even though we show that SMN and HuD interact, it is possible that higher levels of HuD can compensate for low levels of SMN. For example, overexpressing HuD in embryonic rat cortical neurons increased neurite formation and GAP43 expression (Anderson et al., 2001). Our data also support a cell-autonomous role for HuD in motoneuron development. However, driving HuD in motoneurons transgenically did not lead to a complete rescue of mz-smn motoneurons at the cellular level. Thus, there is the possibility that HuD could have a non–cell-autonomous function that affects motoneurons. Because HuD is a neuronal RBP, it would have to be acting through other neurons. This lack of complete rescue could also indicate that other RBPs are functioning in motoneurons and are affected by SMN. This is supported by data showing that SMN interacts with the RBP IMP1 in motoneurons, and this interaction affects GAP43 levels in cultured motoneuronal axons (Fallini et al., 2016).
SMN is involved in facilitating the formation of protein:RNA complexes for splicing. Deficiency in this function correlates with phenotypes in SMA, although how this function directly affects motoneurons has not been resolved. Interestingly, the splicing factor SFPQ was recently shown to have a non-nuclear function that controls transcripts important for motoneuron development (Thomas-Jinu et al., 2017). Like SMN, SFPQ is also found in motor axons and affects splicing of numerous transcripts that could function in motoneuron development, such as cell adhesion, cell polarity, dendritic spine formation, and synapse formation. Compared with the mz-smn mutants, however, SFPQ mutants exhibited much more severe motoneuron developmental defects. Motor axons failed to extend to the distal muscle, often barely leaving the spinal cord, and NMJs were largely absent. This suggests that defects in splicing can have a profound effect on motoneuron development. If SMN deficiency were primarily causing splicing defects, then we would expect many fewer RNAs to be altered because mz-smn mutants have a milder motoneuron phenotype than the SFPQ mutants. Two genes have been identified that exhibit splicing defects caused by low levels of SMN and affect motoneurons. These genes, stasimon and chondrolectin, when introduced transiently into SMN-deficient zebrafish, can partially rescue motoneuron defects (Lotti et al., 2012; Sleigh et al., 2014). In addition, Agrin (Z+ form), a protein involved in NMJ formation, was also shown to be mispliced when SMN levels were low and adding this back to SMA mice rescued the developmental NMJ defect (Zhang et al., 2013; Kim et al., 2017). Thus, low levels of SMN may affect mRNA levels and transport and also splicing of a small number of genes critical for motoneuron development.
SMN is an organizer of RNA protein complexes and, when deficient, leads to a motoneuron disease. We have shown that SMN interacts with HuD in motoneurons to affect motor axon and dendrite development and RNA levels. Based on these data, we hypothesize that SMN interacts with RBPs to traffic mRNAs to growing, branching axons and dendrites for localized translation, a process vital for their formation. Future work will identify the full complement of SMN:RBP interactions in developing motoneurons and their cargo mRNAs.
Footnotes
This work was supported by National Institutes of Health R56NS050414 and RO1NS098780 and CureSMA to C.E.B., and the Greater Milwaukee Foundation Shaw Scientist Award 133-AAA2656 to M.W. We thank Dr. Wilfried Rossoll for the mCherry-HuD construct; and the fish facility staff for fish care.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Christine E. Beattie, Ohio State University, 190 Rightmire Hall, 1060 Carmack Road, Columbus, OH 43210. beattie.24{at}osu.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}