

Abstract
We have previously shown that casein kinase 2 (CK2) negatively regulates dopamine D1 and adenosine A2A receptor signaling in the striatum. Ablation of CK2 in D1 receptor-positive striatal neurons caused enhanced locomotion and exploration at baseline, whereas CK2 ablation in D2 receptor-positive neurons caused increased locomotion after treatment with A2A antagonist, caffeine. Because both, D1 and A2A receptors, play major roles in the cellular responses to l-DOPA in the striatum, these findings prompted us to examine the impact of CK2 ablation on the effects of l-DOPA treatment in the unilateral 6-OHDA lesioned mouse model of Parkinson's disease. We report here that knock-out of CK2 in striatonigral neurons reduces the severity of l-DOPA-induced dyskinesia (LID), a finding that correlates with lowered pERK but unchanged pPKA substrate levels in D1 medium spiny neurons as well as in cholinergic interneurons. In contrast, lack of CK2 in striatopallidal neurons enhances LID and ERK phosphorylation. Coadministration of caffeine with a low dose of l-DOPA reduces dyskinesia in animals with striatopallidal knock-out to wild-type levels, suggesting a dependence on adenosine receptor activity. We also detect reduced Golf levels in the striatonigral but not in the striatopallidal knock-out in response to l-DOPA treatment.
Our work shows, in a rodent model of PD, that treatment-induced dyskinesia and striatal ERK activation are bidirectionally modulated by ablating CK2 in D1- or D2-positive projection neurons, in male and female mice. The results reveal that CK2 regulates signaling events critical to LID in each of the two main populations of striatal neurons.
SIGNIFICANCE STATEMENT To date, l-DOPA is the most effective treatment for PD. Over time, however, its efficacy decreases, and side effects including l-DOPA-induced dyskinesia (LID) increase, affecting up to 78% of patients within 10 years of therapy (Hauser et al., 2007). It is understood that supersensitivity of the striatonigral pathway underlies LID, however, D2 agonists were also shown to induce LID (Bezard et al., 2001; Delfino et al., 2004). Our work implicates a novel player in the expression of LID, the kinase CK2: knock-out of CK2 in striatonigral and striatopallidal neurons has opposing effects on LID. The bidirectional modulation of dyskinesia reveals a central role for CK2 in striatal physiology and indicates that both pathways contribute to LID.
Introduction
Parkinson's disease (PD) is the second most prevalent neurodegenerative disorder, affecting as many as 1% of people 65 years or older (Hirtz et al., 2007). It is characterized by motor symptoms such as tremor, rigidity, bradykinesia, and gait disturbances (Graybiel, 2000), which are caused by striatal dopamine depletion, engendered by a loss of dopaminergic neurons within the substantia nigra pars compacta (Dauer and Przedborski, 2003).
More than 95% of neurons in the striatum are GABAergic projection neurons that are categorized in two classes based on their predominant expression of either D1 or D2 receptors (Gerfen et al., 1990). Neurons projecting directly to the basal ganglia output nuclei (“direct pathway”) express the D1 receptor (D1R), which is positively coupled to cAMP-dependent signaling via Gαs/Gαolf. The other class of projection neurons (“indirect pathway”) expresses the inhibitory, Gαi-coupled D2 receptor (D2R). In these neurons, cAMP signaling is stimulated by the Gαs/Gαolf-coupled adenosine A2A receptor (Rosin et al., 2003). The balance between the two pathways, crucial to movement control, is disrupted in PD. Currently, l-DOPA is the most effective symptomatic treatment for PD. Over time, however, the efficacy of l-DOPA decreases, drug dosage is increased, and debilitating involuntary movements develop in the majority of the patients. l-DOPA-induced dyskinesia (LID) affects up to 78% of patients within 10 years of therapy and represents a major challenge in the clinical management of this disorder (Hauser et al., 2007). Various pharmacological approaches are being developed to delay the need for l-DOPA or mitigate its complications.
CK2 (formerly casein kinase 2), a heterotetrameric kinase consisting of two catalytic subunits (α) and two regulatory (β) subunits, is a major player in the regulation of cell proliferation and apoptosis. Despite high brain expression levels and activity (Lou et al., 2008), its importance in the brain is poorly understood. Brain CK2 substrates have been linked to a variety of disorders including PD (Castello et al., 2017). CK2 is present in Lewy bodies and phosphorylates α-synuclein and synphilin-1 (Ryu et al., 2008). Phosphorylation of the latter seems to impact on the amount of cytoplasmic inclusions (Lee et al., 2004). Constitutive CK2 knock-out (KO) mice are embryonically lethal: CK2α KO mice die at embryonic day (E)10.5 with neural tube and heart defects (Lou et al., 2008), and CK2β KO mice die on day E6.5 (Buchou et al., 2003). We have previously provided evidence that CK2 modulates D1R as well as A2AR signaling (Rebholz et al., 2013). We found that CK2 binds to Gαs/Golf and that inhibition or knockdown of CK2 leads to elevated cAMP concentrations in response to D1R or A2AR agonists (Rebholz et al., 2013). This effect may be mediated through modulation of receptor endocytosis. We have further found that mice with conditional ablation of CK2α in D1- or D2-positive neurons exhibit behavioral phenotypes corresponding to elevated expression/signaling via the D1 or A2A receptor, respectively (Rebholz et al., 2013; Castello et al., 2017).
Due to the importance of D1 and A2A receptors in the cellular responses to l-DOPA, our data lead us to hypothesize that CK2 may also play a role in regulating striatal signaling and behavior in PD. We thus set out to examine behavioral and signaling responses to l-DOPA in conditional CK2α KO mice rendered parkinsonian by a 6-OHDA lesion. We report that the lack of CK2α in direct or indirect pathway striatal neurons has opposite effects on dyskinetic behaviors and on the associated striatal upregulation of ERK signaling.
Materials and Methods
Animals
The generation of the floxed CK2α line was described previously (Gong et al., 2007; Rebholz et al., 2013), mice were continuously kept on a C57BL/6J background. Animals that underwent 6-OHDA lesions were of 25–30 g weight (4–6 months old). The mice were maintained in a 12 h light/dark cycle, with access to food/water ad libitum. Animal procedures were in accordance with the NIH guidelines and approved by CCNY's IACUC. The Drd1a-Cre+/−;CK2αloxP/loxP will be named Drd1a-Cre/CK2KO and the Drd2-Cre+/−;CK2αloxP/loxP will be named Drd2-Cre/CK2KO, whereas their corresponding control littermates Drd1a-Cre−/−;CK2αloxP/loxP or Drd2-Cre−/−;CK2αloxP/loxP will be annotated as WT for both lines. The GENSAT BAC Cre-recombinase driver lines Drd1a-Cre and Drd2-Cre were crossed with the floxed animals (Gerfen et al., 2013). The GENSAT BAC Drd1a-GFP line Tg(Drd1a-eGFPX60Gsat) was also crossed with the Drd1a-Cre+/−; CK2αloxP/loxP line.
Antibodies
Anti-CK2α (RRID:AB_297213), anti-Enkephalin and anti-GFP (RRID:AB_300798) antibodies were purchased from Abcam, anti-Gαs from Calbiochem, anti-tyrosine hydroxylase (RRID:AB_390204) and anti-choline acetyltransferase (RRID:AB_2079751) from Millipore. Anti-tubulin antibody was purchased from Sigma-Aldrich (RRID:AB_477593). Anti-phospho-Thr202/Tyr204-Erk1/2 (RRID:AB_331646), c-Fos (RRID:AB_2247211), and phospho-(Ser/Thr) PKA substrate (RRID:AB_331817) were from Cell Signaling Technology. The anti-Gαolf antibody was a gift from Dr. D. Hervé (INSERM UMR-S 839, Paris). Various AlexaFluor antibodies were used at a 1:500 dilution for immunohistochemistry (Invitrogen).
6-OHDA lesions and l-DOPA treatment
Unilateral 6-OHDA injections into the dorsolateral striatum were performed according to a well established method (Francardo et al., 2011). Mice were anesthetized with a mixture of ketamine (80 mg/kg) and xylazine (12 mg/kg); the local anesthetic Bupivacaine (Marcaine) was subcutaneously injected near the surgery site. 6-OHDA-HCl (3.0 mg/ml; Sigma-Aldrich) was dissolved in a solution containing 0.2 g/L ascorbic acid left striatum at the following coordinates: anteroposterior (AP), +1.0 mm; lateral (L), +2.1 mm; dorsoventral (DV), −3.4 mm; and AP, +0.3 mm; L, +2.3 mm; DV, −3.4 mm. For medial forebrain bundle (MFB) lesion a single injection of 3 μg of 6-OHDA at coordinates: −1.2 AP, −1.3 ML, −4.75 DV from dura was performed. Each injection was performed with a Hamilton needle (33 gauge) connected to a syringe micropump (WPI) by a polyethylene catheter, at a slow rate of 0.4 μl/min to minimize tissue damage. After the injection, the needle was left in place for an additional 4 min before being slowly retracted. After surgery, animals were kept warm with a heated mat and observed three times daily. If necessary, animals received injections of 4% sucrose (10 ml/kg, s.c.) and saline (10 ml/kg, i.p.) as well as hydrogel pouches to avoid dehydration and high-fat chow to reduce weight loss. Mice were allowed to recover for 3 weeks before behavioral evaluation and drug treatment. Lesions were assessed biochemically at the end of experiments by determining the striatal levels of tyrosine hydroxylase (TH) using immunohistochemistry (IHC). Only animals with a TH-depletion of >70% of the striatal area compared with unlesioned side were included in the analyses. If not indicated otherwise, treatment of l-DOPA (20 mg/kg)/benserazide (12 mg/kg) was administered intraperitoneally over 7 consecutive days, starting 3 weeks after the lesion. Drugs were dissolved in physiological saline and administered at the volume of 10 ml/kg body weight.
Sample preparation and Western blot analysis
All animals were killed by decapitation and brains were rapidly dissected. Tissue samples were snap-frozen in liquid nitrogen. Tissues were lysed in 1% SDS (plus phosphatase inhibitors), sonicated for 3 × 10 s, boiled, and cleared by centrifugation for 20 min at 13,000 rpm. Protein concentration was determined with a bicinchoninic acid (BCA) kit (Pierce). Thirty to 40 μg of protein per well were used for SDS-PAGE, proteins were transferred onto nitrocellulose, incubated with antibodies overnight, and detected by enhanced chemiluminescence. Quantification was performed using ImageJ software.
Immunohistochemistry
Thirty minutes after the last intraperitoneal injection, animals were anesthetized with pentobarbital/ phenytoin and transcardially perfused with 50 ml of 4% paraformaldehyde (PFA) in PBS. Brains were incubated overnight in sucrose and sliced at a thickness of 40 μm. Incubations with antibodies and analysis were performed as described previously (Alcacer et al., 2012). Sections were mounted using Prolong Gold (Invitrogen). Confocal microscopy was performed using a Zeiss LMS 710 laser-scanning confocal microscope using the same adjustments for all the sections in a given experiment. For all immunohistochemical analysis, cells were counted manually (in 425 × 425 μm confocal images) and averaged from three slices per animal (unlesioned and lesioned sides under blind conditions concerning the mouse genotype and treatment). TH immunofluorescence intensity was quantified in striata with MacBiophotonics ImageJ, and the data represented as mean gray levels above background value.
Cylinder test
The anti-akinetic effect of l-DOPA was assessed using the cylinder test of forelimb paw placement. Mice were placed in a glass cylinder (10 cm wide × 14 cm high) and recorded for 4 min. The number of supporting paw placements performed independently with the left and right paw was counted. The limb use asymmetry score was calculated by expressing the number of wall contacts performed with the forelimb contralateral to lesion as a percentage of total wall contacts (Lundblad et al., 2002). For tests performed with escalating doses of l-DOPA, the mice were tested with 5–7 d intertrial intervals.
Abnormal involuntary movements
Abnormal involuntary movements (AIMs) were evaluated by an observer blind to the genotype, starting 20 min after administration of l-DOPA/benserazide (± additional drugs as described). Abnormal movements, clearly distinct from natural or stereotypic behaviors (i.e., grooming or sniffing), were classified into four different subtypes as described previously (Lundblad et al., 2005): locomotive [tight contralateral turns (LOC)], axial (contralateral dystonic posture of the neck and upper body), limb (jerky, fluttering movements of the limb contralateral to the lesion), and orofacial (vacuous jaw movements, tongue protrusions) AIMs. Each subtype was scored on a severity scale from 0 to 4: 0, absent; 1, occasional; 2, frequent; 3, continuous; 4, continuous and not interruptible by external stimuli. The total AIMs score corresponded to the sum of individual scores for each AIM subtype. A composite score was obtained by the addition of scores for axial, limb, and orofacial AIMs (ALO score). The ALO score is considered to more closely reflect the human dyskinetic behavior than the locomotive AIMs score (LOC score).
Experimental design and statistical analysis
Male mice were used for all experiments except for the experiment using l-DOPA and caffeine in which females were used.
Experiment 1: lesion and response to acute and chronic l-DOPA
Separate cohorts of mice were lesioned by injection of 6-OHDA into the dorsolateral striatum as described above.
Cylinder test.
One cohort of Drd1a-Cre/CK2KO and one cohort of Drd2-Cre/CK2KO and their WT littermates were tested in the cylinder test to determine the responsiveness to l-DOPA. Successively higher l-DOPA doses (2, 5, and 10 mg/kg, combined with benserazide, 12 mg/kg) were injected intraperitoneally. Injections were performed every 5–7 d. Mice were tested in the cylinder test 60 min after injection. Mice were either killed by rapid decapitation, tissue was quickly dissected and lysed for use in Western blotting (anti-TH antibody; 1:1000), or perfused with 4% PFA for IHC (anti-TH antibody 1:500 in PBST, overnight) as described above.
Chronic l-DOPA treatment.
One cohort of Drd1a-Cre/CK2KO and one cohort of Drd2-Cre/ CK2KO and their WT littermates were used to test the effect of chronic l-DOPA (20 mg/kg/benserazide12 mg/kg, i.p., 7 d) on LID. One day after AIMs assessment (performed as described above), mice were reinjected and killed 30 min thereafter, either by decapitation, quick tissue dissection, and lysis for use in Western blotting (anti-TH, anti-Golf, anti-tubulin), or perfused with 4% PFA for IHC using anti-TH (1:1000), anti-pERK (1:300), anti-c-fos (1:300), and anti-PKA phosphosubstrate antibodies (1:200; all in PBS, 1% serum, overnight). IHC analysis was also performed using anti-pERK and anti-choline-acetyltransferase (ChAT; 1:200 in PBS) antibodies.
To ascertain in which striatal cell type phosphorylation occurs, the Drd1a-Cre/CK2KO was bred with a transgenic Drd1a-GFP line for at least six generations where the GFP allele was kept in a heterozygous state. Using these mice, the same chronic l-DOPA experiment was performed for IHC analysis using anti-GFP antibody (1:500) in addition to the phosphospecific antibodies mentioned above.
Slices from three animals per genotype (of Drd1a-Cre/CK2KO or Drd2-Cre/CK2KO mice and their WT littermates) were probed with anti-CK2α (1:400 in PBS) and anti-ChAT antibodies (1:200 in PBS) to determine whether CK2α knock-out occurs in ChAT-positive cells; for both lines ∼100 ChAT+ cells were counted per genotype.
To ensure specificity of the anti-enkephalin antibody, we stained slices from Drd1a-GFP/Drd1a-Cre/CK2KO mice (1:150 in PBS, 1% horse serum, overnight). If the antibody was specific, then no signal was to be expected in GFP-expressing cells. Once this was confirmed, we stained slices from the chronically l-DOPA-treated Drd2-Cre/CK2KO and WT cohort with this antibody to determine whether pERK occurs in indirect projection neurons. Per animal on average 57 Enk+ cells in the dorsal, dorsolateral and mediolateral striatum were unambiguously identified using z-stacks.
In another variation of this experiment, a Drd2-Cre/CK2KO/WT 6-OHDA lesioned cohort was used for chronic caffeine/l-DOPA coadministration and AIMs assessment. Caffeine, (2 mg/kg, i.p.), coadministered with a high dose of 20 mg/kg l-DOPA did not alter LID scores. We therefore tested a low dose of l-DOPA (caffeine 2 mg/kg; l-DOPA 2 mg/kg/benserazide 12 mg/kg). To determine whether altered LID in the caffeine-treated Drd2-Cre/CK2KO/WT cohort correlated with reduced pERK, immunohistochemical analysis was performed using slices derived from the above cohort treated with caffeine 2 mg/kg; l-DOPA 2 mg/kg /benserazide 12 mg/kg, perfused 30 min after injection.
For all analyses (behavioral and IHC), except for Fig. 1, animals with a TH depletion >70% in the DL striatal area compared with unlesioned side were excluded.
Acute l-DOPA treatment.
A separate cohort of 6-OHDA lesioned Drd1a-Cre/Drd1-GFP/CK2KO and WT mice was acutely treated with l-DOPA (20 mg/kg, i.p./benserazide12 mg/kg, i.p.) and perfused 30 min thereafter. The same antibodies were used for IHC as in the chronic cohort. Similarly, a separate cohort of Drd2-Cre/CK2KO and WT mice was lesioned and acutely treated with l-DOPA, before perfusion 30 min thereafter, followed by IHC.
Statistical analysis.
Statistical analysis was performed using two-way ANOVA with Bonferroni's post hoc comparisons for cylinder tests and unpaired Student's t tests for Western blot and IHC comparisons of Figure 1. Statistical significance was set at p = 0.05 for this and all following experiments. Data are presented as mean ± SEM for all figures.
Statistical analyses of AIMs behavior were performed using Student's t test for (Fig. 2A–F) and two-way ANOVA with Newman–Keuls post hoc comparisons for the time course experiments (Fig. 2D,G). For IHC analyses of phosphorylation status (pERK, pPKA substrate) and c-fos, two-way ANOVA with Tukey's post hoc comparisons was used when two genotypes (WT/KO) and treatment (unlesioned/lesioned) were compared.
Unpaired two-tailed Student's t test was used when WT was compared with KO independently of treatment (Figs. 4D, 5G, 6D).
Experiment 2: haloperidol-induced catalepsy
The catalepsy in response to haloperidol was tested in unlesioned animals of Drd1a-Cre/CK2KO and Drd2-Cre/CK2KO lines. Before the test, mice were injected intraperitoneally with 0.75 mg/kg haloperidol [dissolved with 3% acetic acid in saline, pH adjusted to 5.2 with NaOH (1 m) and sonicated for 10 min]. Mice were tested 30, 60, 90, and 120 min after injection. Catalepsy was assessed by gently placing the paws of the mice on a 4.5 cm high plastic bar of 3 mm diameter. The latency of the mice staying on the bar with both forelimbs extended was measured. Catalepsy was considered to end when both forelimbs were removed from the bar or when the mice engaged in exploratory behavior (strong head movements). A cutoff time of 5 min was imposed.
To determine a potential involvement of the adenosine receptor family, we coadministered caffeine (0.5 mg/kg, in saline) or saline alone with haloperidol (0.75 mg/kg) to the Drd2-Cre/CK2KO/WT cohort and tested mice 30 min thereafter.
Statistical analysis.
Two-way ANOVA with Tukey's post hoc comparisons.
Experiment 3: effect of CK2 inhibitor CX4945
For this experiment, 4- to 5-month-old WT male mice (C57BL/6J) were used. Mice were injected with CX4945 (in 0.9% saline) and killed 30 or 120 min thereafter. To preserve the phosphorylation status of proteins, mice were quickly decapitated and heads dipped in liquid nitrogen until frozen before dissection and lysis in 1% SDS plus phosphatase inhibitors. Tissue was sonicated 3× for 10 s and boiled for 5 min. 40 μg of lysates, quantified by the BCA assay, were separated by SDS-PAGE and phosphorylation levels of pS473Akt and pS129Akt assessed by Western blotting. Three experiments were performed with two animals per treatment, one experiment is shown. For the AIMs assessment, a cohort of WT mice was lesioned by 6-OHDA injection into the medial forebrain bundle as described, and chronically treated for 10 d with l-DOPA (3 mg/kg)/benserazide (10 mg/kg). On day 11 CX4945 (100 mg/kg, i.p.), was pre-injected 15 min before l-DOPA administration and AIMs scored.
Statistical analysis.
Two-way ANOVA.
Results
Effects of 6-OHDA lesion in Drd1a-Cre/CK2KO and Drd2-Cre/ CK2KO mice
To generate a hemi-parkinsonian rodent model, we applied unilateral striatal 6-OHDA lesions to conditional CK2 knock-out mice where CK2 had been ablated by crossing cell type-specific Cre driver lines (Drd1a-Cre+/− or Drd2-Cre+/−) with CK2αloxP/loxP mice. Drd1a-Cre−/− or Drd2-Cre−/− littermates served as controls. We first asked whether the sensitivity to 6-OHDA may be affected by the conditional knock-out of CK2. To this end, we measured the striatal expression of TH in both hemispheres of 6-OHDA-lesioned mice using Western immunoblotting (Fig. 1A,C,E) or IHC (Fig. 1B,D,F). We did not detect any significant differences in TH expression between the two types of KO animals and WT controls by Western immunoblotting or IHC (p > 0.05 for all); neither did we observe a difference in survival rates after lesion between the genotypes. To ascertain lesion severity using a behavioral endpoint, we assessed asymmetries in spontaneous forelimb use in the cylinder test (where the degree of limb use asymmetry correlates with the severity of striatal DA denervation; Boix et al., 2015). The 6-OHDA lesion induced a similar reduction in contralateral forelimb use in all genotypes (Fig. 1G,H; at baseline).

6-OHDA lesion of conditional CK2α KO mice. Total striatal protein lysates (30 μg) from Drd1a-Cre/CK2KO and control 6-OHDA lesioned mice were separated by SDS/PAGE. Immunoblotting analysis with anti-TH antibody was performed and quantified (A, C). Another cohort of animals was used for immunohistochemical analysis using anti-TH antibody (B). Quantification derived from ImageJ density measurements is shown (D). Western blotting and IHC analysis for WT and Drd2-Cre/CK2KO knock-out is depicted in (E) and (F), respectively. The anti-akinetic responses to augmenting doses of l-DOPA were measured in the cylinder test for Drd1a-Cre/CK2KO/WT (G) and Drd2-Cre/CK2KO/WT (H). Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. n = 11–12 (C), n = 13–15 (D), n = 15–17 (E), n = 16 (F), n = 10–11 (G), n = 9–10 (H).
We next asked whether the disuse of the parkinsonian limb could be ameliorated with a therapeutic dose of l-DOPA (Francardo et al., 2011). Mice were thus tested 1 h after the administration of l-DOPA (2, 5, and 10 mg/kg). This treatment dose-dependently improved limb use asymmetry in the WT controls of both KO lines (Fig. 1G,H, empty bars; p < 0.01 for the highest l-DOPA dose vs vehicle in the WT of the Drd1a-Cre/CK2KO and p < 0.05 for the highest l-DOPA dose vs vehicle in the WT of the Drd2-Cre/CK2KO). When treated with l-DOPA, Drd1a-Cre/CK2KO mice showed a trend toward increased contralateral paw use, but the effect did not reach significance. Accordingly, the percentage of contralateral limb use with 10 mg/kg l-DOPA was significantly lower in Drd1a-Cre/CK2KO mice relative to their WT controls (Fig. 1G; p < 0.05). The main effect of dose was significant: F(3,57) = 10.64, p < 0001; and the main effect of genotype was at the cusp of significance: F(1,57) = 4.33, p = 0.05. Mice with CK2 knock-out in D2-MSNs showed a response to l-DOPA and their performance was significantly improved already at the dose of 5 mg/kg (p < 0.05), whereas the effect of this dose was not significant in WT littermates (Fig. 1H). After treatment with 10 mg/kg l-DOPA, contralateral forelimb use tended to be larger in Drd2-Cre/CK2KO mice relative to their WT controls (Fig. 1H), but this difference did not reach significance. The main effect of dose was significant: F(3,45) = 15.04, p < 0001; and the main effect of genotype did not reach significance: (F(1,45) = 1.66, p = 0.21).
Dyskinesia is reduced in Drd1a-Cre/CK2KO and enhanced Drd2-Cre/ CK2KO mice
It is well established that 6-OHDA-lesioned mice rapidly develop AIMs when treated with l-DOPA (Francardo et al., 2011), and ALO AIMs provide a valid behavioral measure of LID in this animal model (Lundblad et al., 2005; Francardo et al., 2011). 6-OHDA-lesioned KO mice were treated for 7 d with a dose of l-DOPA sufficient to induce AIMs (20 mg/kg; Lundblad et al., 2005), and a dyskinesia rating session was performed on the last day of treatment. ALO scores were significantly lower in Drd1aCre/CK2KO than in WT (Fig. 2A; p = 0.005). LOC scores, which correlate with the extent of contralateral rotation (Lundblad et al., 2002), were also significantly lower (Fig. 2B; p = 0.036). Accordingly, the total AIM score was significantly reduced in Drd1aCre/CK2KO compared with controls. This was apparent both on the sum of AIM scores per session (Fig. 2C; p = 0.006) and when comparing the AIM scores per monitoring period (Fig. 2D), the main effect of genotype was significant: F(1,198) = 56.35, p < 0.0001.
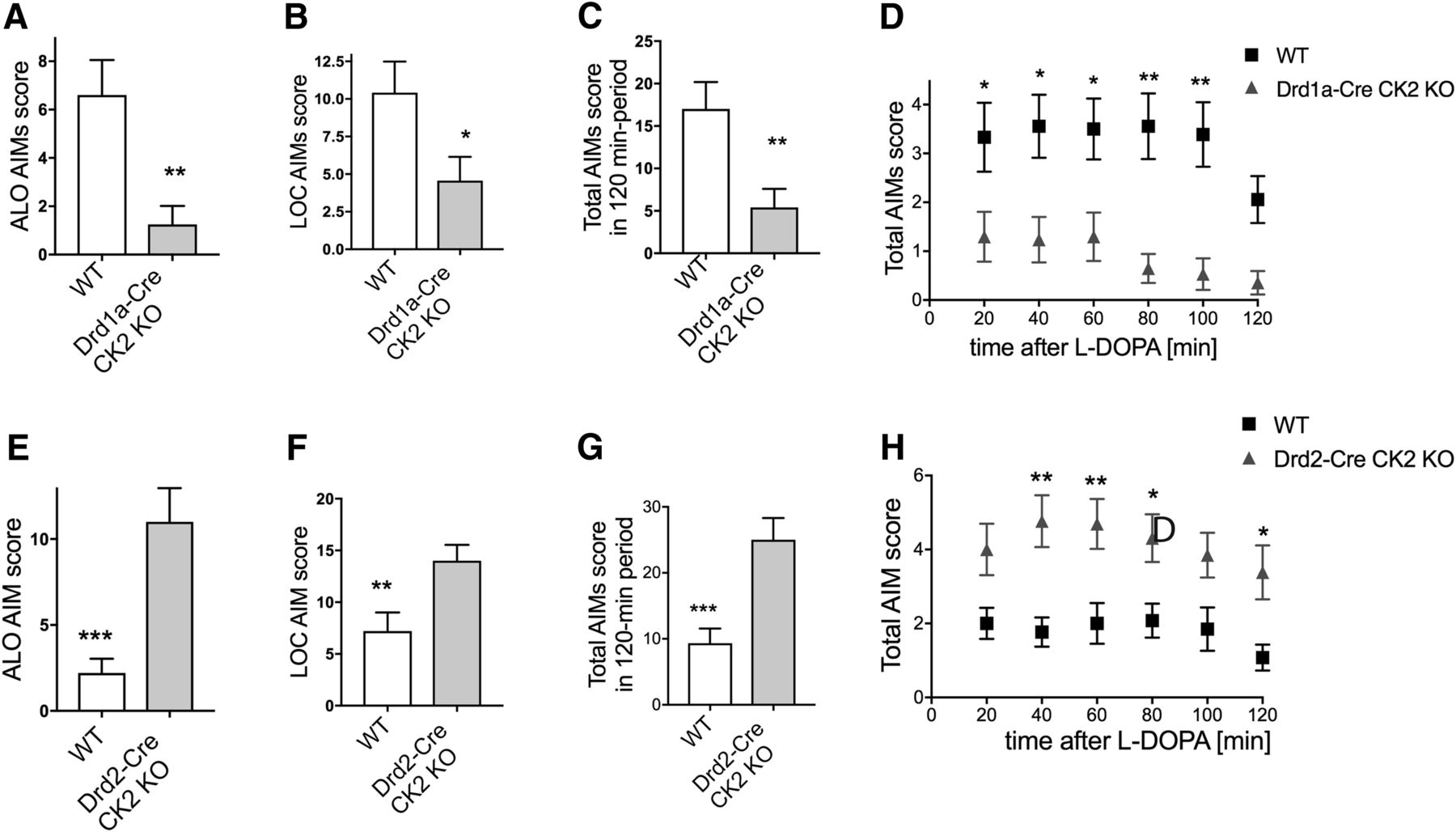
LID in conditional CK2α KO mice. Drd1a-Cre/CK2KO and Drd2-Cre/CK2KO and corresponding WT littermates were treated with l-DOPA/benserazide for 7 d. ALO score (A, E), LOC score (B, F), and total AIMs scores were determined and plotted as summarized score (C, G) or as time course at 20 min intervals for 2 h after l-DOPA injection (20 mg/kg; benserazide 12 mg/kg, i.p.; D, H). Data are mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001. n = 17–18 (A–D), n = 13–15 (E–H).
The development of LID was studied also in Drd2-Cre/CK2KO mice using the same experimental design. After 7 d of treatment with l-DOPA (20 mg/kg/d), ALO AIM scores were significantly elevated in the KO mice compared with their controls (Fig. 2E; p = 0.0002 for KO vs controls). In addition, locomotive scores (Fig. 2F; p = 0.0092) and total scores differed significantly between KO and controls (Fig. 2G), p = 0.0004. When total AIM scores per monitoring period were compared (Fig. 2H), there was a main effect of genotype: F(1,144) = 49.79, p < 0.0001. These data show that, KO of CK2 in D1 and D2 receptor-expressing cells has opposing effects on LID development in the 6-OHDA lesion model.
ERK but not PKA substrate phosphorylation is reduced in the dopamine-depleted striatum of Drd1a-Cre/CK2KO mice after chronic l-DOPA
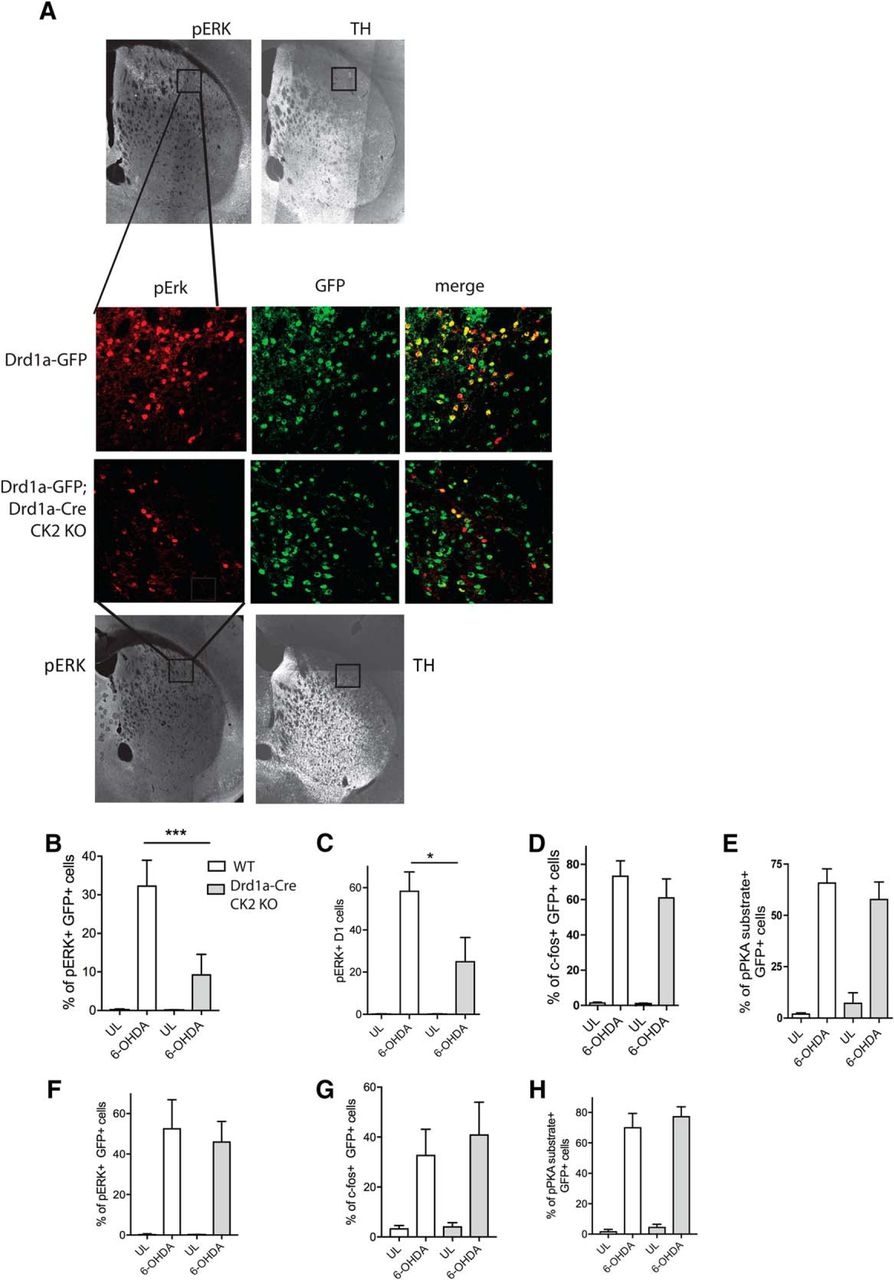
Treatment with l-DOPA activates ERK signaling in the DA-depleted striatum (Gerfen et al., 2002; Pavón et al., 2006), and the levels of phosphorylated ERK correlate with dyskinesia severity (Westin et al., 2007). It was further shown that ERK-dependent plasticity is responsible for the aberrant response to chronic l-DOPA (Cerovic et al., 2015). Moreover, inhibition of striatal ERK signaling improves LID (Santini et al., 2007; Fasano et al., 2010). We used IHC and confocal microscopy to examine the cellular expression of pERK in the dorsolateral striatum. We first used Drd1a-Cre/CK2KO mice crossed with the Drd1a-GFP-reporter mouse line to verify that l-DOPA-induced pERK occurs in D1 but not D2 neurons, as previously reported in this animal model of LID (Santini et al., 2009). The induction of pERK by l-DOPA in GFP-positive cells was significantly attenuated in Drd1a-Cre/CK2KO mice compared with control mice, expressed as percentage of pERK-positive D1 projection neurons as well as the average total number of pERK-positive+ cells per image (Fig. 3A–C), correlating with a behavioral phenotype of reduced dyskinesia severity. Statistically, there was an interaction genotype × treatment for the percentage of pERK in GFP+ cells (F(1,27) = 7.3915, p = 0.011). Adjacent coronal sections were immunostained for c-Fos, a marker of immediate-early genes induced by l-DOPA in DA-depleted striata. Another set of sections was immunostained using an antibody that recognizes the phosphorylated form of the PKA consensus substrate peptide, which provides an indirect estimate of PKA activity (Alcacer et al., 2012). We did not detect significant differences between WT and Drd1a/CreKO on either marker (Fig. 3D,E). Statistically, there was no significant interaction genotype × treatment for c-fos (F(1,20) = 0.491, p = 0.492) or pPKA substrate (F(1,26) = 1.236, p = 0.276). When unilaterally lesioned Drd1a/CreKO and WT mice are acutely treated with l-DOPA, we observe a strong response in ERK phosphorylation in direct pathway neurons, however, no difference between WT and KO in pERK, c-Fos, and pPKA substrate is detected (p > 0.05 for all).

l-DOPA-dependent ERK1/2 phosphorylation in D1-MSNs is reduced in the Drd1-Cre/Drd1a-GFP/CK2KO mice. IHC analysis of the dorsolateral striatum (lesioned side) of coronal slices from WT and Drd1-Cre/Drd1a-GFP/CK2KO mice after l-DOPA treatment (20 mg/kg; benserazide 12 mg/kg, i.p. for 7 d) using pERK antibody (pT202/Y204) and anti-GFP antibody (A). Quantification of immunofluorescence using antibodies directed toward GFP and pERK (B, C), c-fos (D), and pan-pPKA substrate (E). Scale bar, 100 μm. Quantification of immunofluorescence using antibodies directed toward GFP and pERK (F), c-fos (G), and pan-pPKA substrate (H) after acute l-DOPA (20 mg/kg; benserazide 12 mg/kg, i.p.). Data are mean ± SEM. *p < 0.05, ***p < 0.001. n = 6–7 (B–E), n = 8–10 (F–H).
It has been described that ERK phosphorylation also occurs in cholinergic interneurons after chronic (but not acute) l-DOPA administration, and that muscarinic receptor antagonists reduce the development and/or expression of LID both in 6-OHDA lesioned mice and the aphakia mouse model of PD (Ding et al., 2011). We were thus interested in assessing ERK phosphorylation in ChAT-immunoreactive neurons. Drd1a-Cre/CK2KO mice showed a much reduced expression of pERK levels in ChAT cells (Fig. 4A,B), and there was a significant genotype × treatment interaction (F(1,21) = 18.16, p = 0003). To better understand these findings, it was imperative to assess whether CK2 expression in ChAT cells is ablated by Drd1a-Cre or Drd2-Cre. In agreement with published data (Le Moine et al., 1991), we found that CK2 expression is not affected in ChAT cells of Drd1a-Cre/CK2KO mice (Fig. 4C). The percentage of CK2-expressing ChAT cells was determined as 89.4% (±1.5%) for the Drd1a-Cre/CK2KO 88.2% (±2.6%) for the WT mice (p = 0.687; Fig. 4D).

l-DOPA-dependent ERK1/2 phosphorylation in ChAT interneurons is reduced in the Drd1a-Cre/CK2KO mice. Immunohistochemical analysis of the lesioned dorsolateral striatum of coronal slices from WT and Drd1a-Cre/CK2KO mice after l-DOPA treatment (20 mg/kg; benserazide 12 mg/kg, i.p. for 7 d) using pERK (pT202/Y204) and ChAT antibodies (A) and quantification thereof (B). Arrows point to pERK+ cells. Scale bar, 100 μm. IHC analysis using anti-CK2α, anti-GFP, and anti-ChAT antibodies of dorsolateral sections of Drd1-Cre/Drd1a-GFP/CK2KO mice (C). Representative images are shown, arrowheads point to ChAT neurons and arrow to D1-neurons, 3 animals per genotype were analyzed and quantified (D). Data are mean ± SEM. ***p < 0.001. n = 6–7 (B), n = 3 (D).
ERK but not PKA substrate phosphorylation is elevated in the dopamine-depleted striatum of Drd2-Cre/CK2KO mice after chronic l-DOPA
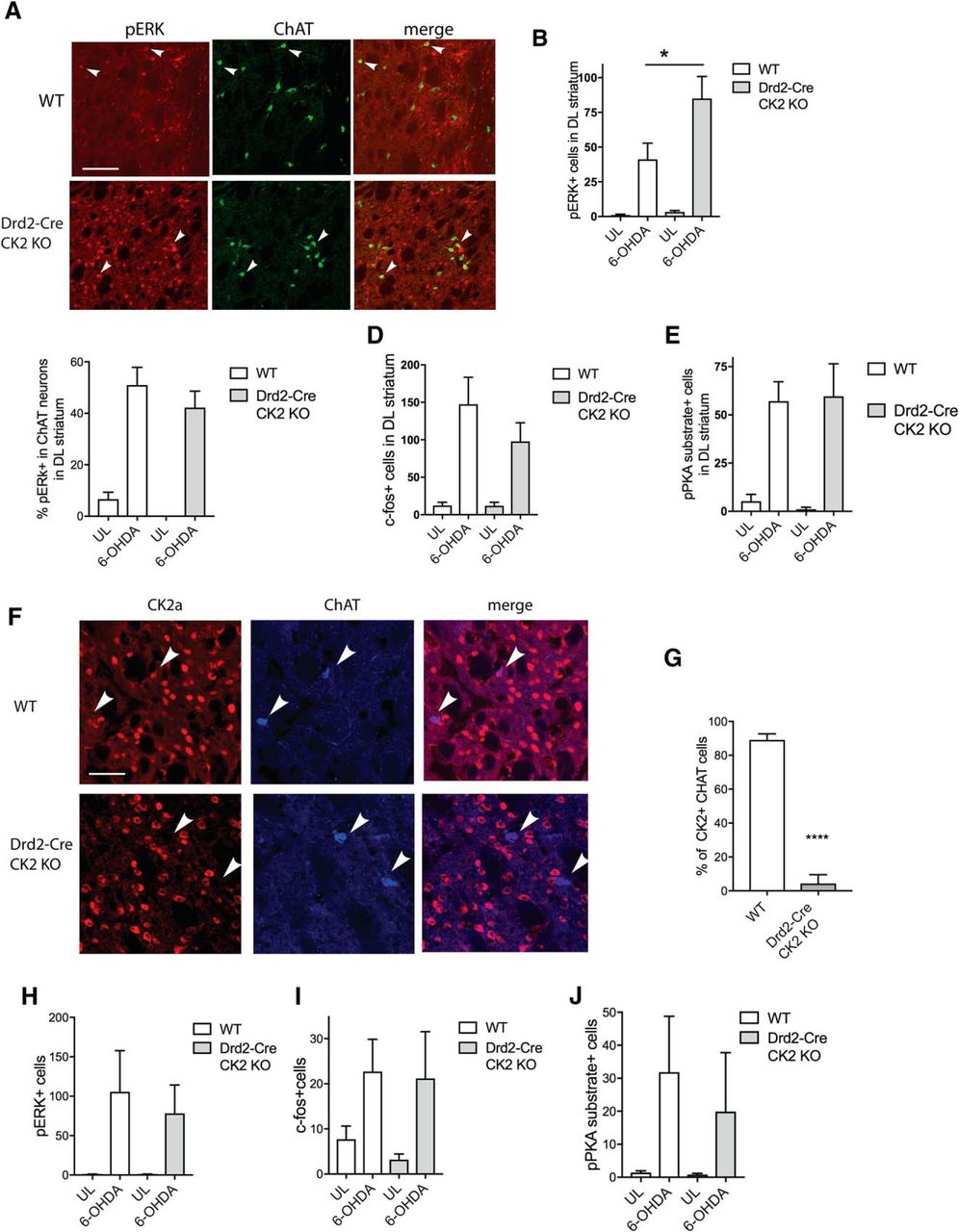
Next, we determined whether pERK levels were affected in the conditional Drd2-Cre/CK2KO mice after chronic l-DOPA treatment. Interestingly, the total number of pERK-positive cells in the 6-OHDA-lesioned striatum was twofold larger in Drd2-Cre/CK2KO mice than WT controls (Fig. 5A,B). Statistically, there was a genotype × treatment interaction (F(1,54) = 4.478, p = 0.039).

l-DOPA-dependent ERK1/2 phosphorylation is reduced in the Drd2/CreKO mice. IHC analysis of the lesioned dorsolateral striatum of coronal slices from WT and Drd2-Cre/CK2KO mice after l-DOPA treatment (20 mg/kg; benserazide 12 mg/kg, i.p. for 7 d) using antibodies against pERK (pT202/Y204) and ChAT, arrowheads point towards pERK+ and ChAT+ cells (A). Scale bar, 100 μm. Quantification of immunofluorescence of pErk1/2-positive cells (B). Quantification of immunofluorescence of cells that are positive for pErk1/2 and ChAT normalized by the total number of ChAT-positive neurons (C). Quantification of cells that are c-Fos-positive (D) or pPKA substrate-positive (E). IHC analysis using anti-CK2α and anti-ChAT antibodies of dorsolateral sections of Drd2-Cre/CK2KO mice. Representative images are shown (F), three animals per genotype were analyzed and quantified (G). Quantification of immunofluorescence using antibodies directed toward GFP and pERK (H), c-fos (I), and pan-pPKA substrate (J) after acute l-DOPA (20 mg/kg; benserazide 12 mg/kg, i.p.). Data are mean ± SEM. ****p < 0.0001. n = 7–8 (B–E), n = 3 (G), n = 5–7 (H–J).
The number of ChAT/pERK-positive neurons did not differ between Drd2-Cre/CK2KO and controls (Fig. 5C) and the interaction genotype × treatment was not significant (F(1,48) = 0.034, p = 0.854). An analysis of sections immunostained for c-Fos and PKA phosphosubstrates did also not detect differences between the two genotypes (Fig. 5D,E), with F(1,45) = 0.853, p = 0.361 and F(1,30) = 0.083, p = 0.775, respectively. We determined that CK2 expression was ablated in ChAT neurons of the Drd2-Cre line, confirming previous data indicating the expression of D2 receptor in cholinergic neurons (Fig. 5F; Aubry et al., 1993). The percentage of CK2-expressing ChAT cells was quantified as 4.3% (±3.1%) for Drd2-Cre/CK2KO and 89.1% (±2.1%) for WT mice, p < 0.0001 WT versus KO (Fig. 5G). This indicates that pERK is activated in these ChAT-positive neurons despite the absence of CK2.
When unilaterally lesioned Drd2-Cre/CK2KO and WT mice were acutely treated with l-DOPA, we did not detect differences between WT and KO in pERK, c-Fos, and pPKA substrate (Fig. 5H–J) or statistically significant interactions (genotype × treatment).
We determined that in the Drd2/CreKO 49.0% (±1.7%) of all striatal cells are depleted of CK2α, indicating that CK2α is depleted in all D2 striatal neurons (data not shown). We wanted to address whether the enhanced pERK signal occurs in the cells depleted of CK2 or whether, as has been described by others, in D1 striatal projection neurons (Gerfen et al., 2002; Santini et al., 2009). To this end, we stained coronal slices from chronically treated mice with anti-enkephalin antibody, a marker of D2 projection neurons. In tile scans, using this antibody, the staining reflects the in situ data (Allen Brain Atlas, RP_060315_01_A07; Fig. 6A). We were able to identify D2 projection neurons by their lack of GFP expression in slices from Drd1a-GFP; Drd1a-Cre/CK2KO mice (Fig. 6B). In slices derived from l-DOPA treated Drd2-Cre/CK2KO, elevated pERK signal was not detected in D2 projection neurons (Fig. 6C). In WT and Drd2-Cre/CK2KO mice no difference in pERK expression was detected: a very low percentage of 2% (±1.5%) and 0.9% (±0.3%) of Enk+ cells expressed pERK in WT and Drd2-Cre/CK2KO, respectively (Fig. 6D; p = 0.265), indicating that the vast majority of ERK phosphorylation must occur in striatonigral neurons. Our findings confirm work by other groups that had shown that these D1R-expressing striatonigral cells are hyper-responsive to l-DOPA and allow us to conclude that lack of CK2 (in D2 projection neurons) has an indirect, non-cell autonomous effect on D1 neurons (Gerfen et al., 2002).

Characterization of anti-enkephalin antibody in the striatum. Tile scan of a coronal slice stained with the Enk antibody (A). Immunohistochemical analysis of the dorsolateral striatum of coronal slices from WT mice that express GFP in direct projection neurons using Enk and GFP antibodies (B). Arrowheads indicate D1 cells that always lack Enk signal. Arrows indicate cells that can be identified as Enk-expressing cells. All of these cells lack GFP signal. Immunohistochemical analysis of the dorsolateral striatum of coronal slices from Drd2/CreKO and WT mice using Enk and pERK antibodies (C). Arrowheads indicate cells that express enkephalin; arrows show pERK-positive cells. Scale bar, 50 μm. Quantification of percentage pERK+ cells that are also Enk+ in Drd2/CreKO and WT (D). Data are mean ± SEM. n = 3 (B), n = 6–8 (D).
Haloperidol-induced catalepsy is altered in opposing ways in Drd1a-Cre/CK2KO and Drd2-Cre/CK2KO mice
Haloperidol is a prototype of antipsychotic drugs that induce extrapyramidal side effects including catalepsy, manifested as extreme hypolocomotion and rigid immobility (Miyamoto et al., 2005). Haloperidol-induced catalepsy has been proposed to result from inhibition of striatal postsynaptic D2 receptors resulting in overactivity of the indirect pathway and decreased movement (Boulay et al., 2000). Antagonists of D1-type receptors (e.g., SCH23390) can also induce catalepsy by reducing the activity of direct pathway neurons (Undie and Friedman, 1988).
To test the involvement of CK2 expressed by D1- or D2-positive neurons in this extrapyramidal disorder, we treated Drd1a-Cre/CK2KO and Drd2-Cre/CK2KO mice with haloperidol (0.75 mg/kg) and quantified catalepsy using the standard bar test (Boulay et al., 2000). Drd1a-Cre/CK2KO mice removed their forelimbs from the bar almost immediately (Fig. 7A), with a main effect of genotype (F(1,54) = 13.49, p = 0.0006). However, in Drd2-Cre/CK2KO, the latency to leave the bar was strongly enhanced (Fig. 7B), the main effect of genotype was significant (F(1,39) = 58.97, p < 0.0001). A2A antagonists have been shown to counteract catalepsy induced by D1 or D2 receptor antagonists (Hauber et al., 2001). Thus, we hypothesized that the elevated catalepsy in Drd2-Cre/CK2KO mice may be mediated by enhanced A2A signaling. To test this hypothesis, we coadministered haloperidol with the A1/A2A antagonist, caffeine. This cotreatment abolished the elevated catalepsy in Drd2-Cre/CK2KO (Fig. 7C) but had no significant effect on the WT group. Statistically, there was a genotype × treatment interaction (F(1,20) = 20.92, p = 0.0002). We had previously determined that caffeine alone, at a dose of up to 2 mg/kg, does not affect locomotor behavior in these mice. Our results confirm that A2AR signaling is elevated in Drd2-Cre/CK2KO mice, engendering a hyperactivity of indirect pathway neurons. Accordingly, caffeine reduces the haloperidol-induced overactivation of indirect pathway neurons preferentially in the Drd2-Cre/CK2KO, rescuing their phenotype.

Haloperidol-induced catalepsy is altered in opposing directions in Drd1a-Cre/CK2KO and Drd2-Cre/CK2KO mice. Latency to remove paws from bar was assessed at three time points (30, 60, and 120 min after haloperidol injection) for Drd1a-Cre/CK2KO (A) and for Drd2-Cre/CK2KO (B). Caffeine (0.5 mg/kg) was injected at the same time than haloperidol and latency to remove paws from bar was measured at 120 min (C). Data are mean ± SEM. *p < 0.05, ***p < 0.001. n = 10–11 (A), n = 7–8 (B, C).
Caffeine has anti-dyskinetic effects in Drd2-Cre/CK2KO mice
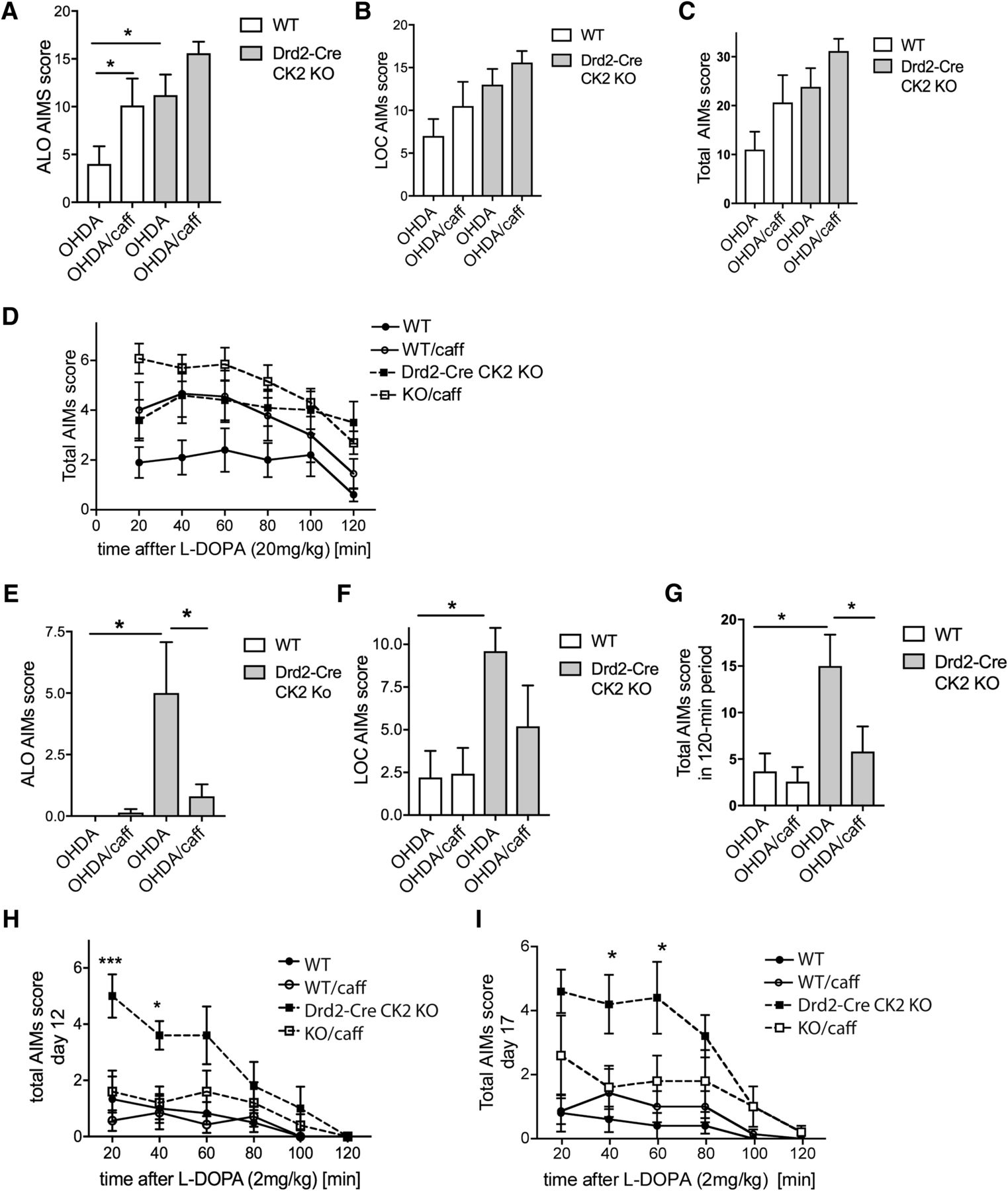
Our results using haloperidol and caffeine prompted the question, whether the enhanced LID score in the Drd2-Cre/CK2KO mice may be caused by elevated A2AR expression and/or signaling. To address this question, we coadministered caffeine with l-DOPA to unilaterally lesioned Drd2-Cre/CK2KO mice and controls. We used caffeine at a dose of 2 mg/kg, intraperitoneally because we had previously shown that at this dose locomotion was not affected. Caffeine antagonizes both A2A and A1 receptors with comparable affinity (Fredholm and Lindström, 1999; Fisone et al., 2004), and it has been shown that at the same dose of 2 mg/kg, administered intraperitoneally, the behavioral phenotype of enhanced wakefulness was completely abolished in conditional A2A receptor KO mice, indicating that at this dose the A2A receptor must be sufficiently affected (Lazarus et al., 2011). Unexpectedly, caffeine with l-DOPA (20 mg/kg) enhanced the ALO score of the WT mice while it did not significantly affect the score of Drd2-Cre/CK2KO mice (Fig. 8A). Statistically, there was an effect of treatment (F(1,36) = 7.109, p = 0.011) and genotype (F(1,36) = 10.32, p = 0.028) and but no interaction genotype × treatment (F(1,36) = 0.1953, p = 0.661). Locomotor AIMs scores were not significantly altered in the presence of caffeine in either genotype (Fig. 8B). Although there was a statistically significant effect of genotype (F(1,36) = 7.981, p = 0.008), there was no interaction genotype × treatment (F(1,36) = 0.055, p = 0.817). Similarly, total AIMs scores were not significantly altered in the presence of caffeine in either genotype (Fig. 8C,D). Statistically, there was no interaction genotype × treatment (F(1,32) = 0.084, p = 0.775) but an effect of genotype (F(1,32) = 8.689, p = 0.006). It was shown that low doses of caffeine exerted anti-dyskinetic effects, whereas higher doses have a motor stimulant action that may exacerbate LID (Xiao et al., 2011). Previous rodent studies have reported that the anti-dyskinetic effects of A2AR antagonists were achieved when AIMs were induced with low doses of l-DOPA (Pinna et al., 2001; Xiao et al., 2011). We thus tested the effects of caffeine on AIMs induced by a low dose of l-DOPA (2 mg/kg). Due to the low dose of l-DOPA used, treatment was performed over a longer time period and AIMs development tested at three time points (5, 12, 17 d). On all days tested, caffeine had a particularly strong effect on ALO AIMs in the Drd2Cre/CK2KO, abolishing the difference between the genotypes (Fig. 8E). Statistically, there was a genotype × treatment interaction (F(1,18) = 4.99, p = 0.038). Neither for WT nor for the Drd2Cre/CK2KO did caffeine affect locomotor scores significantly (Fig. 8F). There was an effect of genotype (F(1,18) = 8.454, p = 0.009) but not treatment (F(1,18) = 1.422, p = 0.249), and no interaction genotype × treatment (F(1,18) = 1.751, p = 0.202). The total AIMs score was reduced in Drd2-Cre/CK2KO but not WT mice (Fig. 8G,H). Statistically, there was an interaction genotype × treatment (F(1,18) = 4.76, p = 0.043) as well as a treatment effect (F(1,18) = 7.03, p = 0.016) and genotype effect (F(1,18) = 7.221, p = 0.015). We found that the effect of caffeine was preserved up to 17 d (Fig. 8I) and was already present after 5 d (data not shown).

LID of Drd2-Cre/CK2KO mice in the presence of caffeine. 6-OHDA lesioned Drd2-Cre/CK2KO mice were treated with l-DOPA/benserazide (20 mg/kg/12.5 mg/kg, i.p.) and caffeine (2 mg/kg, i.p.) for 7 d. ALO score (A), LOC score (B), and total AIMs scores were determined and plotted as summarized score (C) or at 20 min time points over 2 h after l-DOPA injection (D). 6-OHDA lesioned Drd2-Cre/CK2KO or WT mice were treated with low-dose l-DOPA/benserazide/caffeine (2 mg/kg/12.5 mg/kg/2 mg/kg, i.p.) for 17 d and tested at 5, 12, and 17 d. ALO score (E), LOC score (F), and total summarized AIMs scores (G) or total AIMs scores plotted over time (H) after 12 d are shown. 17 d of treatment do no significantly alter the total AIMs scores (I), Data are mean ± SEM. *p < 0.05, ***p < 0.001. n= 8–12 (A–D). n = 5–7 (E–I). caff, Caffeine.
Because the elevated AIMs in the Drd2-Cre/CK2KO can be normalized by caffeine, we hypothesized that striatal pERK levels may be reduced, too. This was indeed the case (Fig. 9A). Upon treatment with 2 mg/kg l-DOPA, Drd2-Cre/CK2KO mice receiving vehicle cotreatment showed a strong upregulation of pERK, which was completely prevented by caffeine cotreatment (Fig. 9B). Statistically, there was a genotype × treatment interaction (F(1,19) = 4.929, p = 0.038).

ERK1/2 phosphorylation is normalized in the presence of caffeine in Drd2-Cre/CK2KO mice. IHC analysis of dorsolateral striatum of coronal slices from WT and Drd2-Cre/CK2KO mice after l-DOPA treatment [l-DOPA/benserazide 2 mg/kg/12.5 mg/kg, with or without caffeine (2 mg/kg, i.p.); A]. Quantification of immunofluorescence of pERK1/2-positive cells (B). Quantification of immunofluorescence of cells that are positive for pERK1/2 and ChAT normalized by the total number of ChAT-positive neurons (C). Scale bar, 100 μm. Data are mean ± SEM. *p < 0.05. n = 6–7 per group. caff, Caffeine.
Caffeine did not affect pERK expression in ChAT-positive cells (Fig. 9C) and there was no significant interaction genotype × treatment (F(1,19) = 0.240, p = 0.630).
Effect of CK2 inhibitor CX4945 on LID
To determine whether acute inhibition of CK2 affects LID in unilaterally lesioned WT mice, we chose to use CX4945, the only known CK2 inhibitor that is bioavailable and has been shown to have an effect on glioblastoma growth in vivo (Zheng et al., 2013). We performed several dose course experiments that lead us to determine that only at very high doses (75 or 150 mg/kg, i.p.), there is a reduction in CK2 phosphorylation site on Akt (S129), accompanied by a dephosphorylation of pS473Akt (Fig. 10A). However, these doses lead to rigidity in the animals for ∼15 min post-injection, suggesting that the use of this inhibitor is not ideal for behavioral experiments. We have injected CX4945 (100 mg/kg) 15 min before l-DOPA injection to 6-OHDA-lesioned medial forebrain bundle lesioned, dyskinetic mice. No significant difference in the LID scores was observed (Fig. 10B,C), and no interaction of time × treatment could be detected (F(5,84) = 0.195, p = 0.984 for ALO and F(5,84) = 0.140, p = 0.982 for LOC AIMs scores). Future experiments will be performed to unambiguously answer the relation of acute CK2 inhibition and LID, to ensure that sufficient CK2 inhibition occurs and that phosphorylation site/s relevant for the LID phenotype is/are dephosphorylated.

AIMs in presence of CK2 inhibitor CX4945. Western blot analysis of lysates derived from mice acutely treated with CX4945 by interaperitoneal injection (A). Mice were lesioned with 6-OHDA targeted to the MFB and treated for 10 d with l-DOPA 3 mg/kg/benserazide (10 mg/kg). Mice were pre-injected with CX4945 (100 mg/kg, i.p.) 15 min before l-DOPA administration. LOC and ALO AIMs scores were assessed (B, C). Data are mean ± SEM. n = 8 per group.
Gαolf expression
We have previously shown that CK2 interacts with the G-protein Gαolf (Rebholz et al., 2009), the major G-protein in the striatum mediating cAMP/PKA signaling in response to D1 receptor stimulation. Moreover, Gαolf expression can be modulated by DA depletion and l-DOPA treatment (Corvol et al., 2007). We therefore tested whether striatal levels of Gαolf were altered in the conditional CK2 KO mice following 6-OHDA lesion and l-DOPA treatment. In Drd1a-Cre/CK2KO mice treated with l-DOPA, Gαolf levels were reduced in the DA-depleted hemisphere but unaltered in the contralateral side (Fig. 11A,B), p = 0.039 for Golf unlesioned versus lesioned side of the Drd1a-Cre/CK2KO and p = 0.983 for the WT. In contrast, in the Drd2-Cre/CK2KO mice, Gαolf levels were not changed under any of the experimental conditions examined (Fig. 11C,D).

Golf levels are reduced in the Drd1a-Cre/CK2KO. Total striatal protein lysates (40 μg) from lesioned and unlesioned hemispheres of Drd1a-Cre/CK2KO and WT mice were separated by SDS/PAGE. Immunoblotting analysis with anti-Golf (A) was performed and quantified (B). Lysates from Drd2-Cre/CK2KO and WT mice were similarly analyzed (C, D). Quantification derived from ImageJ density measurements is shown. Animals were treated with l-DOPA/Benserazide for 7 d (D). Data are mean ± SEM. *p < 0.05. n = 10–11 (B), n = 5–7 (D).
Discussion
Our study investigates the role of CK2 in the development of LID in the mouse 6-OHDA lesion model of PD. Knock-out of CK2α in projection neurons of the direct pathway reduced dyskinesia, whereas knock-out of CK2α in neurons forming the indirect pathway enhanced LID. These results reveal that CK2 regulates signaling events critical to LID in each of the two main populations of striatal neurons.
Dyskinesia and striatal signaling in Drd1a-Cre/CK2KO mice
The reduced LID severity in Drd1a-Cre/CK2KO was at first surprising because we had previously shown that reduction or ablation of CK2 activity/expression enhances D1 receptor signaling (Rebholz et al., 2009, 2013). A preliminary indication for a reduced sensitivity to l-DOPA in these conditional KO mice came from the assessment of contralateral paw use in the cylinder test. Whereas at low doses of l-DOPA (2 and 5 mg/kg) no difference in anti-akinetic response between Drd1a-Cre/CK2KO and WT was detected, at 10 mg/kg the KO mice responded to a lesser extent than WT littermates.
Phosphorylation of ERK in D1-MSNs has been linked to LID development (Gerfen et al., 2002; Santini et al., 2007). Other markers indicative of direct pathway hyperactivity in LID involve an elevated phosphorylation of members of the cAMP/PKA/DARPP-32 cascade, and the expression of c-Fos, FosB, and ΔFosB (Asin et al., 1995; Andersson et al., 1999; Cenci et al., 1999; Lopez et al., 2001). Inhibition of PKA (Lebel et al., 2010) and inactivation or knock-out of DARPP-32 (Santini et al., 2007; Bateup et al., 2010) have shown to reduce dyskinetic behavior in rodent models. However, others have shown that although pERK appears to be absolutely required for AIM development, pPKA substrate phosphorylation (including the PKA-mediated phosphorylation GluR1) can be reduced without affecting LID (Alcacer et al., 2012). Our data show that following l-DOPA treatment, levels of ERK phosphorylation in the DA-denervated striatum were reduced in the Drd1a-Cre/CK2KO, in concordance with the reduced LID phenotype, whereas pPKA substrate phosphorylation and c-fos levels were not affected.
Recent studies have revealed an involvement of cholinergic interneurons in LIDs. Prolonged l-DOPA treatment leads to diminished pERK expression in D1 striatal projection neurons and increased expression in ChAT interneurons (Ding et al., 2011). Inhibition or ablation of ChAT interneurons prevents the development of LID (Ding et al., 2011; Won et al., 2014). In Drd1a-Cre/CK2KO mice, ERK signaling in the ChAT neurons was also reduced. We assessed whether the Drd1a-Cre driver ablates CK2α expression also in ChAT neurons, but this was not the case. In the Drd2-Cre/CK2KO mice, however, CK2 was ablated also in the ChAT-immunoreactive neurons. Despite their lacking CK2, these neurons did not exhibit changes in l-DOPA-induced ERK phosphorylation in the Drd2-Cre/CK2KO. Together, these data suggest that the changes in dyskinesia severity and ERK signaling were not primarily driven by cholinergic interneurons. When these neurons appeared affected, the effect possibly depended on network-level changes primarily driven by the spiny projection neurons.
Gαolf, is a rate-limiting component of the dopamine D1 signaling cascade (Corvol et al., 2004, 2007). Levels of Gαolf increase in DA-denervated striatum of PD patients and rodents (Hervé et al., 1993; Marcotte et al., 1994; Penit-Soria et al., 1997; Rangel-Barajas et al., 2011) and are normalized after l-DOPA (Corvol et al., 2004). We have previously shown that CK2 binds to Gαolf (Rebholz et al., 2009). We therefore examined Gαolf striatal levels, and found them reduced in the 6-OHDA-lesioned side in Drd1a-Cre/CK2KO mice after chronic l-DOPA. This finding appears to parallel the reduced severity of LID in the same animals. Gαolf haploinsufficiency has been found not to affect dyskinesia development, or pERK upregulation, in 6-OHDA-lesioned mice treated with l-DOPA (Alcacer et al., 2012). This may suggest that the reduction in Gαolf levels observed in Drd1a-Cre/CK2KO mice does not play a causal role in their lowered susceptibility to LID. In keeping with this interpretation, Drd1a-Cre/CK2KO mice treated with l-DOPA did not show altered levels of PKA phosphosubstrates, which indirectly indicate that the canonical signaling cascade triggered through Gαolf is intact.
On the other hand, our case is different since the changes in Gαolf expression either stem from 6-OHDA and/or l-DOPA and are thus a response to perturbation of the system whereas in the heterozygous mouse the Gαolf levels are already present prenatally. Furthermore, the conditional KO mice are devoid of CK2 activity in either D1 or D2 projection neurons but not in both cell types. Interestingly, it was recently published that upon chronic l-DOPA treatment the negative regulator of Gαs/Gαolf, RGS6, is upregulated, indicating that compensatory mechanisms are elicited to dampen Gαs/Gαolf signaling (Heiman et al., 2014). Thus, the fact that CK2 depletion reduces the G-protein in the lesioned hemisphere in the Drd1a-Cre/CK2KO may still be relevant for our LID phenotype.
CK2 was shown to phosphorylate ERK within its nuclear translocation signal (NLS; Plotnikov et al., 2011). This site, S246, becomes phosphorylated after ERK is activated on T202/Y204, which are the sites examined in the presented work. Therefore, lack phosphorylation within the NLS of ERK due to CK2 KO may negatively affect longer-term adaptive changes to l-DOPA because it is expected that ERK may not be able to translocate sufficiently to the nucleus to promote transcriptional events, thereby possibly affecting the LID phenotype of both CK2 knock-out lines.
Dyskinesia and striatal signaling in Drd2-Cre/CK2KO mice
Genetic ablation of CK2 in D2 striatal neurons resulted in increased LID severity and enhanced striatal activation of ERK signaling in D1 striatal neurons indicating a non-cell-autonomous regulation of this phosphorylation event. These effects may be mediated, at least in part, via A2A receptors, which are highly expressed in indirect pathway striatal neurons.
There is growing evidence for A2A receptors as a promising drug target for the treatment of PD. Human imaging studies have revealed a link between LID and increased A2A signaling (Ramlackhansingh et al., 2011). A2AR antagonists exert potent anti-akinetic effects in animal models (Chen et al., 2001; Pinna et al., 2001; Bibbiani et al., 2003; Joghataie et al., 2004; Matsuya et al., 2007) and are currently being evaluated in clinical trials (Prediger, 2010; Postuma et al., 2012). The specific A2AR antagonist, KW-6002, alleviates parkinsonian symptoms, including dyskinesia, in MPTP lesioned monkeys treated with low-dose l-DOPA (Kanda et al., 1998; Grondin et al., 1999). Our results implicate A2A signaling in the increased LID susceptibility of Drd2a-Cre/CK2KO mice. As others, we do not detect a reduction of LID with adenosine receptor antagonist caffeine when animals are treated with high-dose l-DOPA. However, the high AIM scores induced in Drd2a-Cre/CK2KO mice by a low dose of l-DOPA were completely eliminated by caffeine.
A2A receptors exert a strong excitatory influence on indirect pathway neurons: A2AR activation leads to an elevation of cAMP and thus antagonizes D2 signaling. D2R and A2AR signaling are negatively coupled and by forming heterodimers with D2 receptors, A2ARs affect D2 receptor affinity for dopamine agonists and G-protein coupling (Díaz-Cabiale et al., 2001; Canals et al., 2003; Fuxe et al., 2014). Furthermore, it was postulated that LIDs may be caused by altered expression of A2A/D2 heteromers versus A2A homomers at the cell membrane (Antonelli et al., 2006). Such a scenario can be imagined to be the case in Drd2-Cre/CK2KO mice where the level of A2AR is elevated (Rebholz et al., 2013). Enhanced A2A signaling in the Drd2-Cre/CK2KO mice may correlate with reduced D2R signaling, which may result in a proportionately larger effect of l-DOPA on the direct pathway. In turn, an imbalance between pathways (with higher activity of the direct one) is likely to favor LID. Furthermore, activation of the A2AR within A2AR/D2R heterodimers will lead to a biased modulation of D2R signaling, decreasing Gi/o signaling and increasing D2R-mediated β-arrestin2/Akt/PP2A signaling (Fuxe et al., 2014). The effect of this signaling cascade on LID has not been well studied. It is however known that chronic l-DOPA elevates Akt phosphorylation and activity in 6-OHDA lesioned rats (Bychkov et al., 2007). Thus, it is tempting to speculate that upregulated A2AR signaling in Drd2-Cre/CK2KO animals may correspond to high Akt/PP2A activity, which in turn leads to the higher severity of LID.
Finally, it is expected that, in Drd2-Cre/CK2KO mice, CK2α is absent from DA neurons and thus there is the possibility that this difference in CK2α expression may also contribute to the phenotype of the KO mice. The extent of the 6-OHDA lesion is similar in WT and Drd2-Cre KO, but since not all of the midbrain neurons are expected to be destroyed by 6-OHDA, we cannot exclude that altered activity of these dopaminergic neurons may play a role in the phenotype.
Concluding remarks
Our data indicate that altered D1 receptor signaling in direct pathway neurons and enhanced A2AR signaling in indirect pathway neurons underlie the opposite changes in susceptibility to LID seen in the two CK2 knock-out lines. The behavioral results of reduced or enhanced dyskinesia severity are consistent with the accompanying changes in ERK activation in the DA-denervated striatum. One important aspect in the development of LID are DA fluctuations induced by standard l-DOPA therapy, which leads to dramatic on-off changes in the activity of DA receptors (Olanow and Obeso, 2000). Against this background, reduced dynamics of DA receptor endocytosis may have important effects (Ahmed et al., 2010). In previous studies, we have shown that ablation of CK2 prevents or slows the endocytosis of D1 receptors, possibly by its interaction with Gαs/olf (Rebholz et al., 2009). Such an effect may counteract fast fluctuations of dopaminergic signaling and thus underlie the behavioral phenotype of reduced LID of the Drd1a-Cre/CK2KO mice.
Together, our results reveal a previously unappreciated role of CK2 in the striatal pathophysiology of motor complications in response to l-DOPA therapy. Moreover, the bidirectional modulation of dyskinesia seen upon ablation of CK2 in either class of striatal projection neurons reveals that both direct and indirect pathways contribute to LID.
Footnotes
This work was supported by the RISE program grant NIGMS R25GM056833 (to M.C. and L.M.), the Rapid Response as well as the Dyskinesia Challenge Awards of the Michael J. Fox Foundation for Parkinson's disease, and the PSC-CUNY45 and 46 awards of the City University of New York (to H.R.). We thank M. Sammon for technical assistance, and Drs. M. Zhou, K. Salas-Ramirez, and A. Kottmann for helpful discussions and equipment.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Heike Rebholz, Department of Molecular, Cellular, and Biomedical Sciences, CUNY School of Medicine, 160 Convent Avenue, New York, NY 10031. hrebholz{at}med.cuny.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}