

Abstract
Midbrain dopamine neuron dysfunction contributes to various psychiatric and neurological diseases, including drug addiction and Parkinson's disease. Because of its well established dopaminotrophic effects, the therapeutic potential of glial cell line-derived neurotrophic factor (GDNF) has been studied extensively in various disorders with disturbed dopamine homeostasis. However, the outcomes from preclinical and clinical studies vary, highlighting a need for a better understanding of the physiological role of GDNF on striatal dopaminergic function. Nevertheless, the current lack of appropriate animal models has limited this understanding. Therefore, we have generated novel mouse models to study conditional Gdnf deletion in the CNS during embryonic development and reduction of striatal GDNF levels in adult mice via AAV-Cre delivery. We found that both of these mice have reduced amphetamine-induced locomotor response and striatal dopamine efflux. Embryonic GDNF deletion in the CNS did not affect striatal dopamine levels or dopamine release, but dopamine reuptake was increased due to increased levels of both total and synaptic membrane-associated dopamine transporters. Collectively, these results suggest that endogenous GDNF plays an important role in regulating the function of dopamine transporters in the striatum.
SIGNIFICANCE STATEMENT Delivery of ectopic glial cell line-derived neurotrophic factor (GDNF) promotes the function, plasticity, and survival of midbrain dopaminergic neurons, the dysfunction of which contributes to various neurological and psychiatric diseases. However, how the deletion or reduction of GDNF in the CNS affects the function of dopaminergic neurons has remained unknown. Using conditional Gdnf knock-out mice, we found that endogenous GDNF affects striatal dopamine homeostasis and regulates amphetamine-induced behaviors by regulating the level and function of dopamine transporters. These data regarding the physiological role of GDNF are relevant in the context of neurological and neurodegenerative diseases that involve changes in dopamine transporter function.
Introduction
Midbrain dopamine neuron dysfunction contributes to various neurological and psychiatric diseases. Because of its therapeutic potential, ectopic application of the dopaminotrophic growth factor glial cell line-derived neurotrophic factor (GDNF) has been investigated extensively in various disease paradigms, including drug addiction and Parkinson's disease. However, the success of GDNF clinical trials has varied, highlighting a need for a better understanding of GDNF's physiological role on dopaminergic system function.
Knock-out mice that lack GDNF, its binding receptor GFRα1, or its signaling receptor RET die within few hours after birth due to kidney agenesis (Marcos and Pachnis, 1996; Moore et al., 1996; Pichel et al., 1996; Sánchez et al., 1996; Enomoto et al., 1998). However, the dopamine system remains largely intact in these animals (Airaksinen and Saarma, 2002). Because the mouse dopamine system matures primarily during the first few postnatal weeks (Jackson-Lewis et al., 2000), GDNF's role in the dopamine system's postnatal development has remained unclear. Recently, using three complementary conditional genetic knock-out approaches, we found that GDNF is not required for either the development or the survival of nigrostriatal dopaminergic neurons in adult animals (Kopra et al., 2015). However, whether GDNF is required for the normal function of dopaminergic neurons is currently unknown.
Here, we first examined whether deleting GDNF selectively in the CNS affects nigrostriatal dopaminergic system function in mice. These mice were generated by crossing Gdnf-floxed mice (Kopra et al., 2015) with Nestin-Cre mice (Tronche et al., 1999; Kramer et al., 2007), resulting in the loss of GDNF expression throughout the entire CNS in embryogenesis (Tronche et al., 1999; Kopra et al., 2015). We then measured the levels of dopamine and dopamine metabolites in various brain areas. We further examined dopamine release and dopamine transporter (DAT)-mediated dopamine reuptake in the dorsal striatum (dSTR). In addition, we examined the stimulatory effect of amphetamine in our GDNF conditional knock-out mice and measured the levels of total and synaptosomal membrane-associated DAT protein in the striatum. Finally, we studied the effect of reducing GDNF levels specifically in adult mice striata via AAV5-Cre delivery. Our results indicate that GDNF is an important regulator of both DAT function and DAT expression patterns and that a loss of brain GDNF reduces amphetamine-induced hyperactivity in mice.
Materials and Methods
Animals.
The generation and genotyping of Gdnf-floxed (F/F) mice and Nestin-Cre mice was described previously (Tronche et al., 1999; Kopra et al., 2015). The mice were bred locally in the animal facility at the University of Helsinki and were maintained on a mixed genetic background (129Ola/ICR/C57BL6). The mice were housed under temperature-controlled conditions at 20–22°C in a 12 h/12 h light/dark cycle. Each cage contained two to five animals, which had ad libitum access to standard chow and water. All animal experiments were approved by the National Animal Experiment Board of Finland or the Umeå Ethics Committee for Animal Studies. Because the Nestin-Cre line generates robust recombination in germ cells, the floxed allele derived from the parent that carries the Nestin-Cre transgene is often constitutively recombined, resulting in a full knock-out “KO” allele (Kramer et al., 2007). Therefore, in homozygous Gdnf conditional knock-out mice, one Gdnf allele is recombined selectively in the CNS and the other Gdnf allele is recombined in all cells. Animals were bred for experiments by crossing homozygous F/F mice with heterozygous wt/F + Nestin-Cre mice. Adult homozygous male Gdnf conditional knock-out mice and littermate controls were used at the age of 10–16 weeks unless stated otherwise. Approximately half of the heterozygous (C/KO) controls (“C” denotes control allele) carried the Nestin-Cre transgene in each experiment. When Nestin-Cre was present, C denoted Gdnf wt allele in the control group. All animals and samples were analyzed together with their littermate controls in a random order.
AAV5-Cre injections.
Gdnf-floxed (F/KO) male mice and heterozygous (Wt/KO) controls aged 5–8 months received bilateral injections of AAV5-Cre viral vector (Johan Jakobsson laboratory, Lund University, Sweden) expressing the Cre recombinase under the human synapsin promoter. The mice were kept under 1.5–2% isoflurane (Attane Vet 1000 mg/g; Piramal Healthcare UK Limited) in oxygen and attached to a stereotactic frame. Lidocaine (10 mg/ml; Orion Pharma) was applied under the skin for local analgesia. A total of 1.7 × 1011 genome copies of the viral vector in Dulbecco's PBS supplied with Mg2+ and Ca2+ were injected in a volume of 1 μl into 2 different coordinates in each dSTR at a 10° angle (relative to bregma: A/P = +0.2 mm and +1.2 mm; M/L = ±2.2 mm; D/V = 3.0 mm) under isoflurane anesthesia using NanoFil syringes (World Precision Instruments) with 33 Ga blunt NanoFil needles (World Precision Instruments). After each injection, the needle was kept in place for 5 min before slowly withdrawn. Each animal received a total of four injections of the viral vector (two for each striatum). A separate group of 5-month-old female animals received only unilateral injections to measure and compare Gdnf expression in the injected and noninjected side. All mice received one injection of buprenorphine (0.1 mg/kg, s.c.) and karprofen (5 mg/kg, s.c.) at the end of the surgery to relieve pain.
Amphetamine-induced locomotion.
To measure amphetamine-induced locomotion, mice were placed individually in transparent plastic cages (24 × 24 × 15 cm) with a perforated plastic lid. The mice were allocated systematically to the experimental sessions to ensure that each session would include both knock-out mice and littermate controls. The cages were located inside an open-field activity monitor (MED Associates) that registered the animal's movements. The mice were habituated for 15 min before receiving an injection of d-amphetamine sulfate (1 mg/kg, i.p.; Division of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Helsinki, Finland) and their locomotor activity was monitored for 60 min. Infrared photo beam interruptions were registered and collected at 5 min intervals.
Animals receiving bilateral AAV5-Cre injections were tested for 60 min of spontaneous open-field activity just before and 40 d after the stereotactic injections. One day after the second session, the mice received an injection of d-amphetamine sulfate (1 mg/kg, i.p.) and their activity was similarly measured. Animals were not habituated in these sessions.
Accelerating rotarod.
The accelerating rotarod test was performed on the Ugo Basile rotarod with an acceleration speed of 4–40 rpm over 5 min. The latency to fall off was recorded with the cutoff time set at 6 min (a constant speed of 40 rpm was applied during the last minute). The test was conducted on 2 consecutive days with 3 trials on each day. The mice were habituated to the room for 1 h before the start of the experiment.
Microdialysis.
A microdialysis guide cannula (MAB 4.1; AgnTho's) was inserted into the dSTR (A/P = +0.6 mm, M/L = +1.8 mm; DV = −2.2 mm) under isoflurane anesthesia. Two screws and dental cement (Aqualox; Voco) were used to affix the guide cannula to the skull. After at least 4 d of recovery, a microdialysis probe (MAB 4.9.1.Cu; AgnTho's) was inserted into the guide cannula and dialysis was started with Ringer's solution containing the following (in mm): 147 NaCl, 1.2 CaCl2, 2.7 KCl, 1.0 MgCl2, and 0.04 ascorbic acid, pH 7.4, at a flow rate of 2 μl/min. After a 2 h stabilization period, samples were collected at 15 min intervals. Extracellular dopamine levels were measured using the mean of four consecutive, stable samples. The concentration of dopamine was measured with high-performance liquid chromatography (HPLC) using a Coulochem II electrochemical detector (ESA). A Kinetex 2.6u column (XB-C18; 50 × 4.6 mm; Phenomenex) was maintained at 45°C using a column heater. The flow rate of the mobile phase (0.1 m NaH2PO4, 0.1 mg/ml octanesulphonic acid, 1.0 mm EDTA, and 8% methanol, pH 4.0) was 1 ml/min. The sample (25 μl) was injected into the chromatographic system using an SIL-20AC autoinjector (Shimadzu). After the experiment, the animal was decapitated and the brain was removed and frozen. Correct placement of the probe was confirmed histologically in 90-μm-thick coronal brain sections.
Fast-scan cyclic voltammetry.
At 8–11 months of age, mice were decapitated, the brain was removed, and 300-μm-thick coronal slices containing the cortex and striatum were cut on a model 7000 smz-2 vibratome (Campden Instruments) in ice-cold cutting saline containing the following (in mm): 125 NaCl, 2.5 KCl, 26 NaHCO3, 0.3KH2PO4, 3.3 MgSO4, 0.8 NaH2PO4, and 10 glucose. The slices were allowed to recover in a holding chamber for 1–2 h at 32°C in oxygen-bubbled (95% O2, 5% CO2) recording saline containing the following (in mm): 125 NaCl, 2.5 KCl, 26 MgSO4, 0.8 NaH2PO4, and 10 glucose. In the recording chamber, the slices were perfused continuously with 35°C oxygen-bubbled recording saline. In the in vitro amphetamine experiments, d-amphetamine hemisulfate (Sigma-Aldrich) was added to the perfusate at a final concentration of 5 μm after a stable baseline of stimulated transient peaks was reached. Fast-scan cyclic voltammetry recordings were performed using cylindrical 5 μm carbon fiber electrodes positioned at the dSTR ∼50 μm below the exposed surface. Striatal slices were stimulated electrically using a stainless steel bipolar electrode placed ∼100 μm from the recording electrode. Square pulses 0.4 s in duration were produced using an Iso-Flex stimulus isolator triggered by a Master-8 pulse generator (A.M.P.I.). Stimulus magnitude was obtained by plotting a current–response curve to single pulse stimulations and selecting the minimum value that produced the maximum response reliably. Triangular voltage ramps from a holding potential of −450 mV to +900 mV over 9 ms (scan rate of 300 mV/ms) were applied to the carbon fiber electrode at 100 ms intervals. The current was recorded using an Axopatch 200B amplifier (Molecular Devices), filtered through a 5 kHz low-pass Bessel filter, and digitized at 40 kHz (ITC-18 board; InstruTech). Triangular wave generation and data acquisition were controlled and the recorded transients were characterized using a computer routine in IGOR Pro (WaveMetrics) (Mosharov and Sulzer, 2005; Mosharov, 2008). Background-subtracted cyclic voltammograms obtained with 1 μm dopamine solution (dopamine-HCl; Sigma-Aldrich) were used to calibrate the electrodes. Dopaminergic terminals were stimulated with: single electrical pulses at 2 min intervals; paired stimulations delivered at fixed intervals of 60, 30, 10, and 5 s (to study paired stimulation depression); or burst stimulations of 5 pulses at 20 Hz (to study release probability at the terminals). In paired stimulations, the subsequent exocytosis is thought to be repressed by both D2 receptor-mediated autoinhibition and the time it takes for reuptake and recycle the released dopamine back into vesicles for next release (Abeliovich et al., 2000; Cragg, 2003; Senior et al., 2008).
In vivo chronoamperometry.
Mice of 7–10 months of age were used in these experiments. Recordings were performed using the Fast Analytical Sensing Technology (FAST-16) system (Quanteon) (Hoffman and Gerhardt, 1998). In brief, a 5 Hz square wave potential of 0.55/0 V was applied to a single fiber carbon electrode, causing oxidation and reduction of electrochemically active substances in close proximity to the electrode tip. Chemical reactions induce a rapid change in current recorded by the electrode and oxidation and reduction currents were integrated and averaged to 1 Hz. An increased extracellular level of electrochemically active compounds results in a detectable peak formation. Because the current recorded is directly proportional to the analyte's concentration (Scatton et al., 1988), the peak amplitude corresponds to the maximum concentration (micromolar) of dopamine recorded. The identity of the analyte recorded was determined by calculating the reduction/oxidation ratio, in which a ratio of ∼0.8 indicates dopamine (Strömberg et al., 1991). The carbon fiber electrodes were coated with Nafion to repel anions and to increase the selectivity for cations (Gerhardt et al., 1984; Nagy et al., 1985). The electrode was calibrated in 0.1 m PBS, pH 7.4, in a beaker containing ascorbic acid (20 mm) and dopamine (2 mm). The carbon fiber electrodes used in this study had a selectivity ratio >200:1 for dopamine over ascorbic acid, a <0.05 μm detection limit for dopamine, and linearity (r2) >0.997. A silver wire plated for 30 min in 1 m HCl saturated with NaCl was used as a reference electrode. The carbon fiber electrodes were mounted together with a glass micropipette (with 130–160 μm between the pipette tip and electrodes) using sticky wax. The micropipette was filled with 200 μm dopamine in 0.9% NaCl containing 20 μm ascorbic acid and connected to a micropressure system. The volume applied was determined using a stereomicroscope fitted with a reticle in one eyepiece to measure the movement of the meniscus in the micropipette (Friedemann and Gerhardt, 1992).
The animals were anesthetized with urethane (1.6–1.9 g/kg; Sigma-Aldrich), tracheotomized, and placed on a heating pad. The skull was fixed in a stereotaxic frame, the scalp was removed, and the skull bone was opened over the striatum using a drill. The Ag/AgCl reference electrode was implanted through a single burr hole caudal to the striatum. The recording electrode/micropipette assembly was lowered into the brain using a microdrive. Striatal recordings were performed at the following coordinates relative to bregma (in mm): A/P + 0.03 and +0.1, M/L +0.18 or −0.18, and at 2 different depths relative to the brain surface (2.5 and 3.0 mm). Dopamine was delivered at each striatal recording site by injecting 25–375 nl of 200 μm dopamine 3–5 times at 5 min intervals.
Brain dissection.
For Western blot analysis, brain monoamine measurements and evaluation of Gdnf mRNA expression levels, samples were obtained from the relevant dissected brain regions using a mouse brain matrix. In brief, mice were killed by decapitation and the whole brain was removed, washed in ice-cold saline for 20 s, and then placed in an ice-cold mouse brain matrix (Stoelting). Slices (2 mm thickness) were prepared to include structures such as the striatum and ventral midbrain. Samples containing the dSTR, ventral striatum (vSTR), substantia nigra (SN), and ventral tegmental area (VTA) were dissected using either a sample corer (inner diameter, 2 mm) or a scalpel. The samples were snap-frozen on dry ice and stored at −80°C until assayed. For HPLC analysis, all samples were weighed.
Estimation of monoamines and their metabolites.
Dopamine and its metabolites were analyzed from the dissected brain samples as described previously (Valros et al., 2015) using HPLC with electrochemical detection.
Crude synaptosome preparations and membrane protein biotinylation assay.
The dSTR was dissected as described above and cut into approximately equal-size pieces. One piece was used to measure total DAT protein levels. Membrane protein biotinylation reactions were performed using the Cell Surface Protein Isolation Kit (catalog number 89881; Pierce) in accordance with the manufacturer's instructions. Crude synaptosomes were prepared as described previously (Hallett et al., 2008). The biotinylated fraction of the synaptosome preparation was then extracted using the Cell Surface Protein Isolation Kit (Pierce) in accordance with the manufacturer's instructions.
Western blot analysis.
The dSTR samples were homogenized on ice in 10 mm HEPES (pH 7.2–7.4), 1 mm EDTA, 0.3 m sucrose, protease inhibitor mixture (Complete Mini-Tabs Protease Inhibitor; Roche), and phosphatase inhibitor (PhosSTOP; Roche). Total protein concentration was measured using the Lowry method (Bio-Rad). A total of 10 or 20 μg of protein was separated by SDS-PAGE to detect DAT and tyrosine hydroxylase (TH), respectively. The proteins were transferred to a nylon membrane, which was washed 3 times in TBS containing 0.1% Tween 20 (TBS-T) for 15 min and then blocked in 5% (w/v) nonfat milk in TBS-T for 1 h at room temperature (RT). To detect TH, the membranes were incubated in mouse anti-TH antibody (1:3000, MAB318; Millipore) at 4°C in blocking solution, followed by anti-mouse-HRP (1:3000, P0447; DAKO) for 2 h at RT. To detect DAT, the membranes were incubated in rat anti-DAT (1:1000, MAB369; Millipore) overnight at 4°C in blocking solution, followed by biotinylated anti-rat antibody (1:500, BA-4000; Vector Laboratories) for 2 h at RT, followed by 2 h at RT with streptavidin-HRP (1:2500, S-911; Invitrogen).
The membranes were stripped for 15 min at 70°C in 50 mm Tris-HCl, pH 7.4, 2% SDS, and 50 mm DTT, and then washed and blocked as described above. The membranes were then incubated in mouse anti-GAPDH antibody (1:10,000 or 1:1000 for total and biotinylated samples, respectively; MAB374; Millipore) for 2 h at 4°C, followed by 2 h at RT in anti-mouse-HRP (1:3000 or 1:1500 for total and biotinylated samples, respectively; P0449; DAKO). The signal was visualized using enhanced chemiluminescence (kit #32106; Pierce), followed by exposure to film. The signal from the gene of interest was normalized to the GAPDH signal using ImageJ software.
RNA isolation, cDNA synthesis, and qPCR.
Total RNA was isolated using TRIzol (Thermo Fisher Scientific) according to manufacturer's recommendations. For complementary DNA synthesis, 500 ng of RNA was treated with DNase I (Thermo Fisher Scientific) for 15 min at 37°C and used immediately in the reverse transcription reaction with RevertAid Reverse Transcriptase (Thermo Fisher Scientific) following the protocol provided by the manufacturer. qPCR was performed with the LightCycler 480 Real-Time PCR System (Roche) using SYBR Green I Master (Roche) supplied with 0.25 mm primers and cDNA corresponding to 62.5 ng of RNA in a final volume of 10 μl. Two replicates of each reaction were included in the qPCR runs. Beta actin was used as the reference gene. The following primers were used: β-actin forward: CCAGTTCGCCATGGATGAC, β-actin reverse: GAGCCGTTGTCGACGACC, Gdnf forward: CGCTGACCAGTGACTCCAATATGC, and Gdnf reverse: TGCCGCTTGTTTATCTGGTGACC. Relative Gdnf expression was calculated according to the formulation in Varendi et al. (2014), assuming that primer efficiencies for both primer pairs were 2.
Statistical analyses.
Data from the three groups were analyzed using a one-way ANOVA followed by the Student–Newman–Keuls post hoc analysis in the event of a significant ANOVA result. For nonhomogeneous variances, a nonparametric Kruskal–Wallis test was used, followed by Mann–Whitney U post hoc tests and Bonferroni correction. Statistical analysis for pairwise comparisons was performed using Student's t test with two-tailed distribution using the equal variance option. A one-way ANOVA for repeated measures was used to analyze amphetamine-induced locomotion. The chronoamperometric data collected from multiple recording sites in the striatum were statistically homogeneous and were therefore pooled for each genotype to provide an estimate of dopamine uptake in the striatum as a whole structure, as described previously (Lundblad et al., 2009; Nevalainen et al., 2011). All summary data are reported as the mean ± SEM.
Results
Reduced response to amphetamine in the absence of brain GDNF
Because Gdnf mRNA and protein expression levels are similar between the wild-type Gdnf and floxed Gdnf alleles (Kopra et al., 2015), we refer to these two alleles collectively as the control (“C”) allele. In the presence of Nestin-Cre, the floxed Gdnf allele is referred to as “floxed allele” or “F” and the conventional Gdnf knock-out allele is referred to as “KO.” First, we challenged animals lacking GDNF in the CNS (i.e., GdnfF/KO + Nestin-Cre mice) with amphetamine, a well known stimulant. Amphetamine reverses the transit of dopamine through DAT from the cytoplasm to the extracellular space (here referred to as “dopamine efflux”), resulting in high levels of extracellular dopamine and increased locomotor activity (German et al., 2015). Heterozygous Gdnf knock-out mice (Gdnfwt/KO) and wild-type mice respond similarly to amphetamine (Gerlai et al., 2001). Interestingly, we found that homozygous mice that lack GDNF in the CNS have significantly reduced locomotion after amphetamine (1 mg/kg) injection compared with Gdnfwt/KO littermate controls (Fig. 1A). To gain mechanistic insight into this phenotype, we quantified dopamine release and reuptake at subsecond resolution using electrical stimulation and fast-scan cyclic voltammetry measurements in striatal slices obtained from adult mice. Consistent with the reduced amphetamine-induced hyperlocomotion response, amphetamine-induced dopamine efflux was significantly reduced in striatal slices from mice that lack GDNF in the CNS (Fig. 1B,C). Furthermore, we found that the DAT “reversal time,” the interval between the addition of amphetamine and the reversal of dopamine transit via DAT, was significantly longer in GDNF-deficient mice (Fig. 1C,D), suggesting that, for these mice, dopamine transporters at dopaminergic terminals are less susceptible to amphetamine-induced reversal. Amphetamine also depletes dopamine from storage vesicles, which is reflected in voltammetric recordings as a steady decrease in stimulated dopamine peak currents. However, the effect of amphetamine on stimulated dopamine peaks was similar between genotypes (Fig. 1E), suggesting that amphetamine is equally effective at driving the release and redistribution of dopamine from storage vesicles regardless of the presence of GDNF.

Loss of GDNF in the brain causes an altered response to amphetamine. A, Locomotor activity was measured in mice following injection of amphetamine (1 mg/kg, i.p.). n = 16 and 9 mice for C/KO and F/KO + Nestin-Cre, respectively. Repeated-measures ANOVA F = 4.447, p = 0.046. B, Nonstimulated DAT-mediated dopamine (DA) release (i.e., DA efflux) was measured in slices during amphetamine perfusion. n = 12, 11, and 14 for C/C, C/KO, and F/KO + Nestin-Cre, respectively; *p < 0.05 versus C/C (Kruskal–Wallis test). C, Example recording traces measured in the continuous presence of amphetamine in a C/C (black) and F/KO + Nestin-Cre (red) mouse; notable events are indicated. The slice was stimulated electrically every 2 min to evoke DA release/reuptake (the rapid DA transients). During continuous amphetamine perfusion, a nonstimulated slow DA signal (i.e., DA efflux) occurred due to amphetamine-induced reversal of the DAT. D, Summary of the delay between the onset of amphetamine perfusion and reversal of DAT transport function. n = 12, 11, and 14 for C/C, C/KO, and F/KO + Nestin-Cre, respectively; *p < 0.05 versus C/C (Kruskal–Wallis test). E, Summary of the effect of amphetamine perfusion on stimulated DA release (normalized to baseline measured before amphetamine addition). n = 12, 11, and 14 for C/C, C/KO, and F/KO + Nestin-Cre, respectively. F, Floxed; KO, knock-out; C, control (including both wild-type and floxed alleles).
Together, the effects of amphetamine measured ex vivo and in vivo led us to hypothesize two nonexclusive consequences of a loss of GDNF in the CNS. The first hypothesis is that the function of the DAT is altered and the second is that total tissue levels of dopamine are altered.
Increased DAT activity in the striatum of GDNF-deficient mice
To test these hypotheses, we studied the details of dopamine release and reuptake ex vivo in striatal slice preparations and in vivo in anesthetized animals.
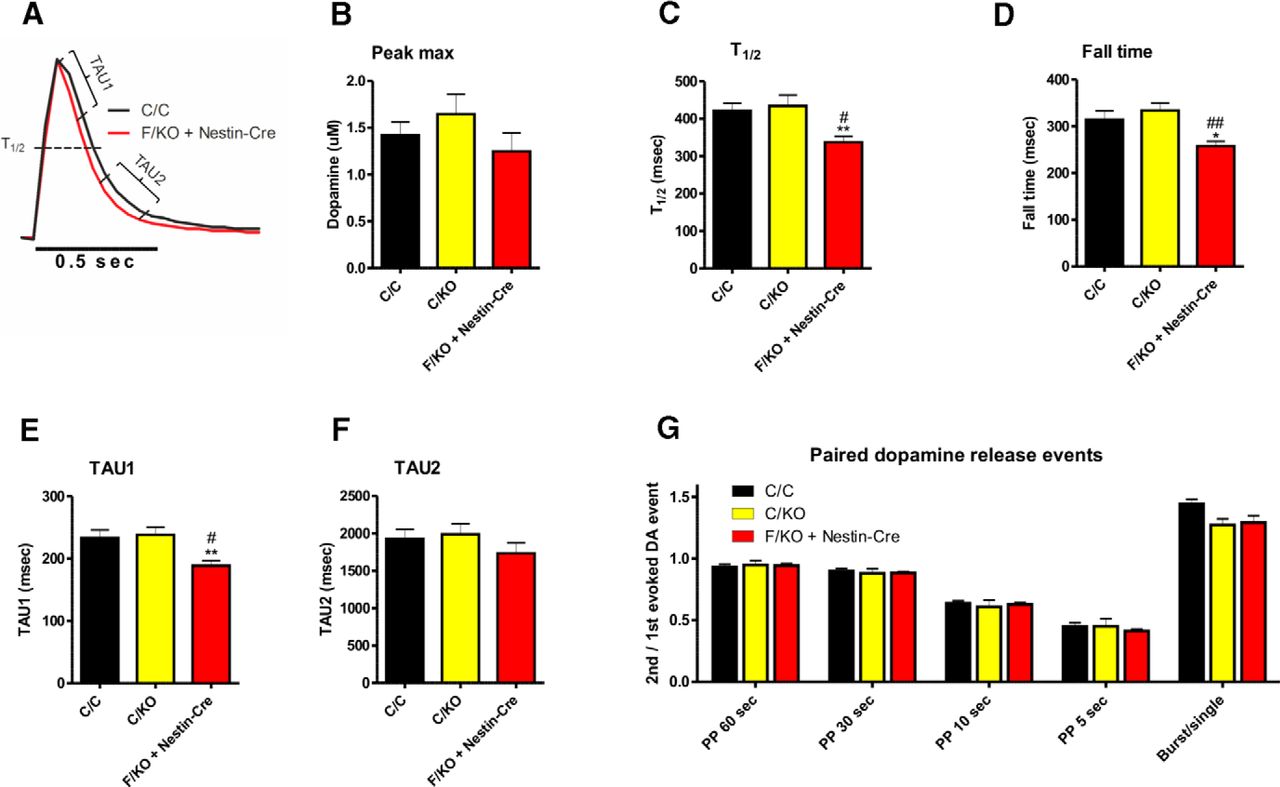
Cyclic voltammetry revealed that the levels of dopamine release induced by individual electrical pulses in the dSTR are similar between genotypes (Fig. 2A,B). However, three measures of kinetics, the half-width of dopamine reuptake (T1/2), the fall time, and TAU1, revealed that GDNF-deficient mice have more rapid DAT-mediated dopamine reuptake compared with control mice (Fig. 2C–E). In contrast, we observed no significant difference between genotypes with respect to the decay constant of the slow phase of dopamine uptake (TAU2) (Fig. 2F), paired stimulus depression of dopamine release (Fig. 2G), or dopamine release probability by bursts of stimuli (Fig. 2G).

Loss of GDNF in the brain increases dopamine uptake, but does not affect dopaminergic terminal recovery or release probability. A, Averaged, normalized single-pulse-stimulated DA release transients were measured in striatal slices obtained from a C/C (black) and F/KO + Nestin-Cre (red) mouse. The parameters shown in B–F are indicated. B–F, Summary of the parameters measured as shown in A. n = 18, 16, and 17 for C/C, C/KO, and F/KO + Nestin-Cre, respectively. TAU1 was measured as reported by Yorgason et al. (2011). G, Striatal dopaminergic terminals were stimulated with paired pulses at the indicated intervals (60, 30, 10, or 5 s) and the ratio of the second peak to the first peak was calculated as a measure of how quickly the terminals recovered from the first stimulus. In the last bar, DA release was stimulated with a single pulse and with a 20 Hz burst of 5 pulses and the ratio of burst-induced peak to the single pulse-induced peak was calculated as a measure of release probability at the terminals. n = 6, 7, and 6 for C/C, C/KO, and F/KO + Nestin-Cre, respectively. F, Floxed; KO, knock-out; C, control (including both wild-type and floxed alleles). *p < 0.05 versus C/C; **p < 0.01 versus C/C; #p < 0.05 versus C/KO; ##p < 0.01 versus C/KO.
To determine whether the increased DAT function measured ex vivo is also present in vivo, we measured striatal dopamine reuptake using chronoamperometry in the intact brains of anesthetized animals. We applied known quantities of dopamine close to the recording electrode. The amplitudes of the currents measured after dopamine injection were correlated with the concentration of dopamine delivered and thus reflect the rate of DAT-mediated dopamine clearance (Cass and Gerhardt, 1995). We found that the rate of dopamine clearance was increased in mice lacking GDNF in the CNS, thereby allowing less dopamine to reach the recording electrode. This was reflected by smaller currents measured during the recording (Fig. 3A).

Loss of GDNF in the brain increases DAT activity in vivo, DAT tissue levels, and DAT localization to the plasma membrane. A, In vivo chronoamperometry was measured after an intrastriatal DA injection. The amplitude of the DA signal was normalized to the amount of DA applied. n = 5 mice per group; Kruskal–Wallis test H = 10.29; p = 0.006. B, C, Total DAT protein in the dSTR was measured by Western blot, normalized to GAPDH, and plotted relative to C/C. n = 6 mice per group. D, E, DAT proteins at the cell surface were biotinylated in dSTR synaptosomes, measured by Western blot, normalized to GAPDH, and plotted relative to C/C. n = 5, 6, and 6 for C/C, C/KO, and F/KO + Nestin-Cre, respectively. F, TH protein in the dSTR was measured by Western blot, normalized to GAPDH, and plotted relative to C/C. n = 5, 4, and 5 for C/C, C/KO, and F/KO + Nestin-Cre, respectively. G, Basal extracellular dopamine concentration was measured in striatal dialysates. The concentration of dopamine was not corrected with in vitro recovery. n = 11, 9, and 7 for C/C, C/KO, and F/KO + Nestin-Cre, respectively. F, Floxed; KO, knock-out; C, control (including both wild-type and floxed alleles).*p < 0.05 versus C/C; **p < 0.01 versus C/C; ***p < 0.001 versus C/C; #p < 0.05 versus C/KO; ##p < 0.01 versus C/KO.
To test whether the increased DAT function in GDNF-deficient mice was due to an increase in DAT protein levels in the striatum, we used Western blot analysis to measure striatal DAT levels in the various genotypes. Compared with control mice, heterozygous GDNF-deficient mice had a slight increase in striatal DAT protein levels and homozygous GDNF-deficient mice had a much larger increase (an increase of ∼70%) (Fig. 3B,C). To investigate the potential effects of deleting GDNF on DAT cellular localization, we biotin labeled the cell surface membrane proteins in dSTR synaptosome preparations and performed Western blot analysis for DAT. We found that mice lacking GDNF in the CNS have more DAT at the cell surface compared with littermate controls (Fig. 3D,E).
Together, these data indicate that a loss of GDNF in the CNS leads to increased DAT activity in the striatum, at least partially due to increased expression and surface localization of dopamine transporters.
Loss of GDNF does not affect striatal dopamine or TH levels
Next, we investigated whether deleting GDNF expression in the CNS affects the levels of dopamine by measuring dopamine and its metabolites in the primary dopaminergic brain areas, including the dSTR and SN of the nigrostriatal tract, as well as the adjacent vSTR and VTA of the mesolimbic tract (Björklund and Dunnett, 2007). We found no significant differences between genotypes with respect to the levels of dopamine or its metabolites 3,4-dihydroxyphenylacetic acid and homovanillic acid (Table 1). Moreover, we found no change in the levels of TH protein, the rate-limiting enzyme in dopamine synthesis, in the striatum (Fig. 3F). Last, we found no difference between genotypes with respect to extracellular dopamine levels measured in vivo using microdialysis (Fig. 3G).
Concentration of dopamine and its metabolites measured in the indicated brain tissues using HPLC
Together, these results indicate that CNS-specific deletion of GDNF does not affect the levels of TH, dopamine, or dopamine metabolites in the striatum nor does it affect the basal levels of extracellular dopamine.
Reduced response to amphetamine upon GDNF reduction in the striatum of adult mice
To investigate the possibility that the observed effects reflect changes in early brain development due to global GDNF deficiency in the CNS since E10.5 (Tronche et al., 1999), we reduced Gdnf expression bilaterally in the dorsal striata of adult Gdnf-floxed mice by AAV5-Cre delivery. Forty days after striatal AAV5-Cre delivery, Gdnf mRNA levels were reduced by 50% in Gdnf conditional knock-out mice (F/KO) (Fig. 4A). We have shown previously that adult GDNF reduction with AAV5-Cre does not affect striatal levels of dopamine or its metabolites (Kopra et al., 2015). Although the baseline locomotor activity was similar in both groups before the AAV5-Cre injections (Fig. 4B), 40 d after the injections, both basal (Fig. 4C) and amphetamine-stimulated (Fig. 4D) open-field activity were reduced in the AAV5-Cre-injected Gdnf-floxed mice. Subtracting the basal activity from the amphetamine-stimulated activity revealed dampened amphetamine response in these mice (Fig. 4E). Accelerating rotarod test revealed no difference between the genotypes, indicating that these mice were not suffering from a motor deficit (Fig. 4F). Cyclic voltammetry recordings in striatal slices demonstrated that, similar to GdnfF/KO + Nestin-Cre animals, amphetamine-induced dopamine efflux was reduced and DAT reversal time was increased in the AAV5-Cre-injected Gdnf-floxed mice (Fig. 4G,H). Striatal dopamine release (Fig. 4I) and reuptake (Fig. 4J–L) were not changed significantly in these mice.

Striatal Gdnf deletion in adult animals reduces responses to amphetamine. A, Unilateral AAV5-Cre injections caused 50% reduction in Gdnf mRNA levels in the dSTR of mice carrying one floxed Gdnf allele (F/KO). Their noninjected dSTR served as control. n = 6 for F/KO; Student's t test p = 0.0008. B, Baseline locomotor activity of F/KO mice did not differ from the heterozygous controls before the AAV5-Cre injections. n = 9 and 15 mice for Wt/KO and F/KO, respectively. C, Baseline locomotor activity of F/KO mice was reduced 40 d after the AAV5-Cre injections. n = 8 and 12 mice for Wt/KO and F/KO, respectively. Repeated-measures ANOVA F = 8.946, p = 0.008. D, Amphetamine-stimulated locomotor activity of F/KO mice was reduced 41 d after the AAV5-Cre injections. n = 8 and 12 mice for Wt/KO and F/KO, respectively. Repeated-measures ANOVA F = 17.027, p = 0.001. E, Subtracting the basal activity (C) from the amphetamine-stimulated activity (D) revealed dampened amphetamine response in F/KO mice. n = 8 and 12 mice for Wt/KO and F/KO, respectively. Repeated-measures ANOVA F = 11.982, p = 0.003. F, Motor performance in accelerating rotarod test 52 d after the AAV5-Cre injections revealed no motor deficit in F/KO mice. n = 8 and 10 mice for Wt/KO and F/KO, respectively. G, H, There was less DA efflux (G) and delayed DAT reversal (H) in response to amphetamine perfusion in striatal slices of F/KO mice. n = 10 and 14 slices, for Wt/KO and F/KO, respectively. Student's t test p = 0.047 and p = 0.017, respectively. The level of electrically stimulated dopamine release (I) and the kinetics of the dopamine reuptake (J–L) did not differ significantly between the groups, n = 11 and 12 slices, for Wt/KO and F/KO, respectively. F, Floxed; KO, knock-out.*p < 0.05, ***p < 0.001 versus C/C.
Collectively, these data indicate that striatal Gdnf reduction in adult mice causes similar alterations in amphetamine responses in vivo and in brain slice preparations to CNS-wide embryonic Gdnf deletion.
Discussion
We reported recently that GDNF is not required for the development or long-term survival of nigrostriatal dopaminergic neurons in mice (Kopra et al., 2015). However, GDNF may regulate the dopaminergic system at the level of neuronal function. Therefore, we first investigated the effects of deleting GDNF in the CNS on the function of the nigrostriatal dopaminergic system using conditional Gdnf knock-out mice, in which GDNF expression is deleted selectively from the entire CNS during embryonic development. We found that GDNF is important for normal DAT function and for mediating a normal response to amphetamine. Specifically, using two independent, complementary methods, ex vivo fast-scan cyclic voltammetry in striatal slices and in vivo chronoamperometry in intact animals, we detected faster dopamine uptake in the dSTR of GDNF-deficient mice. We also measured increased protein levels of DAT in striatal tissue and in the plasma membrane of synaptosome preparations. We found that deleting GDNF from the CNS did not affect the levels of dopamine or TH in the tissue nor did it affect the extracellular levels of dopamine. We further found that local reduction in striatal Gdnf mRNA levels in adult animals resulted in a reduced response to amphetamine, similar to embryonic deletion. These results suggest that the observed changes in DAT reversal function in GdnfF/KO + Nestin-Cre mice are not induced by developmental changes elicited by CNS-wide GDNF deletion during early embryonic development, but instead likely result from more direct GDNF effects on striatal dopaminergic axon terminals. However, dopamine reuptake remained unaltered in animals with adult GDNF reduction, suggesting that either larger reduction in GDNF levels or embryonic GDNF deletion is required to affect the reuptake kinetics.
Recently, GDNF signaling was linked to surface expression of the dopamine transporter (Zhu et al., 2015). In that study, the investigators found that reduced GDNF signaling increases DAT activity by facilitating the transporter's localization to the cell surface and, conversely, increased GDNF signaling reduces DAT activity by facilitating removal of DAT proteins from the cell surface (Zhu et al., 2015). We found increased levels of DAT at the plasma membrane in the dSTR of GDNF-deficient mice even though Zhu et al. (2015) reported that GDNF regulates DAT at the vSTR, but not at the dSTR. This apparent discrepancy may be due to the fact that the previous study did not use a conditional Gdnf knock-out strategy. In addition, we found that endogenous GDNF regulates DAT expression directly. Nevertheless, given previous studies and our current results, it is clear that the functional relationship between GDNF and DAT remains incompletely understood. To illustrate this point, in contrast to expectations based on our data and the study by Zhu et al. (2015), DAT activity is increased, not reduced, in GDNF hypermorphic mice, in which the endogenous Gdnf locus is upregulated (Kumar et al., 2015), as well as in mice that express a constitutively active GDNF receptor RET (Mijatovic et al., 2007, 2008). Therefore, although the evidence to date suggests a link between GDNF signaling and DAT function, the diversity of phenotypes observed among the various mouse models highlights the complexity of this relationship and calls for further research.
Interestingly, the DAT proteins present in the GDNF-deficient striatum were highly effective at transporting extracellular dopamine into the cytosol and were slower to undergo the amphetamine-induced reversal of dopamine transport. The consequences of these changes included reduced DAT-mediated dopamine efflux, reduced extracellular dopamine levels, and reduced amphetamine-induced locomotor responses compared with littermate controls. Given that more DAT proteins are present at the cell surface when GDNF is deleted, it is possible that, in the absence of GDNF, the larger surface DAT population responds more slowly to the reversing effects of amphetamine. Alternatively, some transporters may still reverse direction from uptake to efflux, but a sufficient number of DAT molecules may remain in the conventional (i.e., uptake) transport mode, resulting in reduced hyperlocomotion. Our finding that local GDNF reduction in the adult striatum also makes dopamine transporters less eager to allow dopamine efflux in response to amphetamine without altering dopamine release or reuptake suggests that GDNF reduction hinders DAT reversal specifically. Further insight into the involved mechanism requires future research.
We additionally observed that striatal GDNF reduction in adult mice reduced spontaneous activity in open field without influencing performance in the accelerating rotarod test. Notably, spontaneous open-field activity in GdnfF/KO + Nestin-Cre mice was unaltered (Kopra et al., 2015). Reduced basal activity observed upon adult GDNF reduction could reflect a mild nigrostriatal dopamine system dysfunction that is overcome developmentally in the animals with embryonic GDNF deletion. However, a conclusion on this will require further studies.
Although data regarding the function of endogenous GDNF are scarce, a relatively large body of evidence suggests that intracranial ectopic GDNF has widespread effects on nigrostriatal dopamine metabolism, as reviewed recently by Kramer and Liss (2015). The intracranial delivery of GDNF can increase both stimulated and amphetamine-induced dopamine release and can increase dopamine turnover (Hudson et al., 1995; Hebert et al., 1996; Hebert and Gerhardt, 1997; Georgievska et al., 2004). Our finding that deleting GDNF expression in the CNS alters neither dopamine levels nor stimulated dopamine release suggests that ectopic and endogenous GDNF exert different effects on the function of the striatal dopaminergic system. Similarly, ectopic GDNF can upregulate the expression of TH both in vitro (Theofilopoulos et al., 2001; Xiao et al., 2002) and in vivo in the striatum (Kirik et al., 2000) and this is followed by a downregulation of TH in the striatum during long-term (e.g., several months) GDNF overexpression (Rosenblad et al., 2003; Georgievska et al., 2004). We found that deleting endogenous GDNF expression in the brain did not induce a change in striatal TH protein levels, suggesting that the reported effects of GDNF on TH expression (Theofilopoulos et al., 2001; Xiao et al., 2002; Rosenblad et al., 2003; Georgievska et al., 2004) are likely specific to ectopic GDNF.
In summary, our results suggest that GDNF regulates the normal function of the dopamine transporter in the striatum and that deleting or reducing GDNF has direct functional consequences on amphetamine-induced behavior. Given that DAT function is highly relevant to a wide range of psychiatric and neurological disorders, including Parkinson's disease, ADHD, depression, bipolar disorder, and addiction, future research should investigate the precise role that endogenous GDNF plays in these conditions.
Footnotes
J.J.K. was supported by the Finnish Cultural Foundation and the Emil Aaltonen Foundation. A.P. was supported by the Jane and Aatos Erkko Foundation. K.V. was supported by the University of Helsinki doctoral program Brain and Mind. M.S. was supported by the Sigrid Juselius Foundation. J.-O.A. was supported by the Academy of Finland (Grants 136591, 140983, and 263700), the Sigrid Juselius Foundation, and the Institute of Biotechnology, University of Helsinki. We thank Susanna Wiss for technical assistance.
- Correspondence could be addressed to Dr. Jaan-Olle Andressoo, Institute of Biotechnology, University of Helsinki, Viikinkaari 9 (P.O. Box 56), Helsinki FIN-00014, Finland. jaan-olle.andressoo{at}helsinki.fi





{kind=link}
{kind=link}
{kind=link}
{kind=link}