

Abstract
Astrocytes are highly complex cells with many emerging putative roles in brain function. Of these, gliotransmission (active information transfer from glia to neurons) has probably the widest implications on our understanding of how the brain works: do astrocytes really contribute to information processing within the neural circuitry? “Positive evidence” for this stems from work of multiple laboratories reporting many examples of modulatory chemical signaling from astrocytes to neurons in the timeframe of hundreds of milliseconds to several minutes. This signaling involves, but is not limited to, Ca2+-dependent vesicular transmitter release, and results in a variety of regulatory effects at synapses in many circuits that are abolished by preventing Ca2+ elevations or blocking exocytosis selectively in astrocytes. In striking contradiction, methodologically advanced studies by a few laboratories produced “negative evidence,” triggering a heated debate on the actual existence and properties of gliotransmission. In this context, a skeptics' camp arose, eager to dismiss the whole positive evidence based on a number of assumptions behind the negative data, such as the following: (1) deleting a single Ca2+ release pathway (IP3R2) removes all the sources for Ca2+-dependent gliotransmission; (2) stimulating a transgenically expressed Gq-GPCR (MrgA1) mimics the physiological Ca2+ signaling underlying gliotransmitter release; (3) age-dependent downregulation of an endogenous GPCR (mGluR5) questions gliotransmitter release in adulthood; and (4) failure by transcriptome analysis to detect vGluts or canonical synaptic SNAREs in astrocytes proves inexistence/functional irrelevance of vesicular gliotransmitter release. We here discuss how the above assumptions are likely wrong and oversimplistic. In light of the most recent literature, we argue that gliotransmission is a more complex phenomenon than originally thought, possibly consisting of multiple forms and signaling processes, whose correct study and understanding require more sophisticated tools and finer scientific experiments than done until today. Under this perspective, the opposing camps can be reconciled and the field moved forward. Along the path, a more cautious mindset and an attitude to open discussion and mutual respect between opponent laboratories will be good companions.
Dual Perspectives Companion Paper: Multiple Lines of Evidence Indicate That Gliotransmission Does Not Occur under Physiological Conditions, by Todd A. Fiacco and Ken D. McCarthy
Introduction
Astrocytes are an integral part of our brains, fulfilling multiple disparate roles. Among others, they are thought to maintain brain architecture, nurse neurodevelopment, regulate cerebral metabolism and hemodynamics, and contribute to circuit information processing. Importantly, the exact role played by astrocytes in many of the above processes is still unclear and forms a source of debates and controversies (for review, see Pellerin et al., 2007; Araque et al., 2014; Volterra et al., 2014; Bazargani and Attwell, 2016). The one with perhaps the widest implications is the role in information processing, namely, active information transfer from glia to neurons (dubbed “gliotransmission”). Thus, the ability of communicating bidirectionally with synapses puts astrocytes in the position of actively controlling their short- and long-term properties, of modulating their strength, as well as of influencing the larger network dynamics and, ultimately, behavior (for review, see Volterra and Meldolesi, 2005; Hamilton and Attwell, 2010; Araque et al., 2014). Therefore, more than any other astrocytic property, the functions attributed to these cells by the “gliotransmission theory” have a direct impact on “classical” neuroscience dogmas and beliefs, and, if confirmed, will force revision of current understanding of brain function (and dysfunction).
Astrocyte biologists have made tremendous progress in the last 30 years in deciphering neuron-glial information flow. Today it is near universally accepted that astrocytes receive neuronal information via a wide array of membrane receptors and other sensory mechanisms, and translate it into a complex intracellular Ca2+ code and other signal-transduction pathways. However, does this astrocyte elaboration produce any output signal for the neuronal circuitry? The evidence for signal transmission from astrocytes to neurons in a time-frame of hundreds of milliseconds to minutes comes from multiple independent laboratories (for review, see Hamilton and Attwell, 2010; Zorec et al., 2012; Araque et al., 2014; Sahlender et al., 2014), and comprises several mechanisms, including gliotransmitter release via vesicular (SNARE-mediated) mechanisms (Kang et al., 1998; Bezzi et al., 2004; Zhang et al., 2004; Pascual et al., 2005; Crippa et al., 2006; Bowser and Khakh, 2007; Jourdain et al., 2007; Henneberger et al., 2010; Min and Nevian, 2012; Lalo et al., 2014), channel- and transporter-mediated mechanisms (Lee et al., 2010; Woo et al., 2012), as well as changes in neurotransmitter and ion uptake by the astrocytes (Pannasch et al., 2014; Li et al., 2015). Some of the above mechanisms are Ca2+-dependent (or at least sensitive to Ca2+ manipulations) and involve clostridial toxin-sensitive (TeNT, BoNT) membrane fusion events. This variegated astrocyte signaling was reported to translate into multiple types of synaptic and network modulation, including control of presynaptic transmitter release probability, of postsynaptic excitability, of different forms of activity-dependent and tonic synaptic plasticity as well as in an influence on more complex oscillatory network states (Kang et al., 1998; Jourdain et al., 2007; Fellin et al., 2009; Di Castro et al., 2011; Panatier et al., 2011; Takata et al., 2011; Min and Nevian, 2012; Navarrete et al., 2012; Lalo et al., 2014; Lee et al., 2014; Perea et al., 2014; Martín et al., 2015; Poskanzer and Yuste, 2016; Papouin et al., 2017).
In this context, a watershed moment was triggered in 2010 by simultaneous publication in prominent journals of two papers reporting shockingly contradictory results on the role of astrocytic Ca2+ signaling in LTP at hippocampal CA3-CA1 synapses. One study disrupted LTP by acutely infusing a Ca2+ chelator into CA1 astrocytes, and identified Ca2+-dependent d-serine release as the astrocyte mechanism necessary for LTP (Henneberger et al., 2010) (Fig. 1i), a result in line with those of several other laboratories who observed short- and long-term synaptic changes upon chelating Ca2+ in astrocytes (Kang et al., 1998; Jourdain et al., 2007; Navarrete and Araque, 2010; Di Castro et al., 2011; Panatier et al., 2011; Shigetomi et al., 2011; Min and Nevian, 2012; Navarrete et al., 2012). The other study (Agulhon et al., 2010) used a different approach (i.e., transgenic disruption or activation of the purported astrocyte Ca2+ signaling pathway underlying gliotransmission) and observed no difference in LTP at all! With the benefit of hindsight, we can say that the Agulhon et al. (2010) study was priceless in revealing the following: (1) an incomplete understanding of sources and mechanisms of Ca2+ signaling in astrocytes (see below); and (2) heterogeneity in the astrocytic contribution to LTP depending on the circuit and form of LTP studied. However, at the time, the paper was embraced by gliotransmission skeptics as the ultimate proof of the nonexistence of this form of astrocyte-to-neuron communication.

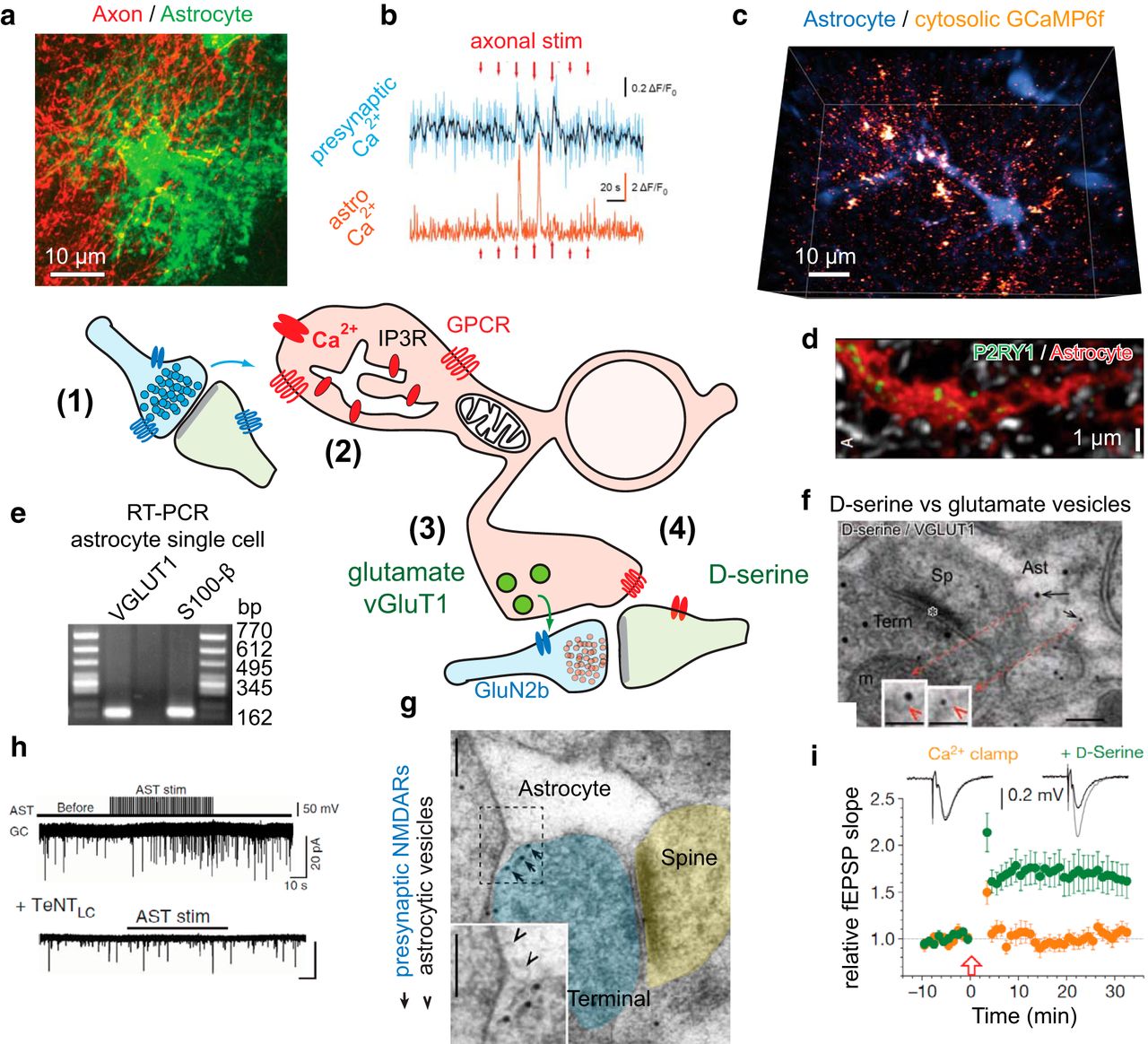
Gliotransmission in context. A simplified schematic of our current view of bidirectional information exchange between neurons and astrocytes via vesicular release of glutamate or d-serine. The cascade begins by (1) synapse activation of the astrocyte (a, b), producing Ca2+ elevations (b, c) (e.g., via astrocytic GPCRs; d). These Ca2+ transients may rely (2) on IP3 receptors on the ER or on other still not well-defined Ca2+ sources, and lead to the release of (3) glutamate (e) or (4) d-serine (f) from the astrocyte, ultimately producing presynaptic (h) or postsynaptic (i) modulatory effects at glutamatergic synapses (g). For clarity, omitted are many other confirmed gliotransmitters (e.g., ATP, lactate, taurine), release mechanisms (e.g., channel mediated and pump release), synaptic effects, and astrocytic signaling receptors and calcium sources (e.g., GPCRs: mGluR5, mGluR3, GABAB, adrenergic α1, CB1, D1/D2; sources: TRPA1, GLT-1/NCX, P2X) discussed in the text and elsewhere. a, Astrocytes interact with a large number of neuronal synapses located within their domains. A subset of the perforant path axons is transiently labeled with tdTomato, whereas the astrocytes are visualized by expression of eGFP under GFAP promoter (G. Carriero, A.V., unpublished observations). b, Minimal electrical stimulation causes confined local calcium elevations both in a targeted axon and in adjacent astrocytic structures, mostly gliapil (axons: jRCaMP1a; astrocytes: GCaMP6f). Modified with permission from Bindocci et al. (2017). c, Astrocytes show large levels of heterogeneous, spatially disconnected local Ca2+ activity in basal condition, particularly at the cell peripheries, in fine processes and gliapil. Shown is a 1 s snapshot of the Ca2+ activity (fire) in a GCaMP6f-expressing astrocyte monitored three-dimensionally. The astrocytic core structure, excluding the gliapil, is visualized by uptake of SR101 dye (blue). Modified with permission from Bindocci et al. (2017). d, Astrocytes express metabotropic receptors, including Gq-coupled receptors, which are among those responsible for Ca2+ transients. Such receptors often display heterogeneous cell distribution. An example is shown of P2RY1 staining (green) along an astrocytic process (red) in adult hippocampal tissue. Modified with permission from Di Castro et al. (2011). e, Astrocytes were also shown to express vesicular glutamate transporters needed for loading glutamate into exocytotic vesicles. Here single-cell RT-PCR data from astrocytes patched in the adult hippocampus. Modified with permission from Bezzi et al. (2004). f, Complementary immunogold EM evidence, showing the presence of both l-glutamate and d-serine particles in astrocytic synaptic-like microvesicle vesicles, apparently in different vesicle pools. Shown is an example of double-immunostaning for d-serine (small gold particles) and VGLUT1 (large gold particles). Scale bars: 100 and 50 nm in insets. Modified with permission from Bergersen et al. (2012). g, An example of EM staining showing the presence of astrocytic synaptic-like microvesicles directly opposed to putative presynaptic NMDARs. Gold particles represent GluN2b staining. Arrowheads indicate docked synaptic-like microvesicles. The receptors appear to be clustered in presynaptic terminal locations far away from the synaptic cleft and in direct vicinity of the astrocytic structures. Scale bars, 100 nm. Modified with permission from Jourdain et al. (2007). h, i, Synaptic consequences of gliotransmitter release. h, Electrical stimulation of astrocytes causes a transient increase in mEPSC frequency recorded in a nearby dentate gyrus granule cell. This increase is abolished by infusing the astrocyte with a tenatus toxin light chain (TeNTLC) that disrupts vesicular fusion by cleaving VAMP2 and VAMP3 SNAREs. Modified with permission from Jourdain et al. (2007). This response is mimicked by local P2Y1 receptor activation, blocked by astrocytic Ca2+ chelation, and modulated by changing glutamate uptake capacity with TBOA (Jourdain et al., 2007; Di Castro et al., 2011; Santello et al., 2011). i, Astrocytic Ca2+ chelation with EGTA-Ca2+ clamp solution in the patch pipette blocks hippocampal LTP in the nearby CA3-CA1 synapses, by preventing release of d-serine. Supplementing the slice with exogenous d-serine in the bath solution fully restores LTP. Adapted with permission from Henneberger et al. (2010). LTP was suppressed also when d-serine synthesis was blocked via infusion of the serine racemase inhibitor HOAsp into the astrocyte or when exocytosis was blocked by perfusing the astrocyte with TeNTLC (Henneberger et al., 2010).
Data vs interpretation
In this wave of skepticism, seminal observations, such as the Ca2+ oscillatory response of cultured astrocytes to neuronal input (Cornell-Bell et al., 1990) and the ensuing Ca2+-dependent neuronal activation (Nedergaard, 1994; Parpura et al., 1994), were dismissed as mere artifacts of the cell culture preparation (Barres, 2008). Likewise, careful follow-up work showing glutamatergic gliotransmission in acute brain slices (Bezzi et al., 1998; Kang et al., 1998; Fellin et al., 2004; Kang et al., 2005; Perea and Araque, 2007) was put aside. Strong cell biological evidence for the existence of a glutamatergic vesicular compartment in astrocytes, based on both specific electron microscopy (EM) immunogold vGlut labeling of vesicular structures and single-cell RT-PCR of vGlut mRNA expression (Bezzi et al., 2004; Bergersen et al., 2012; Ormel et al., 2012), were countered mainly by “negative” transcriptome analysis data for vGlut mRNA expression (Cahoy et al., 2008; Zhang et al., 2014) and by presumed (but unmeasured) insufficiency of glutamate levels in astrocytes due to high glutamine synthase activity (Barres, 2008). Likewise ignored was the related functional evidence of the synaptic effects of astrocyte glutamate release acting on presynaptic NMDA receptors, which comprised the ultrastructural localization of the target receptors at loci in nerve terminals directly apposed to astrocytes (Jourdain et al., 2007).
Eventually, even in vivo data reporting gliotransmission-related behavioral phenotypes in astrocyte-selective transgenic mouse models (Slezak et al., 2012; Clasadonte et al., 2013; Hines and Haydon, 2013; Lee et al., 2014) were attacked both experimentally (Fujita et al., 2014) and conceptually (Sloan and Barres, 2014) as simply due to lack of target specificity (but see counterarguments on PubMed Commons, https://www.ncbi.nlm.nih.gov/pubmed/25505312#comments, as well as more recent studies reaffirming astrocyte-selectivity of the transgenic models, Sultan et al., 2015; Papouin et al., 2017). The criticism of data obtained in culture preparations may have some merit, notably for studies in which the culture data are not supported by companion data in more intact preparations, in view of the property of astrocytes to change their gene expression profile depending on the culture condition (see, e.g., Foo et al., 2011). However, the arguments advanced against gliotransmission in slices and in vivo appear to be flawed by oversimplification and/or preconception about what is required to constitute a gliotransmission system.
Some of the most striking oversimplifications can be listed and discussed as follows:
Focusing on a single neurotransmitter receptor as trigger of gliotransmission
Because most of fast neurotransmission in the brain is glutamatergic, the simplest logical way for astrocytes to receive neuronal inputs would be via glutamate detection, ideally via a single receptor, such as mGluR5. The supposed prominence of mGluR5 signaling in gliotransmission probably stems from the early results in culture showing the role of metabotropic glutamate receptors in causing astrocytic Ca2+ responses, subsequently confirmed by several influential studies in slices and in vivo for mGluR5 (Wang et al., 2006; D'Ascenzo et al., 2007; Panatier et al., 2011; but see below!). The idea also fits with a tantalizingly straight-forward model for tripartite synapse functioning, wherein glutamate release at individual synapses could immediately and accurately trigger rapid Ca2+ responses and feedback gliotransmitter release from astrocytes (D'Ascenzo et al., 2007; Hamilton and Attwell, 2010). On this basis, a report that mGluR5 receptor expression is reduced/absent in adults (Sun et al., 2013; see detailed discussion below) was embraced by skeptics as an argument against the existence of gliotransmission in the adult. However, this is incorrect, as gliotransmission into adulthood is demonstrated by the synaptic, circuital, and behavioral effects seen upon activation of multiple other astrocyte receptors, including purinergic P2Y1 (Santello et al., 2011; Delekate et al., 2014), endocannabinoid CB1 (Han et al., 2012; Martín et al., 2015), and cholinergic muscarinic (Takata et al., 2011; Navarrete et al., 2012) and nicotinic receptors (Papouin et al., 2017).
Focusing on a single Ca2+ source as trigger of gliotransmission
This view combines factual evidence, that buffering Ca2+ levels in astrocytes affects many forms of short- and long-term synaptic plasticity (for review, see Rusakov et al., 2014), and that astrocytes prominently express various GPCRs coupled to PLC-IP3 signaling (while apparently lacking canonical voltage-dependent Ca2+ channels), with the idea that Ca2+ sources in astrocytes are simple, known, and therefore easy to manipulate. Initial studies according to which a single IP3 receptor (IP3R2) was responsible for the absolute majority of astrocyte Ca2+ transients corroborated such a view (Petravicz et al., 2008). Therefore, deleting IP3R2 appeared to provide an ideal, black-and-white model for testing the role of astrocytic Ca2+ transients (and implicitly, Ca2+-dependent gliotransmitter release) on neurons. Negative results in IP3R2ko mice (Agulhon et al., 2010; Nizar et al., 2013; Takata et al., 2013) have strongly alimented gliotransmission skepticism. Later work has, however, shown that this conclusion is incorrect because initial studies in IP3R2KO mice monitored only a small portion of the astrocytic Ca2+ signals, those most easily detectable in standard imaging conditions (see below), but failed to monitor the largest component of the signals, which are fast and local, confined to processes and to their thinner branches, the so-called gliapil (Srinivasan et al., 2015; Bindocci et al., 2017), that is, to the regions most intermingled with synapses (Chao et al., 2002; Volterra et al., 2014). Later, it was likewise shown that such local component is largely IP3R2-independent (Kanemaru et al., 2014; Srinivasan et al., 2015; Rungta et al., 2016) and may instead depend on a variety of additional Ca2+ sources (for review, see Volterra et al., 2014; Bazargani and Attwell, 2016, 2017), or even possibly other IP3 receptor types (Sherwood et al., 2017).
Assuming that the calcium code for gliotransmission is understood and can be reproduced by exogenous manipulations
The assumptions discussed above in Focusing on a single Ca2+ source as trigger of gliotransmission led also to the idea that transgenic expression of a xeno Gq-GPCR (MrgA1) would reproduce the physiological Ca2+ encoding underlying gliotransmission. Experiments showed that MrgA1 activation induces steady, long-lasting (minutes) Ca2+ elevations involving most of the core structure of an astrocyte (Fiacco et al., 2007) that have, however, no effect on the induction of LTP at neighboring synapses (Agulhon et al., 2010). These negative data were taken as additional evidence against gliotransmission. However, there is no experimental proof that relevant astrocyte encoding works this way. Actually, a recent study looking via 3D imaging at the overall endogenous Ca2+ activity of an astrocyte, including in vivo in awake mice, found the signaling to be mostly asynchronous and spatially uncoupled, occurring in myriads of frequent fast local transients, most notably at the cell peripheries (Bindocci et al., 2017). The importance of signal frequency in Ca2+ encoding is highlighted by a vast literature in many cell types (see e.g., Berridge et al., 2000; Berridge, 2007), showing in particular that a single large elevation often produces much lower output response than several small, oscillatory ones (see e.g., Li et al., 1998). This is demonstrated also for astrocyte gliotransmitter release, which occurs in multiple episodes in response to oscillatory Ca2+ patterns, whereas in a single one in response to long-lasting Ca2+ elevation (Pasti et al., 2001; see also Todd et al., 2010 for Ca2+ pattern-dependent glial output responses to synapses). Therefore, it is not surprising that the long-lasting Ca2+ elevation induced by MrgA1 stimulation was not synaptically effective. Likewise, it is not surprising that differences in the amount, tonic activity, and/or localization of transgenic versus endogenous receptors can generate different and functionally nonequivalent patterns of Ca2+ elevation (discussed by Tritsch and Bergles, 2007), the same when using different pharmacological application protocols to stimulate astrocyte receptors, as prominently illustrated by contrasting results upon mGluR5 stimulation with t-ACPD (1-aminocyclopentane-trans-1,3-dicarboxylic acid) agonist in vivo in adults (Sun et al., 2013 vs Nizar et al., 2013; see below). Nonetheless, skeptics would say that the “negative” MrgA1 data still argue against gliotransmission because the large, steady Ca2+ elevation produced by the receptor activation, while not mimicking the endogenous Ca2+ encoding of astrocytes for gliotransmission, should then occlude it, and thereby affect synaptic responses. However, based on the experimental setting used by Fiacco et al. (2007), it is unclear whether MrgA1-evoked long-lasting Ca2+ elevations interfere or not with frequency encoding in the peripheral perisynaptic (gliapil) regions, where astrocytic signals remain highly locally buffered and compartmentalized. Indeed, these authors at the time could not rely on the current genetically encoded Ca2+ indicators which report also gliapil Ca2+ dynamics (Srinivasan et al., 2015, 2016; Agarwal et al., 2017; Bindocci et al., 2017). Therefore, while the MrgA1 receptor seems to be expressed by most (80%–90%) of the astrocytes in the hippocampus (Fiacco et al., 2007), it is not demonstrated what percentage of the gliapil volume is effectively disrupted, and whether this would disrupt ensuing local signaling to synapses by a sufficient number of astrocytes, particularly at those synapses preferentially sampled by the field electrode. One might further argue that a sufficient number of astrocytes remained undisrupted and were able to release d-serine (via volume transmission), or that the astrocytic component of LTP (including d-serine tone) may vary considerably throughout the day, as was recently reported by (Papouin et al., 2017), meaning that LTP readings would have a diurnal component.
Assuming that synaptic plasticity (LTP/LTD) and/or gliotransmission occur in a single flavor
Scientists addressing the role of gliotransmission in synaptic plasticity have a tendency to extrapolate their results obtained in one circuit under highly specific conditions to the entire brain at large. However, one should be aware that there are tens of different forms of synaptic plasticity reported so far (for review, see Citri and Malenka, 2008), with hundreds of individual players involved in what we call “LTP” (discussed by Lisman et al., 2003). These plasticity forms are not mechanistically identical and may involve different forms of astrocyte signaling, and some may not involve astrocyte signaling at all (e.g., at synapses that are not closely surrounded by astrocytic structures). Consequently, reports that at first sight appeared to be contradictory, such as IP3R2 knock-out does not affect tetanic hippocampal CA1 LTP (Agulhon et al., 2010) while affecting cholinergic CA1 LTP (Navarrete et al., 2012), do not indeed need to be contradictory. Likewise, observations using a given plasticity-induction paradigm do not necessarily apply to another paradigm or to the same paradigm when tested in a different brain area or circuit (for review, see, e.g., Araque et al., 2014; Rusakov et al., 2014).
Equating vesicular transmitter release in astrocytes to release at neuronal synapses
Concerning vesicular exocytosis, data in astrocytes have been often compared with data at neuronal synapses and interpreted consequently. The fact that certain components of the canonical exocytotic synaptic release machinery were not found in astrocytes (or where found at very low levels), or that the number of observed vesicles was much lower than in nerve terminals, was considered as evidence questioning the existence of vesicular release, notably for glutamate (e.g., Barres, 2008; Sloan and Barres, 2014). However, this conclusion is not warranted. For instance, ultrastructural EM studies did detect l-glutamate- or d-serine-labeled vesicles in astrocytes (Bergersen et al., 2012; Ormel et al., 2012; but see Nedergaard et al., 2002), albeit in small amounts. This paucity is a problem only if one assumes that release sites in astrocytes must resemble those in nerve terminals and similarly contain a large reserve pool (>90% of the synaptic vesicles). However, this is not expected, given that the signals that activate the reserve pool in neurons, high-frequency firing discharges, are not generated in astrocytes. Moreover, even if astrocytes may express very low levels (no levels for transcriptomic analysis, see below) of canonical SNARE proteins and Ca2+ sensors mediating exocytotic release at neuronal synapses (Cahoy et al., 2008; Zhang et al., 2014; but see Bezzi et al., 2004; Crippa et al., 2006; Martineau et al., 2013; Schnell et al., 2015), they do express alternative SNARE isoforms, which can support vesicular release, albeit with slower kinetics (Verkhratsky et al., 2016). In this context, it is intriguing that clostridial toxins, such as tetanus toxin light chain (TeNTLC), known to abolish release at synapses by cleaving the vesicular SNARE VAMP2, work very effectively also in astrocytes in situ, which apparently contain little VAMP2 (Zhang et al., 2014; but see Chai et al., 2017). A possible explanation is that TeNTLC in astrocytes acts on VAMP3, a VAMP2 isoform that is also a toxin substrate, can functionally substitute for VAMP2 and is abundantly expressed in these cells (Bezzi et al., 2004; Schubert et al., 2011). Alternatively, large levels of VAMP2 are not required, given the lack of a reserve pool. Similar considerations apply to the expression of vesicular glutamate transporters (vGluts) in astrocytes, which was detected at low levels by both EM immunogold labeling and single cell RT-PCR (Bezzi et al., 2004) but not by transcriptome analysis. The latter data were taken as evidence against glutamatergic gliotransmission (Cahoy et al., 2008; Zhang et al., 2014). We will discuss later the issue of sensitivity of the different experimental approaches. However, the “negative” transcriptome results assume that “transcriptomics detectable” levels of vGluts are needed to release glutamate during gliotransmission, whereas this is not the case because functionally relevant glutamate release can occur with <10 vesicles per release site (size of neuronal active pool), each containing ∼10 vGlut molecules (Takamori et al., 2006). Moreover, the synergic argument used against vesicular glutamate release by the same authors (i.e., that conditions to take up glutamate into vesicles are unfavorable in astrocytes because glutamine synthetase lowers the cytoplasmic glutamate concentration to “housekeeping levels”) (Barres, 2008) is also unwarranted. Thus, the argument implies that we know enough about intracellular glutamate in astrocytes to reliably model the interplay between vesicular uptake and enzymatic transformation into glutamine, which is not the case. Indeed, calculations of glutamate levels based on comparative counting of immunogold l-glutamate particles in the cytosol of astrocytes and at synapses (Bergersen et al., 2012) support vGlut-mediated uptake into astrocytic vesicles. Likewise, extrapolation of quantal size of astrocytic vesicles supports functional efficacy of the astrocyte vesicular glutamate release, notably at extrasynaptic NMDA receptors (Hamilton and Attwell, 2010; Araque et al., 2014; Sahlender et al., 2014).
Conceptually out of question? Learning from neurons
Paradoxically, many objections of gliotransmission skeptics seem to stem from pushing the concept too far, as if the authors that proposed the “tripartite synapse” idea implied that astrocytes are to have functions paralleling neurotransmission, that is, that they reliably and faithfully receive all neuronal input, and process and respond to it on a millisecond timescale at every synapse. Obviously, when thus described, the concept of gliotransmission could be invalidated by just challenging a few underlying assumptions (e.g., receiving neuronal input must happen through astrocytic mGluR5, or releasing astrocytic glutamate requires large quantities of vGlut-expressing vesicles (Barres, 2008; Nedergaard and Verkhratsky, 2012). In contrast, if one would come back to the original definition of “tripartite synapse” (for review, see Araque et al., 2014) and read that astrocytic sensing and processing can occur on “slow” temporal scales (e.g., hundreds of milliseconds to minutes), via a variety of mechanisms, and without necessarily requiring a canonical Type I synapse release scenario, one would have to admit that the concept is no longer easy to discard.
Therefore, a key issue is whether the reported features of gliotransmission are conceptually out of the question or not. We argue that they are not and, in particular, that similar examples already exist in the world of neurons! For instance, does neuron-to-neuron communication always rely on a one-to-one, presynaptic/postsynaptic pairing and submillisecond precision? Do neuronal studies exclude noncanonical release mechanisms and sites? First, both fast and slow modes of neurotransmission have been known for decades (for review, see Greengard, 2001): fast transmission involves presynaptic release and postsynaptic action potential response, whereas slow transmission comprises essentially everything that deals with information processing, learning, and memory: LTP/LTD, receptor and channel trafficking and modification, neuromodulatory changes, and so on. In one instance, a parallel between neuronal release of “neuromodulators,” such as catecholamines (e.g., dopamine) and neuropeptides, and astrocytic release of gliotransmitters is quite revealing. Here, the dopamine release from neurons is not targeted to just a single postsynaptic site but may also escape the synaptic cleft and reach extrasynaptic receptors away from the release sites (Floresco et al., 2003). Moreover, remote signaling is not exclusive of dopamine and catecholamines but is seen with many other transmitters, even classical ones, such as GABA and glycine, or with dendritically released neuropeptides that establish communication on large spatial and slow temporal scales (Stern, 2014). Even in the case of glutamate, whose diffusion is highly limited by the high-affinity membrane transporters, activation of extrasynaptic receptors, such as extrasynaptic NMDARs, occurs under some circumstances. In keeping, noncanonical neuronal release sites (e.g., in somas and dendrites) are seen for catecholamines (Rice and Patel, 2015), neuropeptides (Ludwig and Stern, 2015), retrograde messengers, and even for classical neurotransmitters (Koch and Magnusson, 2009; Regehr et al., 2009; Kennedy and Ehlers, 2011). These sites generally do not show large accumulation of vesicles (Rice and Patel, 2015) or the presence of the typical release machinery (Tobin et al., 2012) seen, for example, in glutamatergic terminals. Moreover, in the case of somatodendritic release, evidence that the release is Ca2+- and action potential-dependent is only suggestive, and the identity of the involved Ca2+ source is still debated (Bergquist and Nissbrandt, 2005). Importantly, the temporal scale of neuronal release of neuromodulators can be much longer (seconds to minutes) than normal fast synaptic transmission (1 ms to several milliseconds), and much more comparable with that postulated for gliotransmitters (Schultz, 2007). Finally, does a number of vesicles at astrocytic sites that represents 5%–10% (Bergersen et al., 2012) of the total number of vesicles (∼400) per presynapse (Wilhelm et al., 2014) predict reduced/no functionality? Indeed, in nerve terminals, normal transmission does not require most of the existing vesicles; and under many circumstances, as indicated, it is assured by a readily releasable pool of ∼10 vesicles (Rosenmund and Stevens, 1996), together with the rest of the recycling pool (∼45 vesicles in total) (Marra et al., 2012; for exceptions, see Denker and Rizzoli, 2010). Consistently, the copy number of certain SNARE proteins supports fusion/recycling of only ∼5%-10% of the total number of vesicles in a terminal (Takamori et al., 2006; Wilhelm et al., 2014). Based on the above information, the conditions proposed for astrocytic vesicular gliotransmitter release are clearly not outside the scope routinely encountered in neurons, and therefore not out of the question. Once we consider other more enigmatic forms of transmission in neurons, such as retrograde messaging (CO, NO, lipids) involved in LTP (Zhuo et al., 1993), the proposed gliotransmission mechanisms become “business as usual” for the brain and the remaining question is just “why astrocytes are involved?” which we will address below.
Is “evidence against” methodologically stronger than “evidence for?”
In light of the above data, why are gliotransmission skeptics so adamant in their views? Is their skepticism justified by a few contradictory results? Does not this happen quite commonly in any young, rapidly developing field? Is, perhaps, the negative evidence against gliotransmission methodologically much stronger than the evidence for it? We do not believe so. Let us consider three of the strongest evidences presented against gliotransmission: (1) genetic models like IP3R2KO giving negative results; (2) “absence” of vGluts in the astrocyte transcriptome; and (3) “absence” of mGluR5 in adult brain astrocytes.
As previously mentioned, Agulhon et al. (2010) used the IP3R2KO mouse model to address the role of gliotransmission in synaptic plasticity based on their previous observation that IP3R2 deletion suppresses Ca2+ signaling in astrocytes (Petravicz et al., 2008) and convincingly demonstrated that IP3R2ko mice retain intact tetanic CA1 LTP. This result overturned the expectations and led the field to conclude that “astrocytic Ca2+ signaling is not necessary for LTP.” Does this mean that the studies reporting the opposite result based on astrocytic Ca2+ chelation experiments as well as experiments blocking downstream astrocytic exocytosis via TeNTLC infusion (Henneberger et al., 2010) were wrong? Or were the Agulhon et al. (2010) data wrong? Indeed, neither one was wrong. Years later, a “synthesis” view emerged, wherein IP3R2 may not have been the (main) Ca2+ signaling pathway involved in that particular form of LTP (Volterra et al., 2014). Follow-up studies revealed that astrocytes of IP3R2KO mice largely maintain Ca2+ signals in their small peripheral branches and gliapil (Kanemaru et al., 2014; Srinivasan et al., 2015; Rungta et al., 2016), which was missed by the earlier studies that saw a dramatic reduction of the Ca2+ signals, but looking just in soma and stem processes (Petravicz et al., 2008). The source of the IP3R2-independent Ca2+ signals is still unclear and may be extracellular, through TRPA1 channels (Shigetomi et al., 2011; but see Rungta et al., 2016), mitochondrial (Jackson and Robinson, 2015; Agarwal et al., 2017), or even mediated by other IP3 receptor types (Sherwood et al., 2017). Importantly, as discussed, other forms of LTP, such as cholinergic LTP at the same CA1 synapses, were abolished in IP3R2KO mice (Navarrete et al., 2012). Therefore, the initial conclusion made by the field should be revised to the quite different “astrocytic Ca2+ signaling mediated by a particular isoform of IP3 receptor does not play a role in a particular form of LTP.” The lesson is that the IP3R2KO model, like other “first-generation” models designed to test gliotransmission (dnSNARE, iBot, MrgA1), works under some (not all) conditions (Oliveira et al., 2015). While this supports the existence of gliotransmission, it also highlights that we currently lack proper background knowledge and fine tools to reliably dissect it. In this context, it is important to not forget the major conceptual difference existing in studying Ca2+ dynamics in astrocytes versus neurons: thanks to decades of studies of the electrical encoding, we mostly know what Ca2+ signals in neurons mean in functional terms (e.g., in the reporting of action potential firing), whereas in astrocytes we use Ca2+ imaging and related tools to reveal unknown aspects of the biology of these cells, and this almost inevitably leads to accidents like the one discussed above.
Transcriptome analysis showed below-detection levels of the machinery expected to drive glutamate exocytosis in astrocytes, notably of vGluts (Cahoy et al., 2008; Zhang et al., 2014). Is this negative evidence superior to the positive one obtained with single-cell RT-PCR studies (see, e.g., Bezzi et al., 2004)? Arguably, not, as transcriptome analysis is less sensitive than single-cell RT-PCR and unlikely to detect genes that have low expression in astrocytes. Importantly, transcriptome analysis provides quantitative measures but quantity is not quality, and low abundance expression does not imply functional irrelevance. A striking example of the contrary comes from cell-specific knock-out studies of endocannabinoid CB1 receptors. While the receptor is expressed at much higher level in neurons than in astrocytes, surprisingly, it was removal of the little amount of CB1 protein in astrocytes and not of the large amount in neurons that caused the appearance of a strong phenotype with altered synaptic plasticity and cognitive behavior (Han et al., 2012). The experimental approach used in the above study and in more recent ones (Papouin et al., 2017), that is, comparison of the synaptic and behavioral phenotypes in mouse lines that alternatively carry astrocyte-specific or neuron-specific knock-out of a given protein, is today the most convincing approach for defining functional relevance/irrelevance of astrocytic proteins. In contrast, bulk-level transcriptome data, although immensely useful for large screening studies, should be, in our opinion, treated with much more caution when claiming absence of expression of given proteins and the results of transcriptome analysis confirmed by higher-sensitivity methods, such as mass spectrometric proteomic analysis. Likewise, immunolabeling approaches, notably those relying on optical versus EM detection, are inadequate to define the presence/absence of vGluts in astrocytes due to insufficient resolution in view of the strong intermingling of the astrocytic and neuronal structures, often only tens of nanometers apart, and of the overwhelming vGlut labeling in neuronal terminals. Therefore, ultimate proof for presence/absence of vGluts in astrocytes is not yet available and will require new tools, such as astrocyte-specific vGlut knock-out mice.
A recent study showed decline of astrocyte mGluR5 expression from developmental stages to nearly absence into adulthood (Sun et al., 2013). By reporting lack of somatic Ca2+ responses to mGluR5 agonists in adults in vivo, this influential study implicitly cast grave doubt on the existence of Ca2+-dependent gliotransmission in the adult brain, at least according to the simplified tripartite synapse model discussed above. While the authors carefully avoided any such statement, nonetheless the overwhelming conclusion made by the field (no matter how unjustified) was that gliotransmission is probably just a developmental phenomenon (e.g., see Sun et al., 2013, Editor's summary). However, the study per se has several problems limiting its utility: the scientists did not look beyond somatic Ca2+ responses, which account for no more than 3% of the Ca2+ activity present in astrocytes in the adult brain in vivo (Bindocci et al., 2017). This is problematic for the strength of the study's conclusions, also in view of a previous report that mGluR5 is mainly present in the fine astrocytic processes in the adults (Lavialle et al., 2011). On the other hand, experimental issues should also not be ruled out, as other authors succeeded in observing robust astrocyte Ca2+ responses to mGluR1/5 agonist (t-ACPD) puff applications in adult mice (Nizar et al., 2013). Moreover, those authors puffed t-ACPD at a concentration 10- to 50-fold lower than Sun et al. (2013), which can hardly explain the negative results of the latter authors, unless the higher concentration immediately desensitized mGlu receptors or the downstream signaling.
“Middle-ground?”
To move forward from the present standstill, we suggest that both sides should step back and impartially consider all the existing evidence, positive and negative. What does it mean when the same experimental model/paradigm gives 50/50 evidence, as for the IP3R2KO mice? Does this mean that the underlying phenomenon does not exist, or rather that it is multifaceted and therefore unlikely to be revealed in full using current methods/tools? Should we continue to treat “gliotransmission” as a global, stereotyped On-Off phenomenon, or should we rather start to frame it in the context of age, circuit, stimulation protocol, and paradigm? In our opinion, the emerging picture of “gliotransmission” is that of a highly complex phenomenon, encompassing possibly tens of different mechanisms of tripartite synaptic communication, not unlike the picture we summarize under the term “LTP”. Neuroscience moved on from the simplistic fights over whether “LTP” is presynaptic or postsynaptic: a false binary oversimplification (Kullmann, 2012). It seems time to move on from arguing likewise over “gliotransmission.”
In particular, it is time to stop oversimplifying astrocyte biology. The most striking example is Ca2+ dynamics. A complexity in the distribution of astrocytic receptors, with a resulting heterogeneity of Ca2+ responses, was recognized by several laboratories, including the McCarthy laboratory, already in the 1990s (see, e.g., Lerea and McCarthy, 1989; Shao and McCarthy, 1993; Porter and McCarthy, 1997). However, since then, the field seems to have mostly embraced a conservative tendency, maintaining old-school models and test conditions. For example, until very recently, in vivo studies have kept focusing almost exclusively on somatic Ca2+ responses, despite the mounting evidence that such responses do not represent in anything (frequency, spatial extent, duration) local astrocyte-synapse exchanges occurring mostly at the cell peripheries (Bindocci et al., 2017). While a large subcellular complexity of Ca2+ dynamics with emphasis on local, fast Ca2+ events in processes was recognized already in 2011 (Di Castro et al., 2011; Panatier et al., 2011), several laboratories have nonetheless continued to study astrocyte Ca2+ phenomena with suboptimal space and time resolution (for review, see Volterra et al., 2014). We urge everyone to move forward, and realize that the large complexity of astrocytic signals needs to be taken into account, and catalogued appropriately, even if this requires methodological advances (see below). In summary, we are confident that the field will move out of the present “middle-ground” by avoiding three “deadly sins”: oversimplification, generalization, and overinterpretation.
Astrocytic computation: new view, new tools
Astrocytes were largely ignored by electrophysiologists for half a century. In the last 30 years, particularly thanks to advances in molecular and live imaging techniques, there literally came light: we now know more about these cells than ever seemed possible, and what emerges is complexity, compartmentation, and multifunctionality. Already on an evolutionary perspective, the trend to increased complexity seems quite clear (Verkhratsky and Nedergaard, 2016). Human astrocytes possess larger and more branched structure than rodent astrocytes, with long-range projecting processes endowed with evenly spaced varicosities (Oberheim et al., 2009), as well as unique neuromodulatory properties that can enhance information processing in the neuronal network when transplanted into mice (Han et al., 2013). While these results need confirmation, they support the idea that evolution has worked to complexify astrocytes and their roles in brain function.
Neurons have unique membrane properties and active channel conductances optimized for ultrarapid signal transmission on long distances. In contrast, astrocytes are electrically nonexcitable, highly compartmentalized cells with poor long-distance communication abilities, but with a huge amount of intracellular and extracellular membranes and receptors. Thereby, they are well suited for local chemical signal computation, storage, and processing on different spatiotemporal scales compared with neurons. Neurons, too, perform functions beyond the time and space precision of synaptic (excitatory, inhibitory) transmission, such as dendritic computation, neuromodulation, and retrograde signaling. However, astrocytes could have adapted evolutionarily to further integrate these functions beyond the neuronal domain because of their unique features and their central positioning with respect to all the other brain elements (neuronal, glial, and vascular). Each of these elements is likely to carry a specific set of information (metabolic, immune, etc.) with its own chemical repertoire and spatiotemporal language. Astrocytes may represent a crossroad of such diverse information and be able to integrate them into a language accessible to neurons. This idea is supported by emerging evidence, for instance, that specific products of the brain metabolism formed mainly or exclusively in astrocytes, such as l-lactate or d-serine, may be sent to neurons to produce neuromodulatory effects (Verkhratsky et al., 2016), or that astrocytes use cytokine signaling initiated by microglia to tune their modulatory inputs to synapses (Santello et al., 2011; Habbas et al., 2015). These data suggest that an exclusive function of astrocytes could be to fine-tune neuronal processing according to more general brain states.
The timescales at which neurons (milliseconds) and astrocytes (hundreds of milliseconds to minutes) work are impressively different: a scale difference between 1 ms and 5 min is the same as between 1 min and half a year! Intuitively, a system designed to “remember” things for 1 min might be very different from another one requiring a half-year storage. Analogously, astrocytes may have adapted to act as time integrators in complement to much faster neurons, allowing information to be enriched and processed in a more complex way, as discussed above.
Moreover, astrocytes are organized in individual territories and in subcellular compartments. This organization may underlie a function of astrocytes as integrators of the multiple independent (neuronal and non-neuronal) activities occurring within a given astrocytic territory/compartment (“neighborhood regulation”). In complement, several astrocytes can establish dynamic functional connections between them (e.g., via gap-junction communication). This may lead in turn to the creation of dynamic mosaics of territories, or domains, in which activities are integrated in space and time beyond the level of integration that can be assured by the intrinsic connectivity of the synaptic circuitry (“network regulation”). This dual, local/network, “integrative vocation” of astrocytes is exemplified by the nature of their Ca2+ signals, some of which are ultralocal, whereas others are widespread, at cellular or even multicellular levels.
We suggest that the fil rouge of any future astrocyte study should be complexity. The structural complexity of astrocytes must be intuitively paralleled by a similar molecular and functional complexity. Presumably, interactions with synapses occur mostly locally at the astrocyte peripheries, where processes branch repeatedly forming the gliapil, highly intermingled with the neuronal structures. Understanding the underlying biology, notably the spatiotemporal properties of Ca2+-dependent gliotransmission, requires extending Ca2+ studies to these thin structures (i.e., achieving at least micron-level resolution to capture the smaller faster local Ca2+ events prevalent in these subdomains) (Di Castro et al., 2011; Bindocci et al., 2017). Another key point is that astrocytic interactions with axons and dendrites, as well as with blood vessels and other functional partners, occur three-dimensionally. Yet, until now, two-photon microscopy studies have been performed in 2D, restricting imaging to a single 1-μm-thick focal plane, which captures <5% of an astrocyte volume (Bindocci et al., 2017). Such an approach is inadequate for studying the synaptic interactions of astrocytes and may actually lead to important misinterpretations. Only 3D volumetric Ca2+ imaging correctly spots specific local interactions while, at the same time, offering a complete view on the astrocyte dynamics (Fig. 1c). The latter aspect is critical, as no single locus of an astrocyte is likely to be identical to another locus and embedded in the same microenvironment (Bindocci et al., 2017). This implies that information concerning activity at a given cell locus cannot be held anymore as representative of activity at another locus or, worse, as representative of the overall cell activity. Consequently, studies based on random selection of focal planes and regions of interest as done so far will not be acceptable in the future. An approach combining high-resolution and cell-wide 3D imaging of astrocyte Ca2+ dynamics promises also to help reveal the rules of astrocytic encoding, including the biological determinants and the functions residing in the frequency and spatiotemporal properties of the signals. Together with microscopy advances, parallel availability of better Ca2+ indicators with improved signal-to-noise ratio, speed, and sensitivity, will be needed, particularly to study activity in the very thin peripheral astrocytic structures. This is expected to be very challenging, as the number of Ca2+ atoms involved in a transient in these structures may be so low that, depending on the level of indicator expression, the whole event may be either entirely missed or completely buffered. And, of course, Ca2+ is not the only second messenger for gliotransmission, and future studies will need to focus also on other mediators known to be involved in intracellular and intercellular astrocyte signaling.
Transgenic mouse models have provided crucial breakthroughs in the field of gliotransmission, but also some spectacular controversies. The early Ca2+ disruption models (IP3R2KO, MrgA1) are not as straightforward as originally believed: IP3R2KO mice do not abolish the totality of Ca2+ signals; MrgA1 mouse and DREADD approaches do not faithfully mimic properties and cell location of the endogenous GPCR-mediated transients. Results obtained with disruptable SNARE mediated release (dnSNARE) mice have been criticized by one study that reported transgene expression leakage (Fujita et al. 2014), although this observation could not be replicated by more recent studies, which found the model to be astrocyte-specific as originally described (Sultan et al., 2015; Papouin et al., 2017). In light of the above picture, new efforts are needed in preparing “second-generation” astrocyte-specific mice. A better control on the cell specificity, timing, and location of the changes introduced is required. For example, if an exogenous receptor is expressed, more careful verification is needed as to where the expression occurs, how much the expression level matches that of endogenous receptors, and what changes expression of this xenoreceptor produces in the “tonic” level of cell activity. If a genetically encoded indicator is expressed, proof that it does not unaccountably buffer the ions in the cell is likewise requested.
Restarting the dialogue
Pragmatically, is there anything that we can do at an interpersonal level to restart the dialogue? We believe that open discussion between laboratories (including current opponents) is the way to move forward. When two laboratories obtain contradictory results, an attitude to the dialogue rather than to dogmatic defenses and the willingness to cross-examine differences in experimental approaches, protocol conditions, etc., may help spot shortcomings and pitfalls, and generate new and better experimental designs, most likely with much more chances of success than if the same issues were pursued by each laboratory separately. This attitude is important: just because one laboratory has different results from another laboratory does not mean that one is “right” and the other is “wrong;” most likely, they are both “wrong,” by oversimplifying the picture and interpreting the results according to their preferred view, whereas the truth is more complex. Already understanding this, the principle of Socratic thinking means that both laboratories are now a step closer to grasping the truth. Educational initiatives, such as this Dual Perspectives article series, are welcome as they foster dialogue and mutual respect. Further benefit could come from joint grants and short visit/exchange funds for opponent laboratories to investigate the debated topics. This cultural approach combined with using new tools as detailed above will move the field forward. Ultimate breakthroughs concerning gliotransmission are, in our view, to come via the combination of multiple tools and experiments, ideally across multiple laboratories, up to bridging information at nanodomain (single synapse), subcellular, cellular, and network levels. We believe this is just a matter of years, and the future for astrocyte biology is bright.
Footnotes
This work was supported by European Research Council Advanced Grant 340368 Astromnesis to A.V., Swiss National Science Foundation Grant 31003A 173124/1, and National Center of Competence in Research Synapsy 51NF40-158776 and Transcure 51NF40-160620.
The authors declare no competing financial interests.
- Correspondence should be addressed to Dr. Andrea Volterra, Department of Fundamental Neurosciences, University of Lausanne, Rue du Bugnon, 9, 1005 Lausanne, Switzerland. andrea.volterra{at}unil.ch
References
Response from Dual Perspectives Companion Authors–Todd A. Fiacco and Ken D. McCarthy
Gliotransmission has generally been defined as the Ca2+-dependent release of neurotransmitters (glutamate, d-serine, ATP) by astrocytes. In their perspective, Savtchouk and Volterra assert that a lack of appreciation for the complexity of astrocyte Ca2+ activity underlies the fact that some investigators observe gliotransmission and others do not. While nearly all would agree that astrocyte morphology and Ca2+ activity are complex, to argue that it is the complexity of Ca2+ activity that underlies the different findings ignores a literature where universally noncomplex and nonphysiological forms of stimulating and recording astrocyte Ca2+ are used to induce gliotransmission. These stimuli do not produce a precise or complex “Ca2+ code” exhibiting delicate or localized spatiotemporal dynamics. Approaches that have induced or impaired physiologically and spatiotemporally relevant astrocyte Ca2+ find no evidence for gliotransmission.
Studies in both acute brain slices and in vivo have consistently found that ≥90% of spontaneous Ca2+ activity in hippocampal and cortical astrocytes occurs in the cell processes and not in the soma (Nett et al., 2002; Wang et al., 2006; Shigetomi et al., 2013; Sun et al., 2014; Bindocci et al., 2017).
In addition to this localized baseline Ca2+ activity, astrocytes exhibit widespread, synchronous Ca2+ elevations in vivo in response to sensory input, locomotion, or startle (Dombeck et al., 2007; Ding et al., 2013; Paukert et al., 2014). As discussed in our companion perspective, the vast majority of evoked and spontaneous astrocyte Ca2+ activity is GPCR- and IP3R2-dependent (Srinivasan et al., 2015; Agarwal et al., 2017). Once it was discovered that a small fraction of astrocyte Ca2+ activity persisted in IP3R2−/− mice, proponents of gliotransmission began to assert that these must be the key Ca2+ events essential to gliotransmission. While studies as to the origin of this remaining Ca2+ activity are ongoing, compelling evidence suggests that it is mitochondrial (Agarwal et al., 2017).
Mitochondrial Ca2+ is thought to be released synergistically with activation of IP3R2 (Agarwal et al., 2017). Mitochondrial Ca2+ activity is therefore much higher in the presence of IP3R2. Thus, the Ca2+ activity that is lost in the IP3R2 knock-out is spatiotemporally identical to the activity that remains: there is just much less of it. It is also significantly weaker in amplitude. It is unclear how weaker and significantly less Ca2+ exhibiting the spatiotemporal dynamics deemed essential for gliotransmission is the key to gliotransmission. Less spatiotemporally relevant Ca2+ will certainly not increase the probability of gliotransmitter release or its detection above baseline synaptic chatter in an adjacent neuron. An astrocyte has been estimated to contact upward of 100,000 synapses. Strong and synchronized Ca2+ involving the greatest number of astrocyte microdomains surrounding these synapses has been evoked by stimulating native (endothelin) or transgenic (MrgA1; hM3D DREADD) astrocytic GPCRs in IP3R2-expressing mice, producing the maximal spatiotemporally relevant Ca2+ with no change in neuronal activity. In summary, neither removal nor stimulation of biologically and spatiotemporally relevant astrocyte Ca2+ results in any detectable gliotransmission by astrocytes. Mounting evidence against gliotransmission also includes findings totally independent of Ca2+ (e.g., localization of d-serine to neurons rather than astrocytes; lack of expression of vesicular proteins). We suggest expanding efforts to understand the host of other astrocytic mechanisms and molecules regulating brain physiology and animal behavior to complement what has been an inordinate focus on Ca2+ and gliotransmission.





{kind=link}