

Abstract
Despite the development of numerous novel anticonvulsant drugs, ∼30% of epilepsy patients remain refractory to antiepileptic drugs (AEDs). Many established and novel AEDs reduce hyperexcitability via voltage- and use-dependent inhibition of voltage-gated Na+ channels. For the widely used anticonvulsant carbamazepine (CBZ), use-dependent block of Na+ channels is significantly reduced both in experimental and human epilepsy. However, the molecular underpinnings of this potential cellular mechanism for pharmacoresistance have remained enigmatic.
Here, we describe the mechanism that leads to the emergence of CBZ-resistant Na+ channels. We focused on the endogenous polyamine system, which powerfully modulates Na+ channels in a use-dependent manner. We had shown previously that the intracellular polyamine spermine is reduced in chronic epilepsy, resulting in increased persistent Na+ currents. Because spermine and CBZ both bind use-dependently in spatial proximity within the Na+ channel pore, we hypothesized that spermine loss might also be related to diminished CBZ response. Using the pilocarpine model of refractory epilepsy in male rats and whole-cell patch-clamp recordings, we first replicated the reduction of use-dependent block by CBZ in chronically epileptic animals. We then substituted intracellular spermine via the patch pipette in different concentrations. Under these conditions, we found that exogenous spermine significantly rescues use-dependent block of Na+ channels by CBZ. These findings indicate that an unexpected modulatory mechanism, depletion of intracellular polyamines, leads both to increased persistent Na+ currents and to diminished CBZ sensitivity of Na+ channels. These findings could lead to novel strategies for overcoming pharmacoresistant epilepsy that target the polyamine system.
SIGNIFICANCE STATEMENT Pharmacoresistant epilepsy affects ∼18 million people worldwide, and intense efforts have therefore been undertaken to uncover the underlying molecular and cellular mechanisms. One of the key known candidate mechanisms of pharmacoresistance has been a loss of use-dependent Na+ channel block by the anticonvulsant carbamazepine (CBZ), both in human and experimental epilepsies. Despite intense scrutiny, the molecular mechanisms underlying this phenomenon have not been elucidated. We now show that a loss of intracellular spermine in chronic epilepsy is a major causative factor leading to the development of CBZ-resistant Na+ currents. This finding can be exploited both for the screening of anticonvulsants in expression systems, and for novel strategies to overcome pharmacoresistance that target the polyamine system.
Introduction
Epilepsy is one of the most common and devastating neurological disorders, with ∼50 million affected patients worldwide. Despite the introduction of new antiepileptic drugs (AEDs) to the clinical field, over the past years, a substantial proportion (>30%) of patients remains refractory to AEDs (Blume, 2008). It is thus of great importance to understand why some patients respond well to AEDs, whereas in others treatment fails.
One main idea that has been advanced to explain pharmacoresistance is that molecular targets of AEDs are changed in such a way that their activity is reduced in chronic epilepsy, reducing their clinical efficacy. One such mechanism, described both in human epilepsy and in epilepsy models, is that voltage-gated Na+ channels are altered, displaying a reduced drug response (Remy et al., 2003; Remy and Beck, 2006). Na+ channels are important molecular targets of numerous first-line AEDs, with most AEDs exerting complex voltage- and use-dependent inhibition of Na+ currents. Specifically for carbamazepine (CBZ), a slowing of the fast recovery from inactivation causes a preferential reduction of Na+ channel availability during high-frequency discharges, which is thought to underlie the activity of CBZ in reducing epileptiform activity. Several studies have shown that use-dependent block of transient Na+ channels is lost both in human and experimental epilepsy, suggesting that this is a key mechanism underlying pharmacoresistance to CBZ on the cellular level (Remy et al., 2003; Remy and Beck, 2006).
Following this initial key finding, an intense search has attempted to determine what is the molecular basis for the emergence of “resistant” channels in chronic epilepsy. So far, changes in the expression of pore-forming Na+ channel subunits is not thought to account for altered use-dependent effects of CBZ. Likewise, the known downregulation of accessory β1 and β2 subunits following experimentally induced status epilepticus (SE; Ellerkmann et al., 2003) does not contribute to the generation of CBZ-resistant Na+ channels (Uebachs et al., 2010). This led us to the hypothesis that other molecular mechanisms, such as post-translational modifications of Na+ channel proteins are responsible for loss of efficacy of CBZ. Post-translational modifications could be particularly relevant because they could affect multiple subtypes of Na+ channels that are coexpressed in neurons. In a previous study, we have found that the pronounced upregulation of the persistent Na+ current in chronic epilepsy (Chen et al., 2011), is due to a different mechanism, namely, altered modulation by intracellular polyamines (Royeck et al., 2015). The polyamine system is a powerful modulatory system that use-dependently blocks Na+ channels from the intracellular side via interaction with the ion selectivity filter inside the channel pore (Huang and Moczydlowski, 2001; Fleidervish et al., 2008, Fu et al., 2012). Polyamine block has been shown to particularly affect the persistent Na+ current (Fleidervish et al., 2008). We have found that the polyamine spermine is strongly reduced in chronic epilepsy via increased degradation, leading to unblock of Na+ channels and increased persistent Na+ current (Royeck et al., 2015). We have therefore asked in this study whether altered spermine modulation can also lead to altered anticonvulsant drug pharmacology, and demonstrate that loss of polyamines is a major factor in the generation of CBZ-resistant Na+ channels in chronic experimental epilepsy.
Materials and Methods
Pilocarpine model of chronic epilepsy in rats.
Established protocols were used to develop an acquired refractory chronic state of epilepsy, able to mimic pharmacoresistance observed in human temporal lobe epilepsy (Cavalheiro et al., 1991; Ellerkmann et al., 2003; Remy et al., 2003). Male Wistar rats (200–250 g, age 30 ± 2 d, Charles River Laboratories) were first injected intraperitoneally with methyl-scopolamine (1 mg/kg, Sigma-Aldrich, dissolved in 0.9% NaCl solution) to reduce peripheral muscarinic effects. Thirty minutes after methyl-scopolamine administration, the muscarinic agonist pilocarpine (340 mg/kg body weight, Sigma-Aldrich, dissolved in aqua dest) was injected intraperitoneally to induce SE in most animals (∼80%). Prolonged tonic-clonic seizures were observed permanently and seizure-stages noted. SE was terminated after 40 min and animals were sedated with diazepam (20 mg/kg, i.p.; Ratiopharm). If convulsions could not be terminated, the same dose of diazepam was reinjected. For recovery, surviving animals were tended, hydrated and fed (0.5 ml glucose 5%, subcutaneously) and subsequently video-monitored. Two weeks after SE, most of the animals get spontaneous seizures. Frequency and semiology of seizures were monitored and classified in Racine stages. Only animals with video-documented generalized strong seizures were included in experiments to ensure conditions of chronic epilepsy. Rats with chronic epileptic conditions were used 29–110 d after SE. Sham-control animals were age matched and treated identically, with pilocarpine being substituted by saline. At least six epileptic animals were included in every group; n indicates the number of recorded cells. All animal experiments were conducted in accordance with the guidelines of the Animal Care Committee of the University of Bonn Medical Center. Animals were housed under a 12 h light/dark cycle with ad libitum access to water and food.
Preparation of hippocampal slices.
Rats were deeply anesthetized (ketamine 100 mg/kg, Pfizer, and xylazine 15 mg/kg, Bayer) and perfused through the heart with ice-cold artificial CSF (ACSF) containing the following (in mm): 60 NaCl, 100 sucrose, 26 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 5 MgCl2, 1 CaCl2, and 20 glucose, pH 7.4 (osmolality 305 mOsm). After decapitation, the brain was immediately removed and horizontal slices (300 μm thick) were prepared with a vibrating microslicer (Microm HM 650V). The slices were first transferred to a storage chamber filled with carbogenated (95% O2, 5% CO2) ACSF (the same sucrose-based ACSF as for the perfusion), then gradually warmed to 36°C in a water bath and maintained at this temperature for 30 min. The slices were subsequently transferred to a second storage chamber filled with ACSF containing the following (in mm): 125 NaCl, 26 NaHCO3, 3.5 KCl, 1.25 NaH2PO4, 2 MgCl2, 2 CaCl2, and 15 glucose, pH 7.4 (307 mOsm), where they were maintained for up to 6 h at room temperature. Finally, after an equilibration period of at least 60 min, slices were used for preparation of acutely dissociated neurons for voltage-clamp recordings. If not specifically marked, chemicals were obtained from Sigma-Aldrich.
Dissociation of granule cells.
Neurons were dissociated before starting patch-clamp recordings for optimal voltage control. One slice at a time was transferred to a tube with 5 ml oxygenated (100% O2) titration solution containing the following (in mm): 145 CH3SO3Na, 3 KCl, 1 MgCl2, 0.5 CaCl2, 10 HEPES, and 15 glucose, pH 7.4 adjusted with NaOH (315 mOsm). Pronase (protease type XIV, 2 mg/ml, Sigma-Aldrich) was added to reduce cell adhesion in tissue. Enzymatic incubation was first performed for 12 min at 36°C followed by 10 min incubation at room temperature. Afterward, the slice was washed with Pronase-free titration solution before the dentate gyrus was dissected from the hippocampus and subsequently triturated with fire-polished Pasteur pipettes of decreasing aperture in a Nunc Petri dish (35 mm, Thermo Scientific). The Nunc dish was subsequently mounted on an inverted microscope (Axiovert 100, Zeiss), where cells were allowed to settle down on ground for at least 10 min before perfusion of the chamber was switched on.
Voltage-clamp recordings in isolated neurons.
Patch pipettes were pulled from GB150F-8P borosilicate glass (Science Products) with a Flaming/Brown micropipette puller (P-97, Sutter Instruments). Pipettes with a tip resistance of 5 MΩ ± 1 MΩ were used for recordings and filled with intracellular solution containing the following (in mm): 110 CsF, 10 HEPES-Na, 11 EGTA, 2 MgCl2, 20 tetraethylammonium chloride (TEA), 0.5 GTP-Na2, 5 ATP-Na2, pH 7.25 adjusted with CsOH, (300 mOsm). For measurements with intracellular spermine, different concentrations (1 mm or 100 μm) of spermine were added to the intracellular solution. After settlement of cells, they were superfused with an extracellular solution containing the following (in mm): 40 CH3SO3Na, 90 TEA, 1.6 CaCl2, 2 MgCl2, 10 HEPES, five 4-aminopyridine, 0.2 CdCl2, and glucose 15, pH 7.4 adjusted with HCl (310 mOsm). At least 5 min were allowed for full equilibration with extracellular solution before starting recordings. After break-in, again 5 min were needed for full diffusion of intracellular solution through the patch-pipette (Huang and Moczydlowski, 2001). The anticonvulsant CBZ was dissolved in dimethyl sulfoxide and added to the extracellular solution. CBZ was applied according to established protocols (100 μm CBZ) via a hydrostatic perfusion system for at least 7 min. The effect of wash-in was controlled by reduction of transient Na+ current (INaT) amplitude (48.4 ± 13.2%). All voltage-clamp experiments were performed at room temperature (22°C ± 1°C).
Tight-seal whole-cell recordings were obtained with a seal-resistance of >1 GΩ using a patch-clamp amplifier (Axopatch 200B; Molecular Devices). Series resistance ranged between 4.3 and 6.4 MΩ and was compensated by 70–90%, maximal residual voltage error was <5.3 nA. Currents (filtered at 10 kHz) were sampled at 50 kHz with a Digidata 1440A and recorded by a personal computer using the Clampex 10.2 acquisition software (Molecular Devices). A liquid junction potential of 10 mV was calculated and corrected offline for all measured neurons.
Voltage-clamp paradigms and data analysis.
Established standard protocols (see Fig. 2A) were used to examine the voltage-dependence of steady-state INaT activation and inactivation. Using Ohm's law, obtained current values were converted to conductance G(V) according to:
 where VNa is the Na+ reversal potential, V the command potential and I(V) the peak Na+ current amplitude. G(V) was then fitted with GraphPad Prism software for each set of traces obtained from individual neurons with a Boltzmann equation, expressing conductance G(V) as a function of membrane potential V:
where VNa is the Na+ reversal potential, V the command potential and I(V) the peak Na+ current amplitude. G(V) was then fitted with GraphPad Prism software for each set of traces obtained from individual neurons with a Boltzmann equation, expressing conductance G(V) as a function of membrane potential V:
 where A1 − A0 is range of Na+ conductance, V1/2 represents the voltage at which G(V) is half-maximal, and k represents a slope factor indicating steepness of the curve. V1/2 values of Na+ channel activation and inactivation were determined for quantification.
where A1 − A0 is range of Na+ conductance, V1/2 represents the voltage at which G(V) is half-maximal, and k represents a slope factor indicating steepness of the curve. V1/2 values of Na+ channel activation and inactivation were determined for quantification.
To determine the recovery from fast inactivation of Na+ channels in dentate gyrus granule cells, a double-pulse protocol (Fig. 1A) with increasing interpulse intervals t = 1 ms * 2n {n ∈ ℕ = 0, …, 11} was applied. To ensure full recovery from inactivation, an interval of 5 s at −80 mV holding potential was interspersed between single protocols. Peak INaT amplitude from the second (test) pulse was normalized to the peak INaT amplitude from the first (conditioning) pulse. Time course of recovery was fit with a two-phase exponential association equation:
 with A0 representing the current amplitude at time point zero, τfast and τslow are the time constants of recovery with Afast and Aslow, the corresponding relative amplitude contributions. Fitting was done in GraphPad Prism using a Levenberg–Marquardt algorithm. In all cases, fits were performed on the data points obtained from individual cells and the parameters were averaged for quantification.
with A0 representing the current amplitude at time point zero, τfast and τslow are the time constants of recovery with Afast and Aslow, the corresponding relative amplitude contributions. Fitting was done in GraphPad Prism using a Levenberg–Marquardt algorithm. In all cases, fits were performed on the data points obtained from individual cells and the parameters were averaged for quantification.
Real-time RT-PCR.
mRNA was obtained from micro-dissected hippocampal dentate gyrus of rat brain tissue at different time points after SE using Dynabeads mRNA Direct Micro Kit (Invitrogen) according to the manufacturer's protocol. cDNA was synthesized from purified mRNA by reverse transcription using the RevertAid Premium First-strand cDNA Synthesis Kit (Fermentas) following the manufacturer's manual. Quantification of rat gene transcripts was performed by quantitative real-time RT-PCR (ABI PRISM 9700 HT, Applied Biosystems). For the mRNA quantification of Sat1, Amd1, and Odc1, we used the TaqMan gene expression assays (Sat1 Rn01419247_g1; Amd1 Rn01407917_g1; Odc1 Rn01469805_m1) and synaptophysin as a reference gene (Rn00561986_m1; Invitrogen) together with the Maxima Probe/Rox qPCR Master Mix (Thermo Fisher). Real-time RT-PCR was performed as described previously (Pitsch et al., 2012). After preincubation for 2 min at 50°C and 15 min at 95°C, we performed 40 PCR cycles (15 s at 95°C followed by 60 s at 60°C). We used a 6.25 μl reaction volume containing 3 μl of Master Mix, 0.3 μl of TaqMan gene expression assay, 1.7 μl of DEPC-H2O, and cDNA dissolved in 1.25 μl of DEPC-H2O. Reactions were performed in triplets. Transcript quantification was performed as relative gene expression according to the ΔΔCt method (Fink et al., 1998) compared with the neuron-specific reference gene, as it lacks significant expression changes following pilocarpine-induced SE (Becker et al., 2008). The signal threshold was set within the exponential phase of the reaction for determination of the threshold cycle (Ct).
Data analysis.
All data are presented as mean ± SEM. Data analyses were performed using Clampfit 10.2 software (Molecular Devices), custom made procedures in IGOR Pro 6.3 (WaveMetrics), Prism 7 (GraphPad Software), and Excel (Microsoft). ANOVA was used to analyze the difference between groups where appropriate. Statistic methods including n numbers and post-tests are mentioned for individual statistical comparison throughout this paper. ANOVA results are reported with F values including in-between group and within-group degrees of freedom in parentheses.
Results
Effects of CBZ on the recovery from fast inactivation are diminished in chronically epileptic animals
We first reproduced the findings obtained by Remy et al. (2003), and determined the effects of CBZ (100 μm) on INaT properties in dentate gyrus granule cells from control and epileptic animals. We examined the reduction of INaT maximal amplitudes by CBZ, which was not different in control and epileptic animals (46.5 ± 14.3% and 46.9 ± 15.5% in sham-control and epileptic animals, respectively, n = 21 and 17, voltage steps from −90 to −30 mV for 15 ms). We determined the fast recovery from inactivation with a standard double-pulse protocol (Fig. 1A). CBZ slowed the recovery kinetics in neurons isolated from sham control animals (Fig. 1A,B, left). This is illustrated by the right shift of the time course curve after drug administration (Fig. 1B, blue symbols). As described earlier (Remy et al., 2003), this shift was much less pronounced in post-SE animals (Fig. 1A,B, right). To quantify and compare the CBZ effects during the recovery process, we determined the normalized reduction of the Na+ current at each recovery time point (Fig. 1C). This analysis showed a significant loss of CBZ efficacy in neurons isolated from chronically epileptic animals. Two-way ANOVA revealed a significant effect of pilocarpine-treatment (F(1,432) = 65.5, p < 0,0001), time (F(11,432) = 53.41, p < 0.0001), and interaction (F(11,432) = 5.22, p < 0.0001; Fig. 1C; asterisks indicate Sidak's multiple-comparison post-test, *p < 0.05, ****p < 0.0001).

Effect of CBZ on recovery from fast inactivation of transient Na+ current. A, Double-pulse protocol with increasing time intervals between the two pulses was used to determine the recovery from fast inactivation (A, inset, top right). Representative current traces of INaT elicited by the double-pulse protocol before and after application of 100 μm CBZ are shown for sham-control and epileptic (post-SE) animals. Left current traces (black) are the average response to the first pulse, whereas responses to the second pulse are color-coded according to the time intervals between the two pulses (Δt). B, Averaged time course of recovery of INaT, plotted as mean ± SEM of the fractional peak conductance during the second depolarizing pulse, normalized to the first pulse response. Note the smaller right shift of the CBZ curve in post-SE compared with sham-control animals. C, Quantification of the CBZ effect on recovery from fast inactivation. The difference in peak conductance before and after CBZ application was calculated and plotted as mean ± SEM for responses at the various time points after initial inactivation. Statistical comparison of neurons from sham-control and post-SE animals revealed a highly significant loss of CBZ efficacy in chronic epilepsy (two-way ANOVA followed by Sidak's multiple-comparison post-test). *p < 0.05, ****p < 0.0001 for Sidak's test.
CBZ effects on steady-state voltage dependence of Na+ currents is unaltered by chronic epilepsy
CBZ reduces Na+ channel availability by both use-dependent block of INaT and stabilizing the inactivated state. Therefore, we also examined CBZ effects on voltage-dependence of Na+ channels. We applied holding potentials for 500 ms starting from −100 up to −20 mV in 5 mV steps, directly followed by a test pulse to −10 mV (Fig. 2A). In both sham-control and post-SE animals, 100 μm CBZ shifted the steady-state activation and inactivation curves toward more hyperpolarized potentials (Fig. 2B). The individual curves were fitted and the CBZ-evoked shift in V1/2 was determined for each cell. This analysis showed significant effects of CBZ on V1/2 of both activation and inactivation in both groups of animals (paired t test, sham-control activation: t(21) = 6.16; inactivation: t(21) = 17.17; post-SE activation: t(14) = 5.59, inactivation: t(14) = 10.12, for all four tests ****p < 0.0001). As expected, the effects on the voltage-dependence of activation were quantitatively smaller (Fig. 2C,D). In addition, the analysis revealed no significant differences of CBZ effects on the voltage-dependence of Na+ currents when comparing sham-control and post-SE animals (unpaired t-test with Bonferroni-corrected p < 0.025, n.s.).

Effect of CBZ on steady-state voltage dependence of Na+ currents. A, Voltage-clamp protocol (left) consisted of 500-ms-long pulses to test potentials ranging from −100 to −20 mV, immediately followed by a 15 ms pulse to −10 mV. Representative current traces are shown on the right for activation (recorded at time point a during the protocol) and inactivation (recorded at time point b during the protocol) of INaT, respectively. B, For evaluation of voltage-dependence, peak conductance was calculated from INaT and plotted as mean ± SEM normalized to the maximum peak conductance. Data were fitted to a Boltzmann equation for quantification; fits are shown superimposed on the average data points under ACSF conditions and after application of 100 μm CBZ. C, Membrane potential for half-maximal activation/inactivation (V1/2) of INaT was derived from Boltzmann fits of all neurons. Plots show V1/2 before and after application of 100 μm CBZ for all individual neurons, columns denote mean for ACSF and CBZ. Statistical analysis was performed with paired t test. ****p < 0.0001. D, Horizontal bar graphs show CBZ-induced shifts in V1/2 for channel activation and inactivation. No statistical difference between sham-control and post-SE animals was found (unpaired t-test with Bonferroni-corrected p < 0.025, n. s.).
Spermine metabolism is altered in chronically epileptic animals
Endogenous polyamines act as use-dependent Na+ channel blockers (Fleidervish et al., 2008). We have previously shown that spermine levels were significantly reduced in hippocampal homogenates from post-SE animals. This reduction could be ascribed to an increase in transcription of the spermine degrading enzyme spermidine/spermine N(1)-acetyltransferase (SSAT), determined in CA1 microslices (Royeck et al., 2015). We confirmed that a similar regulation of SSAT is also observed in the dentate gyrus, where the recordings described here were obtained. Indeed, we found that, similar to CA1, the expression of SSAT was increased (Fig. 3; two-way ANOVA, pilocarpine-treatment: F(1,27) = 17.74, p = 0.0003; Sidak's multiple-comparisons post-test, **p < 0.005 at 4 weeks post-SE). The expression of the spermine synthesizing enzyme ornithine decarboxylase was slightly increased (two-way ANOVA, pilocarpine-treatment: F(1,26) = 4.97, p = 0.0346), whereas S-adenosylmethionine decarboxylase was unchanged (two-way ANOVA, pilocarpine-treatment: F(1,27) = 0.146, p = 0.705) after SE.

Expression of key enzymes involved in polyamine metabolism. Results of from hippocampal region dentate gyrus: vertical bar graphs present relative mRNA expression measured by quantitative real-time RT-PCR of the key enzymes involved in polyamine metabolism at different time points after SE. Number of animals are given at the bottom of each column. Asterisks indicate significant impact of pilocarpine treatment (p < 0.005, Sidak's multiple-comparison test following two-way ANOVA).
Intracellular spermine supplementation alters Na+ channel properties
We then performed a series of experiments examining a potential causal relationship between spermine depletion and CBZ efficacy loss in chronic epilepsy. If this would be the case, substitution of spermine should restore CBZ effectiveness. Before studying possible interaction of intracellular spermine supplementation and CBZ effects, we investigated the effects of two different concentrations of intracellular spermine on the functional properties of voltage-gated Na+ channels. Either 100 μm or 1 mm spermine was added to the patch-pipette solution, and the recovery from fast inactivation was examined in interleaved recordings with intracellular solutions lacking spermine. Figure 4A illustrates the time course of recovery after fast inactivation with different concentrations of intracellular spermine for sham-control and post-SE animals. An analysis of the reduction of Na+ currents at the different recovery time points showed that the effects of spermine alone were quite small. However, these small effects on the recovery from inactivation were statistically significant in sham-control neurons (two-way ANOVA, spermine-supplementation: F(2,516) = 6.6675, p = 0.0014; time: F(11,516) = 1834, p < 0.0001; interaction: F(22,516) = 1.701, p < 0.0247) but not in chronically epileptic animals (Fig. 4B).
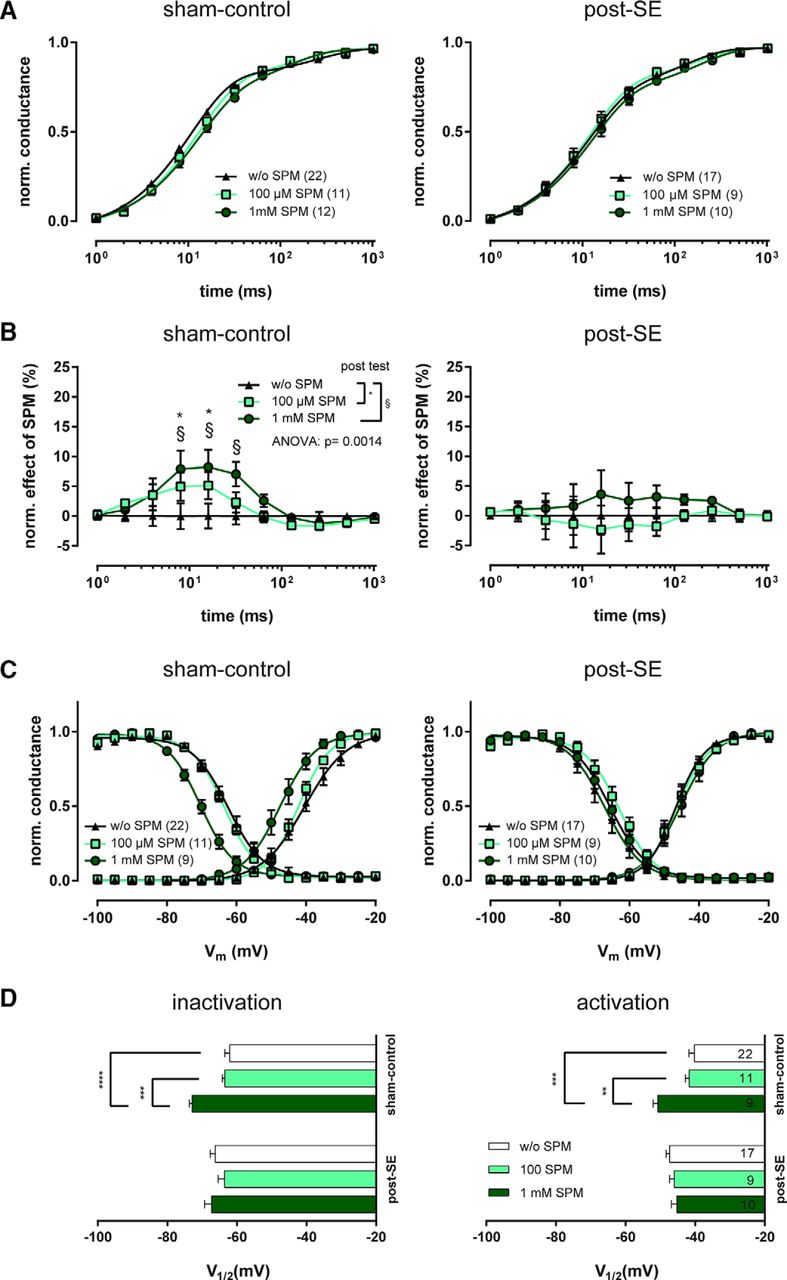
Influence of intracellular spermine on recovery from fast inactivation and voltage-dependence of INaT in dentate gyrus granule neurons. A, Averaged time course of recovery of INaT, plotted as mean ± SEM of the fractional peak conductance during the second depolarizing pulse, normalized to the first pulse response. For voltage-clamp protocol, see Figure 1A. Note the slight right shift of the spermine (SPM) curves only in sham-control animals. The number of cells is given in parentheses in the figure legends. B, For quantitative evaluation of recovery differences by intracellular SPM, we subtracted the peak conductance amplitude values of individual cells recorded with SPM-supplemented intracellular solution from the corresponding mean value measured with spermine-free intracellular solution. Statistical comparison revealed a significant impact of SPM (one-way ANOVA followed by Sidak's multiple-comparison post-test, *p < 0.05, §p < 0.05). C, For evaluation of voltage-dependence of Na+ channel activation and inactivation, a voltage-clamp protocol similar to Figure 2A was used. Peak conductance was calculated from INaT and plotted as mean ± SEM normalized to the maximum peak conductance. Data were fitted to a Boltzmann equation for quantification; fits are shown superimposed on the average data points for neurons recorded with 100 μm, 1 mm, or no SPM added to the intracellular solution. D, Membrane potential for half-maximal activation/inactivation (V1/2) of INaT was derived from Boltzmann fits for all neurons. Horizontal bar graphs show V1/2 for channel inactivation and activation. Asterisks indicate statistically significant differences revealed by Sidak's multiple-comparison post-test following one-way ANOVA. **p < 0.01, ***p < 0.005, ****p < 0.0001.
We then examined the effect of intracellular spermine supplementation on steady-state activation and inactivation. Low concentrations of spermine (100 μm) did not affect activation or inactivation, either in sham-control or in epileptic animals. Higher concentrations of spermine (1 mm) had obvious effects on the voltage dependence of inactivation (one-way ANOVA, F(2,39) = 15.21, p < 0.0001; Sidak's multiple-comparison post-test, p < 0.0005) and activation (one-way ANOVA, F(2,39) = 10.4, p = 0.0002; Sidak's multiple-comparison post-test, p < 0.005) in sham-control animals (Fig. 4C,D). After SE, 1 mm intracellular spermine administration had no significant effect on voltage-dependence (one-way ANOVA, n.s.). In conclusion, 1 mm spermine has effects on Na+ currents similar to the effects of CBZ in sham-control animals. Also, similar to CBZ, the effects of spermine on the recovery from inactivation are diminished in chronically epileptic animals.
Adding intracellular spermine significantly increases effects of CBZ in chronically epileptic animals
Next, we examined how CBZ affects Na+ currents in the presence and absence of spermine. Spermine did not change the reduction of the maximal INaT amplitude (100 μm spermine 49.8 ± 11.4% and 46.9 ± 14.5% in sham-control and epileptic animals, respectively, n = 11 and 9, 1 mm spermine 53.9 ± 9.3% and 50.2 ± 22.3% in sham-control and epileptic animals, respectively, n = 12 and 10, Kruskal–Wallis test, n.s.). We then examined recovery from inactivation, hypothesizing that spermine should reverse the loss of CBZ efficacy.
We found that intracellular spermine affects the CBZ effect on recovery from fast inactivation in a dose-dependent manner (Fig. 5A,B). In sham-control animals (Fig. 5B, left), application of intracellular spermine reduced the effect of 100 μm CBZ on Na+ channel recovery from fast inactivation significantly (two-way ANOVA, influence of spermine on CBZ-effect: F(2,504) = 15.44, p < 0.0001; effect of time: F(2,504) = 67.04, p < 0.0001; interaction: F(11,504) = 2.242, p = 0.0011). Surprisingly, intracellular spermine had an opposite effect in chronically epileptic animals (Fig. 5B, right). In these animals, spermine led to a significant increase in CBZ efficacy, both for 100 μm or 1 mm spermine (two-way ANOVA, influence of spermine on CBZ-effect: F(2,383) = 3.403, p = 0.0343; effect of time: F(2,383) = 24.53, p < 0.0001,). This finding suggests that loss of spermine is a factor in the development of CBZ resistant Na+ channels.

Intracellular spermine supplementation restores efficacy of CBZ on recovery from fast inactivation of Na+ currents. A, Representative current traces of INaT (elicited by the same double-pulse protocol as depicted in Fig. 1A) before and after application of 100 μm CBZ are shown for neurons recorded with and without intracellular spermine (SPM) from sham-control and epileptic (post-SE) animals. Left current trace (black) is the average response to the first pulse, whereas responses to the second pulse are color-coded according to the time intervals between the two pulses (Δt). B, Quantification of the CBZ effect on recovery from fast inactivation. The difference in peak conductance before and after CBZ application was calculated and plotted as mean ± SEM for responses at the various time points after initial inactivation. Note the apparent loss of CBZ efficacy by SPM in sham-control rats, but increase in post-SE animals. Statistical comparison of neurons recorded with or without SPM-supplemented intracellular solution with two-way ANOVA followed by Sidak's multiple-comparison post-test. *p < 0.05, §p < 0.05 for Sidak's test. C, For evaluation of voltage-dependence of Na+ channel activation and inactivation, a voltage-clamp protocol similar to Figure 2A was used. Peak conductance was calculated from INaT and normalized to the maximum peak conductance. Data were fitted to a Boltzmann equation to determine the membrane potential for half-maximal activation/inactivation (V1/2) of INaT. Horizontal bar graphs show CBZ-induced shifts in V1/2 for channel activation and inactivation in neurons recorded with 100 μm, 1 mm, or no SPM added to the intracellular solution. Asterisks indicate statistically significant differences revealed by Sidak's multiple-comparison post-test following one-way ANOVA. *p < 0.05, **p < 0.01.
Finally, we examined how the presence of intracellular spermine affects the voltage-dependence of Na+ currents (Fig. 5C). Intracellular application of 100 μm spermine did not alter the effects of CBZ, 1 mm spermine reduced the effects of CBZ on the voltage-dependence of inactivation (one-way ANOVA, F(2,39) = 8.466, p = 0.0009).
Discussion
Identifying novel treatments for the >30% of the epilepsy population who are pharmacoresistant is a crucial priority in epilepsy research. So far, many novel compounds have been developed by identifying or optimizing drug molecules that act on known AED targets. However, these approaches have not yet led to a breakthrough in treating the group of patients that does not respond to any type of anticonvulsant drug (Kwan and Brodie, 2000). To achieve this goal, it is mandatory to understand the molecular and cellular mechanisms underlying drug resistance.
A key candidate mechanism underlying resistance to CBZ has been previously identified in human and experimental epilepsy, consisting of a loss of use-dependent blocking activity of CBZ (Remy et al., 2003; Doeser et al., 2015). However, the mechanisms underlying this change have remained enigmatic. Transcriptional changes in Na+ channel subunits were unlikely to account for the loss in use-dependent activity. First, individual subunits do not seem to show strong differences in their response to CBZ with regard to use-dependent block (Qiao et al., 2014). Thus, it is unlikely that α subunit expression changes underlie the emergence of resistant channels. Likewise, auxiliary β1 and β2 subunits do not modulate the CBZ sensitivity of Na+ channels (Uebachs et al., 2012). We have addressed the mechanisms underlying the development of a CBZ-resistant Na+ current using the pilocarpine model, because this model replicates the resistance of Na+ currents in human temporal lobe epilepsy to carbamazepine (Remy et al., 2003; Doeser et al., 2015).
We propose that loss of the polyamine spermine occurs in chronic epilepsy, as described previously (Royeck et al., 2015). We further propose that loss of spermine causes a decrease in the efficacy of CBZ, and that the efficacy of CBZ can be restored if intracellular spermine levels are substituted. Compared with transcriptional mechanisms, this is a plausible mechanism because spermine likely acts on different subtypes of Na+ channels present in neurons.
We examined intracellular substitution with a lower (100 μm) and higher (1 mm) concentration of spermine. Interestingly, 100 μm spermine did not lead to any change in Na+ current properties per se, but did significantly enhance the efficacy of CBZ in chronically epileptic animals. This suggests that a concentration range exists at which spermine reconstitution has beneficial effects on AED responses without affecting steady-state Na+ current properties. At the very high concentration of 1 mm, spermine had effects on Na+ channel properties that were very similar to the actions of CBZ, consistent with previous studies (Huang and Moczydlowski, 2001).
How could spermine and CBZ interact at Na+ channels? We considered what is known about the receptor sites involved in the actions of both spermine and anticonvulsants. Early site-directed mutagenesis studies have provided first insights into channel drug binding sites, suggesting that a common receptor site for anticonvulsants and related drugs is formed by amino acids in the S6 segments (Ragsdale et al., 1994, 1996; Yarov-Yarovoy et al., 2001, 2002). More recently, the structure of the bacterial voltage-gated Na+ channel NavAb has been determined at high resolution (Payandeh et al., 2011). This has triggered simulation studies that have provided important information on the access pathways and binding of local anesthetic and anticonvulsant drugs (Boiteux et al., 2014). The polyamine spermine, on the other hand, can insert into the Na+ channel from the intracellular side, controlling the magnitude of persistent Na+ influx. The binding site for polyamines is less well characterized, but is strongly altered by mutation of a lysine residue within the selectivity filter of the Na+ channel, implicating a pore-blocking site (Huang and Moczydlowski, 2001). Moreover, block of Na+ channels by polyamines also exhibits use-dependence (Huang and Moczydlowski, 2001). These biophysical similarities, as well as the relatively close proximity of putative spermine and anticonvulsant binding sites are consistent with the view that spermine and anticonvulsant binding might interact. This idea is also supported by the findings of Fu et al. (2012), showing that intracellular spermine supplementation affects lidocaine efficacy in blocking Na+ currents.
In our case, the hypothesis would be that spermine binds in a cooperative manner with CBZ, thus promoting its actions. Molecular modeling of drug interactions with Na+ channels may constitute a valuable approach to generate a molecular level model of such interactions (Boiteux et al., 2014). An understanding of AED-channel interactions may then enable the rational computational design of newer-generation AEDs that have a high probability to overcome loss of use-dependent block. This does not mean that the absence of spermine renders channels completely insensitive to use-dependent channel blockers, as effects of carbamazepine and lidocaine on Na+ current voltage-dependence are observed in outside-out patches (Jo and Bean, 2014), showing that both compounds can act on voltage-dependence in the absence of polyamines.
The decrease in spermine levels in epileptic hippocampus (Royeck et al., 2015) may be due to changes in polyamine metabolism. This is suggested by the upregulation of the spermine catabolizing enzyme SSAT in hippocampal CA1 (Royeck et al., 2015) and dentate gyrus (this paper). SSAT overexpression alone has been demonstrated to decrease spermine levels in different tissues (Vujcic et al., 2000; Pegg and Casero, 2011). SSAT catalyzes the conversion of spermine to N1-acetylspermine or N1,N12-diacetylspermine, which are consequently increased when SSAT is upregulated. How the presence of acetylspermine isoforms acts on Na+ channel pharmacology remains to be revealed.
What are further implications of our findings? First, the identification of a resistant Na+ channel may permit the generation of simple cell culture models based on e.g., SSAT overexpression that replicate this resistance mechanism. This may permit the development of a cell-line-based, fast and simple, high-throughput identification and validation of promising novel AEDs. Additionally, these results implicate the polyamine system itself as a potentially interesting target in chronic epilepsy. Loss of polyamines leads both to an increased excitability through increased persistent Na+ current (Royeck et al., 2015) and to a loss of use-dependent block by CBZ (this paper). Because extracellular polyamines exert multiple, and potential proconvulsant effects, e.g., via NMDA receptors (Kirby and Shaw, 2005), the therapeutic exploitation would require a more specific approach, perhaps using inhibitors of spermine catabolism.
Footnotes
This work NMB was supported by the BONFOR Program of the medical faculty of the University of Bonn (N.M.B.), and the SFB 1089, the EU Integrated Project Epitarget, and the ERANET DeCipher (H.B.).
The authors declare no competing financial interests.
- Correspondence should be addressed to Heinz Beck, Institute for Experimental Epileptology and Cognition Research, University of Bonn, Sigmund-Freud-Str. 25, 53105 Bonn, Germany. Heinz.Beck{at}ukbonn.de





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}