

Abstract
Sphingosine 1-phosphate (S1P) is a bioactive signaling lipid associated with a variety of chronic pain and itch disorders. S1P signaling has been linked to cutaneous pain, but its role in itch has not yet been studied. Here, we find that S1P triggers itch and pain in male mice in a concentration-dependent manner, with low levels triggering acute itch alone and high levels triggering both pain and itch. Ca2+ imaging and electrophysiological experiments revealed that S1P signals via S1P receptor 3 (S1PR3) and TRPA1 in a subset of pruriceptors and via S1PR3 and TRPV1 in a subset of heat nociceptors. Consistent with these findings, S1P-evoked itch behaviors are selectively lost in mice lacking TRPA1, whereas S1P-evoked acute pain and heat hypersensitivity are selectively lost in mice lacking TRPV1. We conclude that S1P acts via different cellular and molecular mechanisms to trigger itch and pain. Our discovery elucidates the diverse roles that S1P signaling plays in somatosensation and provides insight into how itch and pain are discriminated in the periphery.
SIGNIFICANCE STATEMENT Itch and pain are major health problems with few effective treatments. Here, we show that the proinflammatory lipid sphingosine 1-phosphate (S1P) and its receptor, S1P receptor 3 (S1PR3), trigger itch and pain behaviors via distinct molecular and cellular mechanisms. Our results provide a detailed understanding of the roles that S1P and S1PR3 play in somatosensation, highlighting their potential as targets for analgesics and antipruritics, and provide new insight into the mechanistic underpinnings of itch versus pain discrimination in the periphery.
Introduction
Sphingosine 1-phosphate (S1P) is a bioactive sphingolipid associated with a variety of skin disorders, including psoriasis (Checa et al., 2015; Myśliwiec et al., 2016), atopic/allergic contact dermatitis (Kohno et al., 2004; Sugita et al., 2010), and scleroderma (Castelino and Varga, 2014), as well as neuropathic pain (Patti et al., 2012; Janes et al., 2014) and other inflammatory diseases (Rivera et al., 2008; Chiba et al., 2010; Tränkner et al., 2014; Donoviel et al., 2015; Roviezzo et al., 2015). S1P signaling via S1P receptor 1 (S1PR1) facilitates the migration, recruitment, and activation of immune cells (Matloubian et al., 2004; Cyster and Schwab, 2012). S1P is also thought to influence epithelial cell differentiation and cell division via the action of several other S1PRs (Schüppel et al., 2008; Japtok et al., 2014). Intriguingly, expression of both S1PR1 and S1PR3 has been reported in the somatosensory ganglia (Camprubi-Robles et al., 2013; Usoskin et al., 2015). Furthermore, S1P modulates sensory neuronal excitability (Zhang et al., 2006a,b; Mair et al., 2011; Camprubi-Robles et al., 2013; Li et al., 2015; Wang et al., 2017) and has been implicated in pain and pain hypersensitivity (Mair et al., 2011; Camprubi-Robles et al., 2013; Finley et al., 2013; Hou and Fang, 2015; Weth et al., 2015; Hill et al., 2018). We and others have recently shown an important role for S1P signaling via S1PR3 in cutaneous pain sensation in rodents (Camprubi-Robles et al., 2013; Hill et al., 2018); however, the downstream mechanisms by which S1PR3 triggers neuronal excitation and pain are unclear. Likewise, it is not known whether S1P also acts as a pruritogen to cause itch.
S1P has been examined in a variety of inflammatory skin diseases and disease models. There is evidence supporting both protective and pathogenic effects of S1P signaling in mouse and human chronic itch and inflammatory diseases. In humans, elevated serum S1P is correlated with disease severity in both psoriasis and systemic sclerosis (Castelino and Varga, 2014; Checa et al., 2015; Thieme et al., 2017). Furthermore, ponesimod, an S1PR modulator and blocker of S1P signaling, appears to be promising for psoriasis treatment in humans (Brossard et al., 2013; Vaclavkova et al., 2014; Krause et al., 2018). In contrast, in the imiquimod mouse model of psoriasis, it was observed that topical S1P exerts protective effects (Schaper et al., 2013) and, in dogs with atopic dermatitis, it was found that S1P levels are decreased in lesional skin (Bäumer et al., 2011). However, in genetic and chemical mouse models of dermatitis, the S1PR modulator fingolimod, which downregulates the activity of S1PR1, S1PR3, S1PR4, and S1PR5, was found to attenuate inflammation (Kohno et al., 2004; Sugita et al., 2010). In light of these studies, we investigated whether S1P can act as an acute pruritogen in mice.
We set out to answer this question by examining the role of S1P in acute itch and by determining the downstream molecular mechanisms in somatosensory neurons responsible for S1P-evoked behaviors and neuronal activation. Here, we show that S1P can act as both a pruritogen and algogen. Our study is the first to identify a role for S1P signaling in acute itch and to elucidate the downstream molecular mechanisms by which nociceptive and pruriceptive somatosensory neurons detect and respond to S1P. Our findings demonstrate the contribution of S1P signaling to cutaneous itch and pain and have important applications for the design and use of S1PR modulators as therapeutics for chronic itch and pain diseases.
Materials and Methods
Mice.
S1pr3mCherry/+ (Sanna et al., 2016; B6.Cg-S1pr3tm1.1Hrose/J; The Jackson Laboratory catalog #028624; RRID:IMSR_JAX:028624) and S1pr3−/− (Kono et al., 2004; B6.129S6-S1pr3tm1Rlp/Mmnc; MMRRC Stock #: 012038-UNC; RRID:MMRRC_012038-UNC) mice were obtained from The Jackson Laboratory and backcrossed to C57BL/6J (WT); The Jackson Laboratory catalog #000664, RRID:RRID:IMSR_JAX:000664), Trpv1−/− (V1 KO; Trpv1tm1Jul, MGI catalog #4417977, RRID:MGI:4417977) and Trpa1−/− (A1 KO; Trpa1tm1Jul, MGI catalog #3696956, RRID:MGI:3696956) mice were described previously (Caterina et al., 2000; Bautista et al., 2006) and Trpv1−/−/Trpa1−/− mice were bred from crossing Trpv1−/+ and Trpa1−/+ mice. Mice were housed in 12 h light/dark cycle at 21°C. All experiments were performed under the policies and recommendations of the International Association for the Study of Pain and approved by the University of California–Berkeley Animal Care and Use Committee.
Correlation of gene expression with itch.
A previous study examined the correlation of transcript expression of individual genes in dorsal root ganglion (DRG) neurons with itch behavior among BXD mouse strains (Morita et al., 2015). One of the top correlated genes from the screen was Fam57b (r = 0.57), a recently identified ceramide synthase (Yamashita-Sugahara et al., 2013) and component of the S1P pathway that is robustly expressed in somatosensory neurons (Gerhold et al., 2013). To assess whether expression of the S1P pathway genes as a group was correlated with somatosensory behaviors across the mouse strains of the BXD population, we first tabulated the absolute value of the Pearson's correlation r between expression of each S1P pathway gene in turn (Fam57b, Ppargc1a, Spns1, Spns2, Sphk2, S1pr3, S1pr1, Esrrb, Esrrg, and Lrp2) and itch behavior (Morita et al., 2015) and calculated the median of these correlation values, rtrue. We then drew 10 random genes from the set of all 16,220 genes with detected expression and computed the median correlation as above using this null set, rnull. Repeating the latter 10,000 times established a null distribution of median correlations; we took the proportion of resampled gene groups that exhibited (rtrue ≥ rnull) as an empirical p-value reporting the significance of enriched correlation between expression and itch in the genes of the S1P pathway.
Mouse behavior.
Itch and acute pain behavioral measurements were performed as described previously (Wilson et al., 2011; Morita et al., 2015). Mice (20–30 g; 8–10 weeks) were shaved 1 week before itch behavior. Doses of S1P injected were as follows: 0.2, 2, and 10 μm (Tocris Bioscience), in PBS with methanol-PBS vehicle controls. Fresh S1P was resuspended in methanol and single-use aliquots were prepared and dried under nitrogen gas before use. Compounds were injected using both the neck/rostral back model (20 μl) and the cheek model (20 μl) of itch, as described previously (Wilson et al., 2011). Behavioral scoring was performed with the investigator blinded to experimental condition and mouse genotype. All scratching and wiping behavior videos were recorded for 1 h and scored for either the first 30 min (scratching) or the first 5 min (wiping). Bout number, time spent scratching, and bout length were recorded for scratching behavior. Wiping was recorded as number of wipes.
For radiant heat hypersensitivity behavior, S1P was injected intradermally into the plantar surface of the hindpaw (15 μl). Radiant heat paw withdrawal latencies before and after application of compound or vehicle were performed as described previously (Tsunozaki et al., 2013; Morita et al., 2015) using the Hargreaves test system (IITC Life Science). Mice were injected with compound of interest into the hindpaw and paw withdrawal latencies were measured 15 min before and 20–30 min after injection. Heat-evoked responses included fast paw withdrawal, licking/biting/shaking of the affected paw, and flinching. Mice were allowed to acclimate on a platform for 1 h before injection. The radiant heat source raised the platform temperature to 39.8°C within 5 s and to 60°C within 10 s, as measured by a fast temperature probe (Physitemp).
Wherever possible, WT littermate controls were used in behavioral experiments. Mice were singly housed 1 week before all behavioral experiments. All mice were acclimated in behavioral chambers on the 2 subsequent days for at least 1 h before treatment for itch/pain behavior and radiant heat. Age-matched or littermate male mice were used for all behavioral studies. Mice were tested in four-part behavior chambers (IITC Life Science) with opaque dividers (Tap Plastics). Itch and acute pain behavior was filmed from below using high-definition cameras.
In situ hybridization (ISH).
ISH was performed as described previously (Hill et al., 2018). Fresh DRG were dissected from 8- to 12-week-old mice, flash frozen in optimal cutting temperature embedding medium and sectioned at 14 μm onto slides. ISH was performed using Affymetrix Quantigene ViewISH Tissue 2-plex kit according to manufacturer's instructions with type 1 and type 6 probes. The following probes against mouse mRNAs were created by Affymetrix and used for ISH: S1pr3 and Mrgpra3. Slides were mounted in Fluoromount with number 1.5 coverglass. Imaging of ISH experiments and all other imaging was performed on an Olympus IX71 microscope with a Lambda LS-xl light source (Sutter Instruments). Images were analyzed using ImageJ software. Briefly, 4′, 6-diamidino-2-phenylindole-positive cells were circled and their fluorescence intensity for all channels was plotted against cell size using Igor Pro software (WaveMetrics). Colabeling analysis was performed using ImageJ. Intensity thresholds were set based on the negative control staining slide. Cells were defined as “coexpressors” if their maximum intensities were greater than the threshold for both the type 1 and type 6 probe channels.
Cell culture.
Cell culture was performed as described previously (Wilson et al., 2011). Briefly, neurons from dorsal root ganglia (DRG) of 2- to 8-week-old male and female mice or trigeminal ganglia (TG) from postnatal day 0 (P0)–P4 neonates, where indicated, were dissected and incubated for 10 min in 2 mg/ml Collagenase P (Roche) in Hanks' calcium-free balanced salt solution, followed by incubation in 0.25% standard trypsin (v/v)-EDTA solution (Invitrogen) for 2 min with gentle agitation. Cells were then triturated in MEM Eagle's with Earle's BSS medium, supplemented with 10% horse serum (v/v), MEM vitamins, penicillin/streptomycin, and l-glutamine (Invitrogen). DRG cultures were plated onto poly-d-lysine- (Sigma-Aldrich, 1 mg/ml) and laminin-coated (Corning, 1:300), clean German glass coverslips (Chemglass) and used within 24 h. TG cultures were plated onto poly-d-lysine-coated coverslips and used within 24 h.
Ca2+ imaging.
Ca2+ imaging experiments were performed as described previously (Wilson et al., 2011). DRG neurons from 2- to 8-week-old male and female mice or P0–P4 TG from mice of unknown sex were used for all experiments. An age-matched WT control was also prepared and imaged the same day for experiments on knock-out neurons. No significant differences were observed in S1P responses due to the age or sex of animals used [page = 0.4115 (F(2,21) = 0.9266); psex = 0.5280 (F(1,21) = 0.4119); two-way ANOVA; pinteraction = 0.5781 (F(2,21) = 0.5625)]. Cells were loaded for 60 min at room temperature with 10 μm Fura-2AM supplemented with 0.01% Pluronic F-127 (w/v, Life Technologies) in a physiological Ringer's solution containing the following (in mm): 140 NaCl, 5 KCl, 10 HEPES, 2 CaCl2, 2 MgCl2, and 10 d-(+)-glucose, pH 7.4. All compounds were purchased from Sigma-Aldrich. Acquired images were displayed as the ratio of 340 nm/380 nm. Cells were identified as neurons by eliciting depolarization with high-K+ Ringer's solution (75 mm) at the end of each experiment. Responding neurons to compounds were defined as those having a >15% increase from baseline ratio and neurons were defined as cells having suprathreshold responses to high-K+ Ringer's or as cells that responded to allyl isothiocyanate (AITC) or capsaicin with a >50% increase in ratio even if the high-K+ response was minimal because these compounds can cause intracellular Ca2+ levels to peak. Image analysis and statistics were performed using automated routines in Igor Pro (WaveMetrics). Fura-2 ratios were normalized to the baseline ratio F340/F380 = (ratio)/(ratio t = 0).
Electrophysiology.
Current-clamp experiments were performed as described previously (Hill et al., 2018). Briefly, gap-free current-clamp recordings were collected at 10 kHz and filtered at 2 kHz (Axopatch 200B; pClamp software). Electrode resistance ranged between 2 and 5 MΩ. Internal solution contained 140 mm KCl, 2 mm MgCl2, 1 mm EGTA, 5 mm HEPES, 1 mm Na2ATP, 100 μm GTP, and 100 μm cAMP, pH 7.4. Bath solution was physiological Ringer's solution. The pipette potential was canceled before seal formation. Experiments were performed only on cells with a series resistance of <30 MΩ and membrane capacitance of <40 pF, in accordance with parameters previously used to discriminate S1PR3+ putative small- versus medium-diameter neurons (Hill et al., 2018). Current injection was used to stabilize cells to ∼−60 mV before the experiment. Action potentials that occurred within ±1 s of drug addition or a gravity perfusion artifact were not counted as responses to the drug. For experiments in which two drugs were added in succession, an increase in spike frequency was considered a response for the second drug. For recordings from S1pr3mCherry/+ animals, S1PR3+ DRG neurons were identified using a standard fluorescence microscope. Analysis of electrophysiology data was performed in pClamp and IgorPro. The experimenter was blinded to the antagonists applied until after data analysis was completed.
Experimental design and statistical analyses.
All statistical tests were performed using Prism (GraphPad). Values are reported as the mean ± SEM (where n = number of mice used) for Ca2+ imaging experiments where multiple independent days of imaging were performed and mean ± SD for all other experiments (n = number of wells for imaging or number of mice for behavior). A one- or two-way ANOVA followed by the Sidak's, Dunnett's, or Tukey's post hoc tests (where appropriate) was used. The number of mice or samples required to attain significance was not calculated beforehand and was based on numbers used in similar behavioral studies. For behavioral experiments, mice were randomly assigned to treatment groups by a separate individual; the experimenter was blinded to groups. For behavioral experiments, every effort was made to ensure that equal numbers of mice of each genotype were used for each experiment (where appropriate) and that treated and control groups were of identical or near-identical size. Significance was labeled as follows: n.s., not significant, p ≥ 0.05, *p < 0.05, **p < 0.01, ***p < 0.001.
Results
S1P triggers itch via S1PR3
Our group previously harnessed natural variation in somatosensory behaviors among genetically distinct mouse strains to identify candidate transducers in DRG somatosensory neurons (Morita et al., 2015). Analysis of this dataset revealed that members of the S1P synthesis and signaling pathways (Fig. 1a) were expressed in somatosensory neurons in a manner that correlated with itch behaviors (Fig. 1b). Previous studies have shown that S1P promotes acute pain (Camprubi-Robles et al., 2013) and thermal sensitization via S1PR3 and that S1P is required for normal mechanical pain sensitivity (Hill et al., 2018). Here, we sought to address the specific contribution of S1P/S1PR3 signaling to S1P-evoked itch behaviors.

S1P triggers itch via S1PR3. a, Tabular list of the top 10 most highly expressed S1P pathway genes in DRG neurons. b, Histogram showing median absolute r-value for gene expression versus itch plotted against frequency for 10,000 permutations of random subsets of 10 genes in the BXD DRG transcriptome. Red line indicates median r-value for S1P genes (p = 0.0116, rmedian = 0.399, see Materials and Methods). c, Intradermal cheek injection of 20 μl 10 μm S1P, 2 μm S1P, 0.2 μm S1P, and 1% methanol-PBS (VEH; concentration of methanol was chosen based on amount in 10 μm S1P solution), with quantification of time spent scratching over the 30 min postinjection interval [p < 0.0001, one-way ANOVA (F(3,30) = 18.29); n = 7–9 mice per condition]. Dunnett's multiple-comparisons p-values are represented on graph: **p10 μM vs. VEH = 0.002; **p2 μM vs. VEH = 0.001; *p0.2 μM vs. VEH = 0.03. d, Time spent scratching in response to intradermal injection of 20 μl of 0.2 μm S1P or 0.1% methanol-PBS vehicle in rostral back of age-matched S1pr3+/+ (WT) and S1pr3−/− (KO) mice; p = 0.0089 (one-way ANOVA (F(3,9) = 7.274); n = 3–4 mice per treatment). Tukey's multiple-comparisons p-values are represented on graph : ***pWT = 0.0007; pKO = 0.83. Error bars indicate mean ± SD.
We first investigated whether exogenous S1P can trigger itch using the cheek assay, which allows for simultaneous discrimination of pain-evoked wiping and itch-evoked scratching in mice (Shimada and LaMotte, 2008). Injection of 2–10 μm S1P triggered acute nocifensive behaviors (pain-evoked cheek wiping), previously shown by our group (Hill et al., 2018). However, we also observed robust scratching in these animals (Fig. 1c). Intriguingly, 0.2 μm S1P elicited scratching behaviors (Fig. 1c), but no pain behaviors. Scratching behaviors developed with an average latency of 4 min 29 s ± 1 min 37 s and persisted for at least 30 min. We also observed that injection of 0.2 μm S1P into the rostral back triggered itch behaviors with an average scratching time of 66 ± 18.1 s, which was significantly greater than scratching evoked by vehicle (0.1% methanol-PBS; 14.5 ± 2.88 s; Fig. 1d). Bout times and average time per bout also increased in response to S1P (boutsS1P vs Vehicle: 8.5 ± 5.12 vs 3.33 ± 1.5; time per bout: 11.39 ± 8.66 vs 4.23 ± 1.44 s). To begin to elucidate the mechanisms underlying these effects of S1P, we focused on the S1P receptor S1PR3, which is required for S1P-evoked pain and pain hypersensitivity (Camprubi-Robles et al., 2013; Hill et al., 2018). Assessment of itch behavior in S1pr3−/− mice revealed significantly attenuated S1P-evoked scratching indistinguishable from vehicle injection (10.6 ± 12.3 s vs 6.3 ± 6.2 s, respectively; Fig. 1d). Furthermore, in a previous report, we observed no defects in itch responses to the pruritogens chloroquine and histamine in S1pr3−/− mice (Hill et al., 2018). These data suggest that S1PR3 is the primary receptor by which S1P signals in somatosensory neurons to drive itch. Our findings show that S1P can act as a pruritogen, selectively triggering itch at nanomolar concentrations, whereas at micromolar concentrations, S1P acts as a pruritogen and as an algogen, triggering itch and pain. Our discovery that S1PR3 is required for S1P-evoked itch is consistent with previous studies showing that S1P-evoked acute pain (Camprubi-Robles et al., 2013), heat hypersensitivity, and mechanical pain (Hill et al., 2018) are absent in S1pr3−/− mice. These results add S1P to a growing list of endogenous molecules that act as both pruritogens and algogens (Shimada and LaMotte, 2008; Akiyama and Carstens, 2013; Moore et al., 2018; Esancy et al., 2018).
S1PR3 is functional and expressed in a subset of pruriceptors
We next investigated whether S1PR3 is functional and expressed in itch neurons. Ca2+ imaging of S1P responses in WT mouse somatosensory neurons revealed that 1 μm S1P activates 34.2% of all neurons, which fall into two main populations: 22.6% (66.1% of S1P+ neurons) are TRPV1+/TRPA1+ neurons, which are sensitive to the TRPV1 agonist capsaicin and the TRPA1 agonist AITC, and 11.6% (32.5% of S1P+ neurons) are TRPV1+/TRPA1− neurons, which are capsaicin-sensitive and AITC-insensitive (Fig. 2a,b). Neurons that were responsive to both AITC and S1P exhibited a half-maximal effective concentration (EC50) for S1P of 102 nm and were activated by lower S1P concentrations (10 nm) than neurons that were responsive to capsaicin and S1P (EC50 = 155 nm) and showed robust responses to 100 nm S1P (Fig. 2c). Although we observed a small number (1.4% of total neurons) of S1P responses in neurons that were unresponsive to AITC or capsaicin, the proportion of these responses were similar to those evoked by vehicle. Therefore, we conclude that S1P-responsive neurons exhibit near-complete overlap with capsaicin-responsive neurons, consistent with roles for S1P in itch and pain. Within this population, we observed several subsets of itch- and pain-sensing neurons that have been described previously (Caterina et al., 2000; Jordt et al., 2004; Bautista et al., 2006; Imamachi et al., 2009; Liu et al., 2009; Wilson et al., 2011; Dong and Dong, 2018). For example, previous studies have shown that the MrgprA3+ subpopulation of TRPA1+ neurons is required for many forms of non-histaminergic acute and chronic itch (Liu et al., 2009; Wilson et al., 2011; Han et al., 2013; Reddy et al., 2015; Zhu et al., 2017). In keeping with our hypothesized role for S1P in itch, 80% of chloroquine-responsive MrgprA3+ neurons responded to S1P (Fig. 2b). Likewise, we also observed a population of S1P-sensitive neurons that were sensitive to the pruritogen histamine (23.6% of S1P-sensitive neurons; Fig. 2b). We conclude that S1P activates a variety of thermal nociceptors that express TRPV1 and pruriceptors that express TRPA1 and TRPV1.

S1PR3 is functional and expressed in a subset of pruriceptors. a, Representative Ca2+ imaging traces showing normalized Fura-2 signal (Norm. F340/F380) over time before and after addition of indicated compounds in cultured DRG neurons (CQ, 1 mm chloroquine; S1P, 1 μm S1P; CAP, 1 μm capsaicin; K+, Ringer's with 75 mm K+). Left, Putative pruriceptor that responds to both chloroquine and S1P. Center, Putative pruriceptor or nociceptor that responds to both AITC and S1P. Right, Putative thermal nociceptor that responds to S1P and capsaicin but not AITC. b, Venn diagram demonstrating overlap of neuronal subpopulations activated by capsaicin, AITC, histamine (HIS; 100 μm), chloroquine, and S1P. Relative proportion of overlap represents percentage overlap as calculated from ratiometric Ca2+ imaging data (n = 4000 neurons). c, Dose–response curve showing proportion of DRG neurons responding to varying concentrations of S1P. Concentrations used: 1, 10, 50, 100, 200, 1000, and 10,000 nm (n = 2 animals). Colored traces indicate proportion of neurons responding to S1P and AITC or S1P and capsaicin at indicated concentrations. d, S1P-evoked Ca2+ transients in DRG neurons are inhibited by the selective S1PR3 antagonist TY 52156. Black dotted line indicated sigmoidal fit from which IC50 was derived (n = 2 animals). Error bars indicate mean ± SEM. e, S1P-evoked Ca2+ transients in DRG neurons are not inhibited by the selective S1PR1 antagonist W146 (n = 2 animals). Error bars indicate mean ± SEM. f, Co-ISH of S1pr3 (green) with Mrgpra3 (magenta) in sectioned whole DRG from adult WT mice, with overlay (right) and inset (far right). Scale bar, 100 μm. Images were acquired using a 10× air objective. Arrows indicate cells showing coexpression of both markers.
To pursue further the mechanism by which S1P acts in somatosensation, we examined the effects of pharmacological blockade of S1PR3 and S1PR1 on S1P-evoked Ca2+ responses. Incubating cells with the S1PR3-selective antagonist TY 52156 attenuated S1P-evoked Ca2+ transients with an IC50 of 0.4 μm (Fig. 2d). In contrast, the S1PR1-selective antagonist W146 had no discernable effect on S1P responses at a range of concentrations that exceed reported inhibitory concentrations (70–80 nm) for this drug (Finley et al., 2013; Janes et al., 2014; Fig. 2e). These data support previous studies showing that cultured somatosensory neurons from S1pr3−/− mice exhibit no Ca2+ responses to 1 μm S1P (Camprubi-Robles et al., 2013; Hill et al., 2018) and suggest that S1PR3, but not S1PR1, is required for activation of pruriceptors and nociceptors by S1P.
Although S1PR3 is expressed (Hill et al., 2018) and functional in distinct subsets of TRPV1+ and TRPV1+/TRPA1+ DRG neurons, whether this receptor is expressed in pruriceptors was unknown. To investigate this, we performed co-in situ hybridization (co-ISH) of S1pr3 with Mrgpra3, which encodes the chloroquine receptor that marks a neuronal subpopulation required for some forms of acute and chronic itch (Liu et al., 2009). We found that 35.1% of Mrgpra3+ cells express S1pr3 and 11.6% of S1pr3+ cells express Mrgpra3 (Fig. 2f). The differing proportions of S1pr3+/Mrgpra3+ neurons in our ISH and of S1P+/chloroquine+ neurons in our Ca2+ imaging studies (Fig. 2b) may reflect a difference between intact DRG versus cultured DRG neurons and/or the sensitivity of mRNA expression versus Ca2+ imaging assays. In summary, our Ca2+ imaging and co-ISH data, along with our previous findings, reveal two main populations of small-diameter S1pr3+ neurons: Trpv1+ thermal nociceptors and Trpa1+ cells, including a subset of Mrgpra3+ pruriceptors. These results agree with our finding that S1PR3 is required for S1P-evoked itch (Fig. 1d).
S1P triggers neuronal activation in distinct subsets of nociceptors and pruriceptors
We next sought to identify molecular components downstream of S1PR3 underlying S1P-evoked Ca2+ transients in cultured mouse somatosensory neurons. S1P responses were mediated solely by Ca2+ influx because chelation of extracellular Ca2+ abolished all responses (Fig. 3a), suggesting that S1P triggers the opening of plasma membrane cation channels. We also observed similar results with application of ruthenium red (Fig. 3b), a blocker of a number of Ca2+ permeable ion channels, including transient receptor potential (TRP) channels.
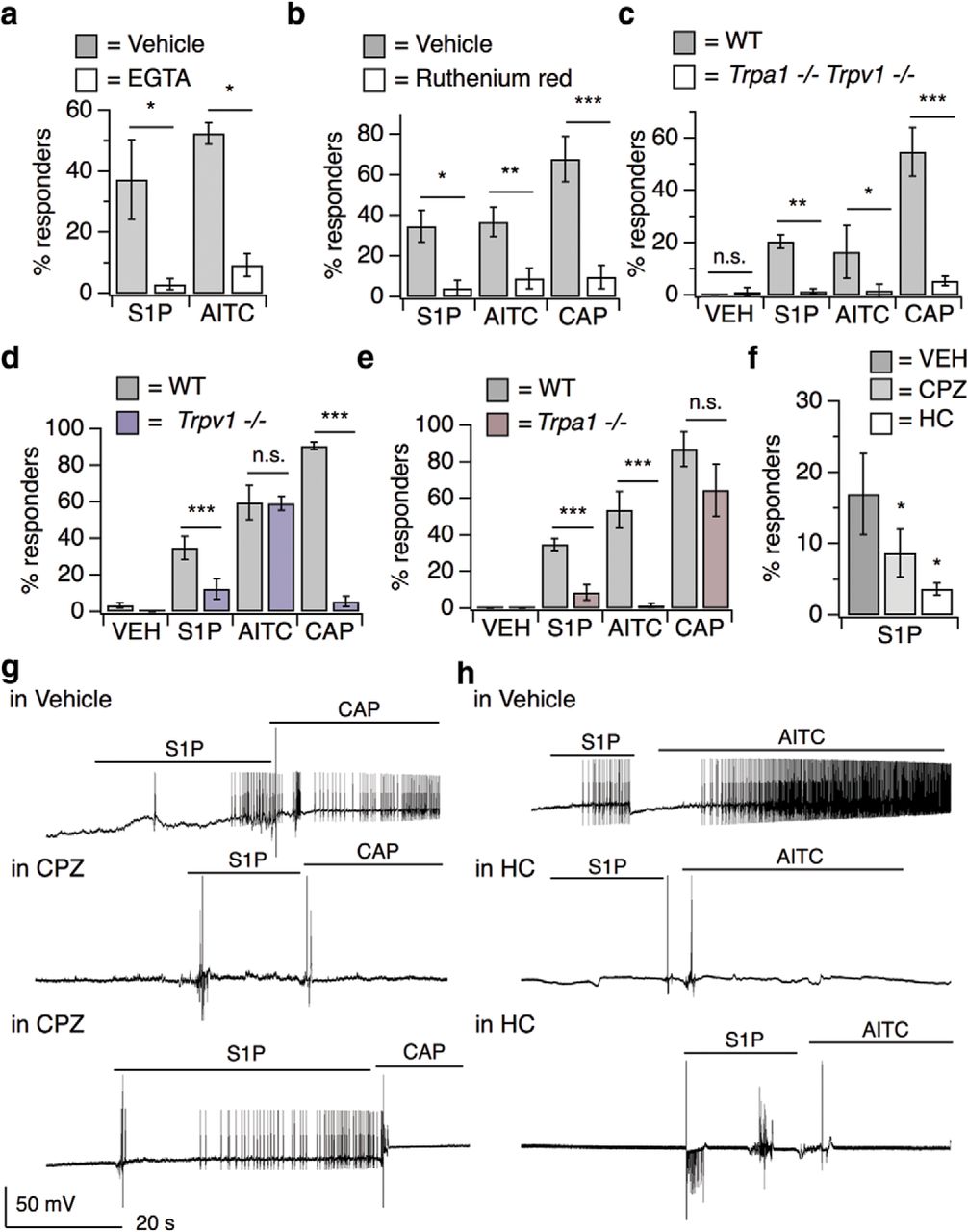
S1P triggers neuronal activation in distinct subsets of nociceptors and pruriceptors. a, Percentage DRG neurons responding to S1P and AITC in Ca2+ Ringer's and 10 mm EGTA Ringer's; p < 0.0001 [one-way ANOVA (F(3,11) = 8.291); n = 4 wells of ∼100 neurons each per treatment]. Error bars indicate mean ± SD. Sidak's multiple-comparisons p-values are represented on graph: *pS1P = 0.01; *pAITC = 0.0163. b, Percentage responders to 1 μm S1P, AITC, and capsaicin in vehicle and 10 μm ruthenium red; p < 0.0001 [one-way ANOVA (F(5,17) = 17.42); n = 5 wells of ∼100 neurons each per treatment]. Error bars indicate mean ± SD. Sidak's multiple-comparisons p-values are represented on graph: *pS1P = 0.048; **pAITC = 0.004; ***pCAP < 0.0001. c, Percentage responders to vehicle, S1P, AITC, and capsaicin in Trpa1/Trpv1+/+ and −/− DRG neurons; p < 0.0001 [one-way ANOVA (F(7,40) = 8.555); n = 2 age-matched mice per genotype]. Error bars indicate mean ± SD. Sidak's multiple-comparisons p-values are represented on graph: pVEH = 0.99; **pS1P = 0.0075; *pAITC = 0.04; ***pCAP = 0.0003. d, Percentage responders to vehicle, S1P, AITC, and capsaicin in Trpv1+/+ and Trpv1−/− neurons; p < 0.0001 [one-way ANOVA (F(7,179) = 72.67); n = 3 age-matched mice per genotype]. Error bars indicate mean responses ± SEM. Sidak's multiple-comparisons p-values are represented on graph: ***pS1P < 0.0001; pAITC = 0.80; ***pCAP < 0.0001. e, Percentage responders to vehicle, S1P, AITC, and capsaicin in Trpa1+/+ and Trpa1−/− neurons; p < 0.0001 [one-way ANOVA (F(7,210) = 105); n = 3 age-matched mice per genotype]. Error bars indicate mean responses ± SEM. Sidak's multiple-comparisons p-values are represented on graph: ***pS1P < 0.0001; ***pAITC < 0.0001; pCAP = 0.07. f, Effect of 50 μm CPZ (TRPV1 antagonist) and 50 μm HC-030031 (HC; TRPA1 antagonist) versus DMSO-Ringer's vehicle on S1P-evoked Ca2+ responses in cultured DRG neurons; p < 0.0001 [one-way ANOVA (F(8,30) = 84.32); n = 3 wells of ∼100 neurons each per treatment]. Error bars indicate mean responses ± SD. Dunnetts's multiple-comparisons p-values are represented on graph: *pCPZ vs. VEH = 0.04; *pHC vs. VEH = 0.03. g, Whole-cell current-clamp recordings of S1PR3+ DRG neurons from S1pr3mCherry/+ animals pretreated for 5–10 min with vehicle (1% DMSO in Ringer's) or 50 μm CPZ and subsequently exposed to 1 μm S1P followed by 1 μm capsaicin (CAP), indicated by lines. Top, Recording from a neuron that responded to S1P and CAP. Middle, Recording from a neuron that responded to neither S1P nor CAP. Bottom, Recording from a neuron that responded to S1P but not CAP. In these experiments, 5 of 6 S1PR3+ cells that received vehicle responded to S1P and 5 of 6 responded to CAP; 3 of 8 cells that received CPZ responded to S1P and 0 of 8 cells responded to CAP (n = 3 animals). h, Whole-cell current-clamp recordings of S1PR3+ DRG neurons from S1pr3mCherry/+ animals pre-treated for 5–10 min with vehicle (1% DMSO in Ringer's) or 50 μm HC-030031 (HC) and subsequently exposed to 1 μm S1P followed by 100 μm AITC, indicated by lines. Top, Recording from a neuron that responded to S1P and AITC. Middle, Recording from a neuron that responded to neither S1P nor AITC. Bottom, Recording from a neuron that responded to S1P but not AITC. In these experiments, 4 of 5 S1PR3+ cells that received vehicle responded to S1P and 2 of 5 responded to AITC; 4 of 11 cells that received HC responded to S1P and 0 of 11 cells responded to AITC (n = 3 animals).
Because we observed that all S1P-responsive neurons expressed TRPA1 and/or TRPV1 and these channels have been shown to functionally couple to a variety of GPCRs (Chuang et al., 2001; Prescott and Julius, 2003; Bandell et al., 2004; Dai et al., 2007; Shim et al., 2007; Kwon et al., 2008; Rohacs et al., 2008; Imamachi et al., 2009; Schmidt et al., 2009; Wilson et al., 2011; Moore et al., 2018), we investigated whether either channel mediated S1P-evoked neuronal activation. Neurons isolated from mice lacking both TRPA1 and TRPV1 exhibited greatly attenuated S1P-evoked Ca2+ responses, similar to those evoked by vehicle (Fig. 3c). Intriguingly, we found the percentage of S1P-responsive neurons was partially attenuated in neurons from Trpv1−/− (Fig. 3d) and Trpa1−/− single knock-out mice (Fig. 3e). Such genetic effects were mirrored by pharmacological blockade of these channels; the TRPV1 antagonist capsazepine (50 μm; CPZ) or the TRPA1 antagonist HC-030031 (50 μm; HC) were sufficient to fully block capsaicin or AITC responses, respectively, and resulted in a significant, although partial, attenuation of neuronal S1P responses (Fig. 3f).
To determine whether TRPA1 and/or TRPV1 are required for S1P-evoked neuronal excitability, we turned to current-clamp whole-cell electrophysiology to examine action potential (AP) firing. For this purpose, we isolated and cultured DRG neurons from the S1pr3mCherry reporter mouse, which produces a functional S1PR3-mCherry fusion protein (Sanna et al., 2016), and examined changes in membrane potential in response to S1P in small-diameter (capacitance < 40 pF) somatosensory neurons. Our experiments revealed that 1 μm S1P elicited AP firing in both the capsaicin-sensitive and AITC-sensitive S1PR3+ populations of sensory neurons (Fig. 3g,h, top). We next used a pharmacological approach to investigate the role of TRPV1 and TRPA1 in S1P-evoked AP firing. TRPV1 blockade with the antagonist CPZ decreased the proportion of neurons that fired APs in response to S1P and, as expected, completely blocked capsaicin-evoked AP firing in these neurons (Fig. 3g, Table 1). Likewise, blockade of TRPA1 with HC resulted in a decreased proportion of cells which fired in response to S1P and the expected complete loss of AITC-evoked firing (Fig. 3h, Table 1). These results suggest that TRPV1 and TRPA1 directly contribute to S1P-evoked neuronal activation and AP firing. Combined with our Ca2+ imaging studies using TRP channel inhibitors and A1/V1 knock-out neurons, our data show that TRPA1 and TRPV1 are required for S1P-evoked neuronal activation in distinct populations of somatosensory neurons.
S1P triggers neuronal excitation in distinct subsets of TRPA1+ and TRPV1+ somatosensory neurons
S1PR3 uses distinct G-protein-coupled pathways to activate subsets of somatosensory neurons
We used a pharmacological approach to investigate the mechanisms by which S1PR3 signals to TRPA1 and TRPV1. We first focused on phospholipase C (PLC), a known target of the S1PR3 signaling partner Gq (Kim et al., 2011; Flock et al., 2017). Given that GPCRs have been shown to activate TRPV1 and TRPA1 channels via PLC activity in somatosensory neurons (Chuang et al., 2001; Prescott and Julius, 2003; Bandell et al., 2004; Dai et al., 2007; Shim et al., 2007; Kwon et al., 2008; Rohacs et al., 2008; Imamachi et al., 2009; Schmidt et al., 2009; Wilson et al., 2011; Paulsen et al., 2015; Gao et al., 2016; Moore et al., 2018), we hypothesized that PLC signaling could be required for S1P-evoked Ca2+ responses in sensory neurons. This notion bore out, in that inhibition of PLC signaling using the drug U-73122 significantly decreased the percentage of neurons that displayed S1P-evoked Ca2+ responses (Fig. 4a). This effect was most pronounced in the TRPA1− population of S1P-responsive neurons (Fig. 4b,c), but had only a small effect on the TRPA1+ population (Fig. 4b,c). Next, given that GPCR signaling via Gβγ can activate TRPA1 (Wilson et al., 2011), we investigated whether Gβγ signaling may also influence S1P-evoked Ca2+ responses. We observed a significant reduction in the percentage of neurons responding to S1P upon blockade of Gβγ activity using the drug gallein (Fig. 4a). Further, in contrast to PLC inhibition, gallein's effects were robust in the TRPA1+ population and were minimal in the TRPA1− population (Fig. 4b,c). Blockade of both pathways using U-73122 and gallein resulted in a significant loss of all neurons responsive to S1P (Fig. 4a) regardless of population (Fig. 4b,c). Therefore, S1PR3 can signal within two different somatosensory neuronal subtypes (TRPA1+/TRPV1+ and TRPA1−/TRPV1+ neurons) in a concentration-dependent manner using distinct molecular signaling molecules (Gβγ and PLC, respectively).
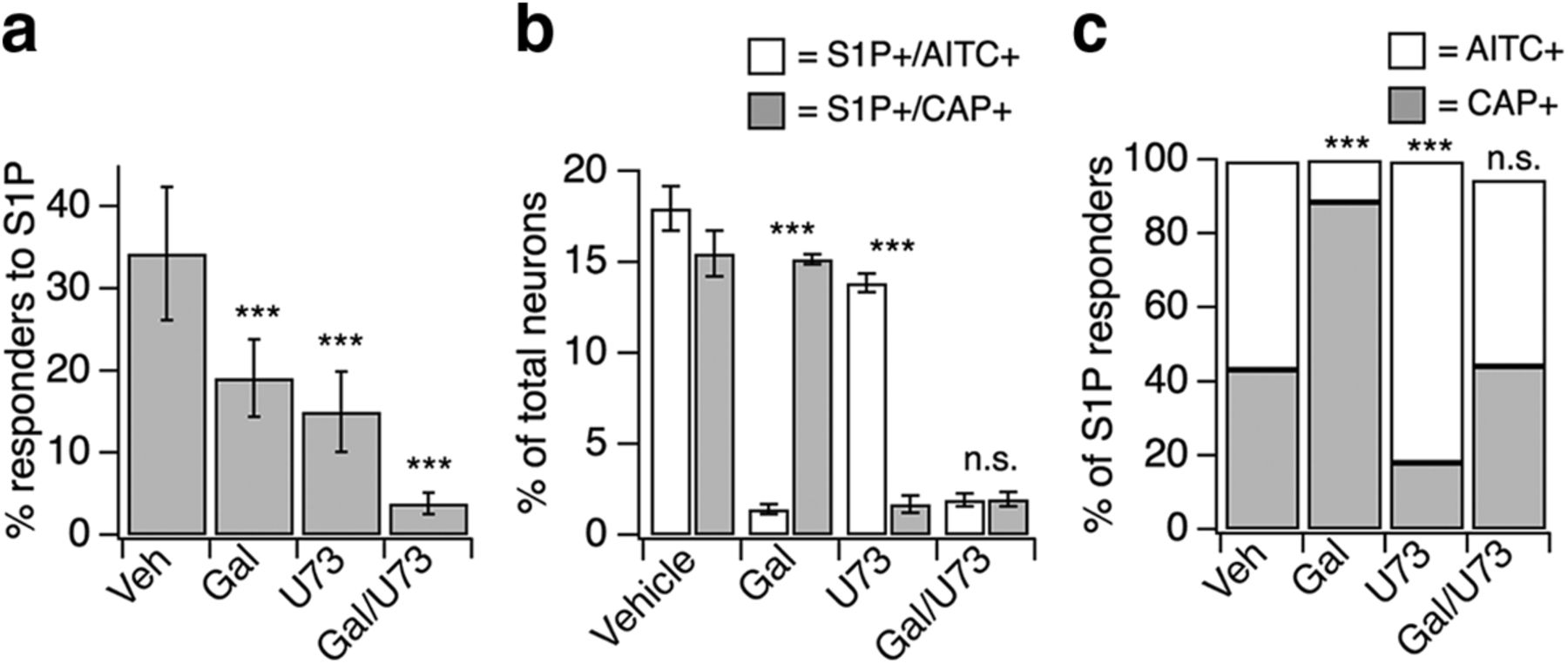
S1PR3 uses distinct G-protein-coupled pathways to activate subsets of somatosensory neurons. a, Percentage of DRG neurons responding to 1 μm S1P after 15 min incubation with either DMSO-Ringer's vehicle (Veh), 100 μm gallein (Gal; Gβγ blocker), 1 μm U-73122 (U73; PLC blocker), or U-73122 + gallein; p < 0.0001 [one-way ANOVA (F(3,28) = 31.46); n = 4–10 wells of ∼50 neurons each per treatment from 2 animals]. Error bars indicate mean responses ± SD. Dunnett's multiple-comparisons p-values are indicated on graph: ***pGal vs. VEH < 0.0001; ***pU73 vs. VEH < 0.0001; ***pGal/U73 vs. VEH < 0.0001. b, From experiments in a, percentage of total neurons sensitive to S1P, AITC, and capsaicin (white, left) or S1P and capsaicin only (gray, right). Error bars indicate mean responses ± SD; p < 0.0001 [one-way ANOVA (F(3,28) = 41.48]. Dunnett's multiple comparisons p-values are represented on graph: ***pGal < 0.0001; ***pU73 < 0.0001; pGal/U73 = 0.98. c, Data displayed in b now plotted as percentage of S1P-responsive neurons that were sensitive to AITC and capsaicin (white) or capsaicin only (gray); p < 0.0001 [one-way ANOVA (F(3,28) = 41.48]. Dunnett's multiple-comparisons p-values are indicated on graph: ***pGal < 0.0001; ***pU73 < 0.0001; pGal/U73 = 0.98.
S1P evokes itch and pain behaviors via distinct TRP channels
Here, we have shown that TRPA1 and TRPV1 are required for S1P-evoked neuronal excitation and Ca2+ responses in pruriceptors and nociceptors. We thus tested the requirement of TRPA1 and TRPV1 to S1P-evoked itch and pain behaviors. We observed that S1P evoked robust heat hypersensitivity in WT and Trpa1−/− mice, but not in Trpv1−/− mice (Fig. 5a). Similarly, 10 μm S1P triggered acute nocifensive behaviors (wiping) in WT and Trpa1−/− mice, but not in Trpv1−/− mice (Fig. 5b). Finally, we examined S1P-evoked itch behaviors in WT, Trpa1−/−, and Trpv1−/− mice using the rostral back model. In contrast to our pain data, 0.2 μm S1P evoked robust scratching behaviors in WT and Trpv1−/− mice, but not in Trpa1−/− mice (Fig. 5c). We also observed a selective loss of itch-evoked scratching in Trpa1−/− mice even with 10 μm S1P, which triggers both itch and pain behaviors in WT mice in the cheek model (time spent scratching to 10 μm S1P: WT: 113 ± 47.57 s; V1 KO: 157.66 ± 42.01 s; A1 KO: 10.66 ± 6.66 s; pgenotype = 0.0102; two-way ANOVA (F(2,9) = 7.962); n = 2–3 mice per group). To conclude, our data support a model wherein TRPV1 selectively mediates S1P-evoked acute pain and heat hypersensitivity (Fig. 6a) and TRPA1 selectively mediates S1P-evoked acute itch (Fig. 6b).

S1P evokes itch and pain via distinct TRP channel-dependent pathways. a, Radiant heat paw withdrawal latencies 20–30 min after injection of 15 μl of 10 μm S1P or 1% methanol-PBS vehicle intradermally into the hindpaw of age-matched WT, Trpa1−/−, and Trpv1−/− mice; p < 0.0001 [one-way ANOVA (F(5,66) = 14.13); n = 7 or more age-matched mice per condition]. Sidak's multiple-comparisons p-values are represented on graph: ***pWT < 0.0001; ***pTrpa1 < 0.0001; pTrpv1 = 0.193. b, Number of wipes in response to intradermal injection of 20 μl of 10 μm S1P or 1% methanol-PBS vehicle in cheek of age-matched WT, Trpa1−/−, and Trpv1−/− mice; p < 0.0001 [one-way ANOVA (F(5,11) = 33.98); n = 3 age-matched mice per condition]. Sidak's multiple-comparisons p-values are represented on graph: ***pWT = 0.0001; ***pTrpa1 < 0.0001; pTrpv1 = 0.99. c, Time spent scratching in response to intradermal injection of 20 μl of 0.2 μm S1P or 0.1% methanol-PBS vehicle in rostral back of age-matched WT, Trpa1−/− and Trpv1−/− mice; p < 0.0001 [one-way ANOVA (F(5,91) = 14.13); n = 8 or more age-matched mice per condition]. Sidak's multiple-comparisons p-values are represented on graph: ***pWT = 0.0002; pTrpa1 = 0.99; ***pTrpv1 < 0.0001; ***pWT S1P vs. Trpv1 S1P = 0.0005. Error bars indicate mean ± SD.

Effects of S1P on thermal hypersensitivity and acute pain and itch. a, Elevated S1P elicits thermal hypersensitivity and acute pain via S1PR3-dependent activation of TRPV1, which requires PLC activity. b, S1P also elicits itch-evoked scratching via S1PR3-dependent activation of TRPA1, which requires Gβγ.
Discussion
Recent studies have linked aberrant S1P signaling to a variety of diseases, including asthma (Chiba et al., 2010; Tränkner et al., 2014; Roviezzo et al., 2015), multiple sclerosis (Brinkmann et al., 2010; Choi et al., 2011), cancer (Liang et al., 2013), psoriasis (Checa et al., 2015; Myśliwiec et al., 2016), and chronic pain (Patti et al., 2012; Janes et al., 2014; Grenald et al., 2017). However, the cellular and molecular targets of S1P in these disorders are largely unknown. Here, we show that S1PR3 is required for S1P-evoked pain and itch, highlighting the potential for a direct contribution of S1P to sensitization, pain, and/or itch in these diseases.
There is much interest in exploring the therapeutic potential of S1PR signaling in chronic pain and itch. However, little was known about the role of S1PRs in sensory neurons and their role in itch. We now show that S1P activates pruriceptors and triggers itch via S1PR3 and TRPA1. In mice, we found that injection of 0.2 μm S1P triggered acute itch behaviors, whereas concentrations ≥ 2 μm triggered both itch and pain. S1PR3-deficient mice displayed a complete loss of S1P-evoked acute itch (Fig. 1d) and pain (Hill et al., 2018).
Our findings, along with other recent studies, support a role for sensory neurons in disorders linked to S1P signaling. Others have shown that sensory neurons innervating the lung express S1pr3, that administration of a nonselective S1P receptor agonist can enhance asthma-like airway hyperreactivity after acetylcholine challenge (Tränkner et al., 2014), and that sensory neurons of the nodose ganglia are directly activated by S1P (Wang et al., 2017). It is also known that TRPV1+ sensory afferents are required for airway hypersensitivity (Rogerio et al., 2011). Furthermore, mast cells, which secrete S1P when activated (Idzko et al., 2002; Rivera et al., 2008; Olivera et al., 2013; Roviezzo et al., 2015; Saluja et al., 2017), play important roles in allergic asthma as well as itch. Therefore, we propose that S1PR3 presents an attractive target for the study of itch and/or airway hyperreactivity under these conditions.
We have thus identified two distinct populations of sensory neurons that are excited by S1P: the TRPA1+/TRPV1+ and TRPA1−/TRPV1+ populations, which have been shown to differentially contribute to itch and pain behaviors (Caterina et al., 2000; Bandell et al., 2004; Bautista et al., 2006; Dai et al., 2007; Shim et al., 2007; Gerhold and Bautista, 2009; Imamachi et al., 2009; Wilson et al., 2011; Moore et al., 2018). Our observations that the TRPA1+ population is activated by lower doses of S1P than the TRPA1− population (Fig. 2c) and that itch behaviors are triggered by doses that are nonpainful (Fig. 1c) supports a recent study showing that both zebrafish and mouse pruriceptors are significantly more sensitive to chemical stimuli than nociceptors. This increased sensitivity results in selective recruitment of pruriceptors over nociceptors by the same agonist in a dose-dependent manner, such that lower concentrations of agonist selectively trigger itch and higher concentrations trigger both itch and pain (Esancy et al., 2018). Although that study observed these effects using the weak TRPA1 activator imiquimod and the strong TRPA1 activator AITC, we saw similar effects using varying concentrations of S1P (Fig. 1). It is interesting that our results in the cheek model mirror theirs and lend support to a peripheral “population coding” model wherein low-intensity stimuli can selectively drive pruriceptor activation and itch, whereas high-intensity stimuli activate both nociceptors and pruriceptors to drive pain and itch. We show that more S1P/S1PR3 signaling is required to activate nociceptors and drive pain behaviors than is required to activate pruriceptors and trigger itch.
But how, mechanistically, does this dose-dependent and selective activation of itch versus pain by S1P occur? A number of studies have generally implicated Ca2+ activated Cl− and voltage-dependent K+ and Na+ channels as being important for S1P-evoked neuronal excitation (Zhang et al., 2006a; Camprubi-Robles et al., 2013; Li et al., 2015). Here, we show for the first time that both TRPA1 and TRPV1 play key roles in S1P-evoked AP firing in distinct subsets of somatosensory neurons and in itch and pain behaviors. TRP channels are nonselective cation channels that trigger Ca2+ influx and depolarize the cell and serve as important upstream transducers that trigger the activation of Ca2+ and voltage-sensitive channels. Although S1P may activate multiple channels in somatosensory neurons, we have used pharmacological and genetic tools to show that TRPA1 and TRPV1 are essential for S1P-evoked excitation and are differentially required for S1P-evoked itch and pain, respectively. Our experiments using G-protein pathway inhibitors suggest that S1PR3 uses distinct G-protein pathways to activate TRPA1 and TRPV1, which may contribute to differential initiation of itch and pain behaviors, and support a recent study suggesting that S1PR3 can couple to a number of downstream G-protein-signaling pathways (Flock et al., 2017). Although it is difficult to probe G-protein-coupled pathways in vivo or with high specificity due to the pleiotropic effects of the available pharmacological tools, it is tempting to speculate that the different signaling pathways downstream of S1PR3 in vitro may account for the differential engagement of pain and itch pathways by S1P.
While S1P signaling in general has been implicated in a variety of inflammatory skin diseases, our data suggest that S1PR3 may be an important contributor to cutaneous itch associated with these disorders. Previously, S1P receptor modulators have demonstrated efficacy in reducing inflammation in mouse models of dermatitis (Kohno et al., 2004; Sugita et al., 2010). Similarly, ponesimod, which inhibits S1PR1, decreases disease severity scores in psoriasis patients (D'Ambrosio et al., 2016). However, one study found that topical S1P can exert protective effects in mouse models of psoriasis and allergic contact dermatitis (Schaper et al., 2013). This is surprising because one would expect S1P to promote inflammation rather than inhibit it. These contrasting effects may be due to the diverse roles S1PRs play in different cell types. For example, S1PR2 exerts proliferative effects on keratinocytes (Japtok et al., 2014), whereas S1PR1 directs immune cell migration into tissues (Matloubian et al., 2004). Our findings suggest that S1P/S1PR3 signaling may play an important role in itch sensations associated with these skin disorders, although S1P signaling in general may contribute in complex ways via the actions of other S1PRs in skin.
Several S1PR modulators are currently in use or in clinical trials for diseases linked to pain and/or itch, including multiple sclerosis, inflammatory bowel disease, and psoriasis (Brinkmann et al., 2010; Kunkel et al., 2013; Degagné and Saba, 2014). Of particular note is ponesimod, which is in clinical trials for psoriasis treatment (Vaclavkova et al., 2014). Whereas ponesimod acts on the immune system and targets S1PR1 to combat inflammation, we propose neuronal S1P/S1PR3 signaling as a potential target for the treatment of inflammatory pain and itch. We showed previously that loss of S1PR3 can block inflammatory pain without affecting immune cell recruitment and that a selective S1PR3 antagonist can ameliorate inflammatory hypersensitivity, suggesting that S1PR3-specific blockers may be effective for treating pain (Hill et al., 2018). Our finding that S1P can act as a pruritogen also suggests a role for S1P/S1PR3 in chronic itch, in which circulating S1P levels have been found to be significantly increased in human psoriasis patients (Checa et al., 2015; Myśliwiec et al., 2016). Although we and others have demonstrated the relevance of S1PR3 signaling to chronic pain, it will be essential to explore the role of S1P/S1PR3 signaling in chronic itch models to ascertain its pathological relevance. Our data support a key role for S1PR3 in activation of pain and itch neurons and may inform rational design of S1P and S1PR modulators to treat pain, itch, and inflammatory skin diseases.
Footnotes
We thank Z. Rifi for assistance with scoring mouse behavior, N. Kucirek for critical reading of the manuscript, and all members of the D.M.B. laboratory for constructive feedback and criticism.
R.Z.H., T.M., R.B.B., and D.M.B. are co-inventors on a patent based upon several of the findings of this manuscript.
- Correspondence should be addressed to Diana M. Bautista, Department of Molecular and Cell Biology, University of California, 142 Life Sciences Addition #355, Berkeley, CA, 94720. dbautista{at}berkeley.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}