

Abstract
Although the reduction of viral loads in people with HIV undergoing combination antiretroviral therapy has mitigated AIDS-related symptoms, the prevalence of neurological impairments has remained unchanged. HIV-associated CNS dysfunction includes impairments in memory, attention, memory processing, and retrieval. Here, we show a significant site-specific increase in the phosphorylation of Syn I serine 9, site 1, in the frontal cortex lysates and synaptosome preparations of male rhesus macaques infected with simian immunodeficiency virus (SIV) but not in uninfected or SIV-infected antiretroviral therapy animals. Furthermore, we found that a lower protein phosphatase 2A (PP2A) activity, a phosphatase responsible for Syn I (S9) dephosphorylation, is primarily associated with the higher S9 phosphorylation in the frontal cortex of SIV-infected macaques. Comparison of brain sections confirmed higher Syn I (S9) in the frontal cortex and greater coexpression of Syn I and PP2A A subunit, which was observed as perinuclear aggregates in the somata of the frontal cortex of SIV-infected macaques. Synaptosomes from SIV-infected animals were physiologically tested using a synaptic vesicle endocytosis assay and FM4–64 dye showing a significantly higher baseline depolarization levels in synaptosomes of SIV+-infected than uninfected control or antiretroviral therapy animals. A PP2A-activating FDA-approved drug, FTY720, decreased the higher synaptosome depolarization in SIV-infected animals. Our results suggest that an impaired distribution and lower activity of serine/threonine phosphatases in the context of HIV infection may cause an indirect effect on the phosphorylation levels of essential proteins involving in synaptic transmission, supporting the occurrence of specific impairments in the synaptic activity during SIV infection.
SIGNIFICANCE STATEMENT Even with antiretroviral therapy, neurocognitive deficits, including impairments in attention, memory processing, and retrieval, are still major concerns in people living with HIV. Here, we used the rhesus macaque simian immunodeficiency virus model with and without antiretroviral therapy to study the dynamics of phosphorylation of key amino acid residues of synapsin I, which critically impacts synaptic vesicle function. We found a significant increase in synapsin I phosphorylation at serine 9, which was driven by dysfunction of serine/threonine protein phosphatase 2A in the nerve terminals. Our results suggest that an impaired distribution and lower activity of serine/threonine phosphatases in the context of HIV infection may cause an indirect effect on the phosphorylation levels of essential proteins involved in synaptic transmission.
Introduction
Despite successful combination antiretroviral therapy in people with HIV (PWH), the viral reservoir can persist in the brain, leading to neuronal dysfunction and impairment of the molecular machinery required for proper neuronal excitability and communications (Haughey et al., 2001; Musante et al., 2010; Fitting et al., 2013; Gelman, 2015). There is no evidence that HIV-1 infects neurons; however, non-neuronal HIV-infected cells, such as macrophages, microglial cells, and astrocytes, release viral neurotoxic proteins that may lead to aberrant synaptodendritic pruning and neuronal injury (Eggers et al., 2017). On the other hand, HIV-mediated neuroimmune dysfunction plays a significant role in dysregulating neurotransmission and neuroplasticity in PWH. Thus, with successful suppression of viral replication by combination antiretroviral therapy (ART), neuroinflammation continues to perturb the CNS (Burdo et al., 2013). Although many studies have investigated HIV-associated neuronal dysfunction, the molecular pathways leading to the impairment of synaptic connectivity and neuronal transmission by HIV are understudied.
Calcium-dependent synaptic vesicles (SVs) release in the “active zone” of presynaptic terminals is responsible for tuned neuronal activities in both inhibitory and excitatory synapses, governing the synaptic plasticity, learning, and memory formation (Südhof, 2012). Neurotransmission machinery consists of SV-associated proteins that regulate neurotransmitter uptake, vesicles release, and recycling to ensure the efficient transfer of signals to the postsynaptic terminals (Greengard et al., 1993). Among SV-associated proteins is the small family of phosphoproteins called synapsins (Syn), which play a pivotal role in timely release of the neurotransmitters and neuronal differentiation (Bähler et al., 1990). Syn I undergoes multiple phosphorylation and dephosphorylation cycles at several amino acids during neurotransmission modulating the interactions with Syn I, actin, SVs, and phospholipids, thus allowing the SVs reorganization and trafficking at the synaptic terminals. The phosphorylation cycling confers tight regulation of SV trafficking within the presynaptic terminals, maintaining a releasable pool and ultimately facilitating SV fusion to the presynaptic terminals (Cesca et al., 2010).
Syn I interacts with the presynaptic components through its conserved domains. Multiple kinases, including protein kinase A (PKA), Ca2+/calmodulin-dependent kinase (CaMK) I/II/IV, mitogen-activated protein kinase (MAPK), and cell division protein kinase 1 and 5 (cdk1/5) are known to specifically phosphorylate amino acid residues of Syn I domains, altering the Syn I interaction with synaptic components following the depolarization of neurons, thus promoting the timely trafficking of SVs for membrane fusion and neurotransmitter release to the synaptic clefts. (Onofri et al., 1997; Hosaka and Südhof, 1999; Cheetham et al., 2001; Angers et al., 2002; Yamada et al., 2009).
Various mutations in synapsins are associated with neurological disorders. Nonsense and missense mutations and single nucleotide polymorphisms have been reported in patients with epileptic seizures and autism (Fassio et al., 2011; Lignani et al., 2013), schizophrenia (Saviouk et al., 2007), bipolar disorder (Lachman et al., 2006), and multiple sclerosis (Liguori et al., 2004). Dysregulation of SUMOylation and phosphorylation of Syn I occurs in patients with autism and Huntington's diseases (Xu et al., 2013; Tang et al., 2015).
Here, we examined the state of synapsin I phosphorylation in the frontal cortex of an accelerated simian immunodeficiency virus (SIV)-infected rhesus macaque model of NeuroHIV. We found that Syn I (S9, site 1) phosphorylation is significantly higher in the frontal cortex of SIV-infected rhesus macaques compared with uninfected controls. Interestingly, we found restoration of Syn I (S9) phosphorylation in SIV-infected ART-treated animals to the baseline levels of uninfected animals. We further demonstrated a reduction in the synaptosomal expression of protein phosphatase 2A (PP2A) A subunit in SIV-infected animals. Together with an impaired cellular distribution and lower PP2A activity in the frontal cortex of SIV-infected animals, we propose that an impaired PP2A activity in the context of SIV infection leads to the functional dysregulation of PP2A substrates at the synaptic levels. Using an endocytosis assay of synaptosomes isolated from SIV-infected macaques, we showed that an FDA-approved drug can mitigate high levels of the depolarization in synaptosomes from SIV-infected animals.
Materials and Methods
Animals.
Male Indian rhesus macaques (Macaca mulatta) were inoculated intravenously with SIVmac251 viral swarm (5 ng p27; Tulane National Primate Research Center's Viral Core) and subsequently CD8-depleted through administration of 10 mg/kg of anti-CD8 antibody subcutaneously at 6 d postinfection (dpi) and 5 mg/kg of antibody intravenously at 8 and 12 dpi (Nonhuman Primate Reagent Resource) (Lakritz et al., 2015). The SIV+ animals were necropsied according to humane endpoints consistent with the recommendations of the American Veterinary Medical Association Guidelines for the Euthanasia of Animals. The development of simian AIDS was determined postmortem by the presence of Pneumocystis carinii-associated interstitial pneumonia, Mycobacterium avium-associated granulomatous enteritis, hepatitis, lymphadenitis, and/or adenovirus infection of surface enterocytes in both small and large intestines. For ART treatments, animals were not only SIV-infected and CD8-depleted but also received a triple ART regimen of Raltegravir (22 mg/kg oral twice daily, Merck), Tenofovir (30 mg/kg subcutaneous once daily, Gilead), and Emtricitabine (10 mg/kg subcutaneous once daily, Gilead) at 21 dpi until the timed necropsied at 118–120 dpi. Animals were anesthetized with ketamine-HCl and necropsied by intravenous pentobarbital overdose. Animals used in the study were housed at the Tulane National Primate Research Center. All animals used in this study were handled in strict accordance with American Association for Accreditation of Laboratory Animal Care with the approval of the Institutional Animal Care and Use Committee of Tulane University.
Immunoblotting.
Whole protein lysates were prepared using a lysis buffer containing 6 m urea, 0.025% SDS, 5 mm β-mercaptoethanol, and mechanical homogenization. Synaptosomes were isolated from frontal cortices of uninfected, SIV+, and SIV+ ART animals, using Syn-PER synaptic protein extraction reagent (Thermo Fisher Scientific) as described by the manufacturer in the presence of Halt proteinase inhibitors mix (Thermo Fisher Scientific) and phosphatase inhibitors cocktail 3 (Sigma-Aldrich). Protein assays were performed using Bradford reagent (Bio-Rad) according to the manufacturer's protocol. A total of 60 μg of protein lysates or 30 μg of synaptosomes per condition was analyzed on SDS-PAGE gels. The following primary antibodies were used: Cell Signaling Technology: phospho-CaMKII (1:1000, 12716S; RRID:AB_2713889), Synapsin I (1:1000, 5297S; RRID:AB_2616578), phospho-Synapsin I (S9) (1:1000, 2311S; RRID:AB_2200427), PP2A A subunit (1:1000, 2039S; RRID:AB_2713889), and PKA RI α/β (1:1000, 3927S; RRID:AB_1658217). Invitrogen: Phospho-Synapsin I (S549) (1:1000, PA1-4697; RRID:AB_2175503), phospho-Synapsin I (S603) (1:1000, PA1-4604; RRID:AB_560615), phospho-CaMKI (Thr177) (1:1000, PA5-38434; RRID:AB_2555035), phospho-CaMKIV (Thr196/200) (1:1000, PA5-37504; RRID:AB_2554113). R&D Systems: Phospho-CREB (1:1000, 702710; RRID:AB_10972977), CaMKII (1:1000, MAB7280). EMD Millipore: Phospho-Synapsin I (S62/S67) (1:1000, AB9848; RRID:AB_673006). ProteinTech: GAPDH (1:3000, 60004-1-Ig; RRID:AB_2107436). The signals were detected using ODYSSEY CLx Imaging system (LI-COR). Protein band intensities were quantified using Image Studio Software and normalized against corresponding actin or GAPDH signals.
Immunohistochemistry.
Paraffin-embedded sections (5 μm) were prepared and immunolabeled using a mouse Synapsin I (Synaptic System: 1:200, 106001; RRID:AB_887805) and the PP2A A subunit (1:200) antibodies. The sections were deparaffinized by 20 min incubation in xylene. They were then hydrated for 2 min with serial changes in 100%, 90%, and 75% of ethanol, each two times for 2 min and washed with distilled water. For heat-induced antigen unmasking, the sections were heated up at 95°C for 20 min in an antigen unmasking solution (Vector, H-3300), cooled down, and washed in DPBS (Invitrogen). The sections were then incubated in 0.25% Triton X100 in DPBS for 5 min and blocked in 0.1% Triton X-100, 10% FBS in DPBS for 1 h. The primary antibody mix were prepared in the blocking solution, and the sections were incubated for overnight at 4°C. Primary antibody-treated sections were washed three times with DPBS and incubated in a mix of secondary antibodies; goat anti-rabbit Alexa-488 (1:1000, Invitrogen) and goat anti-mouse Alexa-555 (1:1000, Invitrogen) for 1 h in the presence of DAPI to stain nuclei. The sections were imaged using a Keyence microscope at 20× or 40× objective. Corresponding images were then quantified using BZ-X Analyzer software and the Hybrid Cell Count module.
Protein kinase A assay.
We assayed the PKA activity of synaptosome preparations of uninfected, SIV+, and SIV+ ART rhesus macaques using PKA colorimetric activity kit (Invitrogen) according to the manufacturer's instructions in 15 ng of final protein concentrations. The synaptosomes were prepared as previously described in the presence of phosphatase and proteinase inhibitors.
PP2A activity assay.
PP2A activity was assayed in synaptosome preparations of uninfected, SIV-infected, and SIV-infected with ART rhesus macaques; 100 μg of the synaptosomal proteins (without phosphatase inhibitors) were subjected to immunoprecipitation of PP2A using an antibody against PP2A A subunit (1:50, Cell Signaling Technology) in the immunoprecipitation buffer (50 mm Tris, pH 7.4, 1% NP-40, 150 mm NaCl, 2 mm EDTA) plus proteinase inhibitors. The complex was then pulled down using protein A Sepharose 4B (Invitrogen) and washed three times with the immunoprecipitation buffer. The PP2A assay was performed as described previously (McAvoy and Nairn, 2010) in colorimetric assay buffer (20 mm Tris, pH 7.5, 5 mm MgCl2, 1 mm EGTA, 0.02% β-mercaptoethanol, and 0.1 mg/ml BSA) using 4-nitrophenyl phosphate (pNPP, Sigma-Aldrich) as a substrate. The absorbance was read at 405 nm using Epoch2 microplate reader (BioTek). The protein phosphatase inhibitor fostriecin (Cayman Chemical) was added to some wells at 1 μm to specifically inhibit PP2A (Swingle et al., 2007).
SV endocytosis assay.
Pellets containing synaptosomes were analyzed for SV endocytosis assay as described previously (Daniel et al., 2012). In summary, the pellets were resuspended into SET buffer (0.32 m sucrose, 1 mm EDTA, 5 mm Tris, pH 7.4). Synaptosomes (20 μg/ml) were then attached to polyethyleneimine-coated glass bottom 96-well microplates (Greiner bio-one) at 4°C, as described previously (Daniel et al., 2012). Synaptosomes were revived at 30°C for 15 min. In some experiments, synaptosomes were labeled with 1 μm calcein blue-AM (Invitrogen) for 30 min at 30°C in HBK buffer (143 mm NaCl, 4.7 mm KCl, 1.3 mm MgSO4 1.2 mm CaCl2, 20 mm HEPES, 0.1 mm NaH2PO4, and 10 mm d-glucose, pH 7.4). SV endocytosis was assayed using FM4-64FX (Invitrogen) at 1 mm. Extraneous FM4-64FX was then washed with 1 mm Advasep-7 (Biotium) for 2 min. Hydroxy dynasore was added at 100 μm to some wells for 30 min before FM4-64FX labeling. For stimulation, synaptosomes were depolarized in the presence 40 mm KCl for 2 min at 30°C. The plates were either immediately imaged or fixed using 4% PFA in HBK buffer. The imaging was performed using a Keyence microscope at 20× or 40× objective. Four images per well from three wells were captured using the same exposure times. Total fluorescence intensities of objects between 3 and 75 μm in area were measured and compared.
Statistical analysis.
The data analysis comparing the intensities between the samples were performed either with one-way ANOVA with post hoc Tukey HSD (honest significant difference) or with Student's t test. A criterion of α = 0.05 was used to establish statistical significance. The mean values ± SEM were results of analysis with the indicated number (n) of experiments.
Results
Synapsin I is hyperphosphorylated at site 1 serine 9 in the frontal cortex of SIV+ rhesus macaques
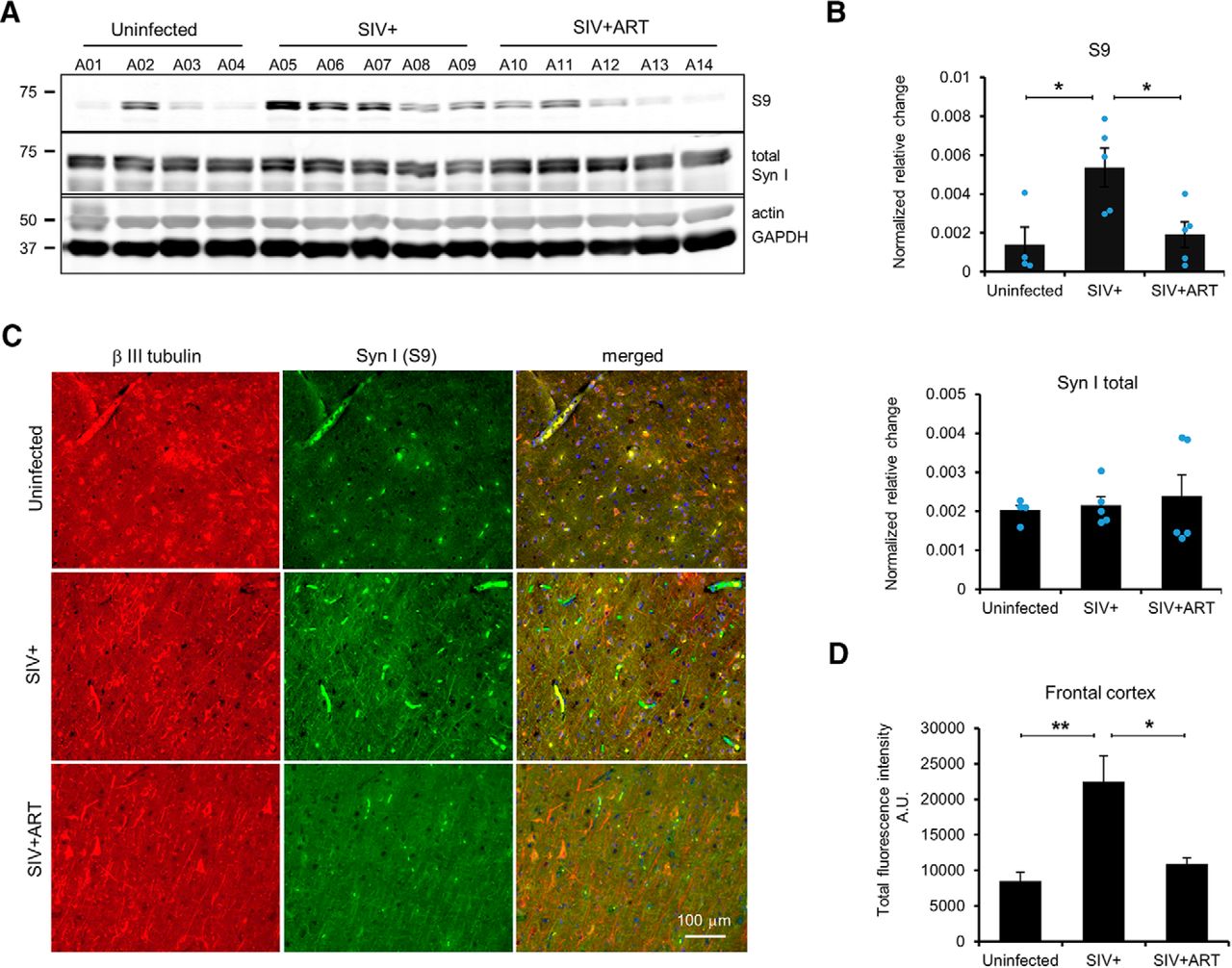
To investigate mechanisms of SIV-associated CNS dysfunction, 10 male Indian rhesus macaques were intravenously infected with SIVmac251 followed by depletion of CD8+ cells by anti-CD8 antibody to accelerate SIV CNS pathology (SIV+) (Lakritz et al., 2015). Five SIV-infected rhesus macaques received a triple ART regimen at 21 dpi to suppress the viral load (SIV+ ART). SIV+ animals (A05-A09) were necropsied according to humane endpoints consistent with the recommendations of the American Veterinary Medical Association Guidelines for the Euthanasia of Animals, and SIV+ ART animals (A10-A14) were time-sacrificed at 120 dpi. Four uninfected rhesus macaques (A01-A04) served as experimental controls (Table 1). We prepared brain lysates from the frontal cortex of uninfected, SIV+, and SIV+ ART animals and examined Syn I phosphorylation at several phosphorylation sites using Syn I phosphor-specific antibodies. The phosphorylation of Syn I (S9) was significantly different among uninfected, SIV+, and SIV+ ART animals (Fig. 1A,B, top; one-way ANOVA, F(2,11) = 6.3849, p = 0.01). Syn I (S9) phosphorylation was significantly higher in SIV+ cohort compared with uninfected animals (Fig. 1B; p < 0.05). Furthermore, Syn I (S9) phosphorylation was lower in SIV+ ART animals compared with SIV+ animals, suggesting that ART mitigated SIV-associated increased phosphorylation (Fig. 1B; p < 0.05). Total Syn I protein expression was not significantly different among groups indicating a higher phosphorylation at S9 than an increased expression of Syn I in SIV+ animals (Fig. 1B, bottom). Next, we examined sections of the frontal cortex of SIV+ animals by immunofluorescence using anti-p-Syn I (S9) and β III tubulin antibodies and quantified as the fluorescent intensity. Phosphorylated Syn I (S9) significantly differed among the groups (Fig. 1C,D; one-way ANOVA, F(2,21) = 8.3541, p = 0.002). These experiments further confirmed higher p-Syn I (S9) in the cortex of SIV+ animals. We also examined sections of parietal and occipital lobes for p-Syn I (S9); both lobes displayed significantly higher S9 phosphorylations in SIV+ compared with sections of uninfected and SIV+ ART animals (Fig. 2A,B; one-way ANOVA, F(2,41) = 4.97, p= 0.01 and F(2,28) = 8.87, p= 0.001, respectively).
Information about the animals used in this studya

Hyperphosphorylation of Syn I (S9) in the frontal cortex of SIV+ rhesus macaques. A, Representative Western blots of whole-tissue protein lysates from the frontal cortex of uninfected, SIV+, and SIV+ ART animals using an antibody against Syn I (S9). B, Bands were quantified and normalized against actin or GAPDH signals. There was a significant difference among groups. A higher Syn I (S9) phosphorylation was detected in the frontal cortex lysates from SIV+ compared with uninfected or SIV+ ART. No changes were observed in the expression levels of total Syn I (B, bottom). C, Immunohistochemistry of sections of uninfected, SIV+, and SIV+ ART probed with anti-Syn I (S9, green) and β III tubulin (red) antibodies. D, Quantification of experiments illustrated in C revealed a significant difference among groups. A significant increase in the intensities of the positive Syn I (S9) signals was detected. **p < 0.005 (one-way ANOVA with Tukey HSD). *p < 0.05 (one-way ANOVA with Tukey HSD). Error bars indicate SD of mean; ± SEM.

Hyperphosphorylation of Syn I (S9) in parietal and occipital lobes of SIV+ rhesus macaques. Representative sections of parietal (A) and occipital (B) lobes of uninfected, SIV+, and SIV+ ART animals were immunostained with antibodies against β III tubulin (red) and p-Syn I (S9, green). P-Syn I (S9) signal intensities were measured and quantified. Significant increases in the immunopositive p-Syn I (S9) signals were observed in SIV+ sections compared with uninfected or SIV+ ART sections. PL, Parietal lobe; OL, occipital lobe. *p < 0.05 (one-way ANOVA with Tukey HSD). **p < 0.005 (one-way ANOVA with Tukey HSD). Error bars indicate SD of mean; ± SEM.
We further assessed Syn I phosphorylation on residues located in the other Syn I domains. Phosphorylation-specific antibodies against p-Syn I (S62/67), p-Syn I (S549), and p-Syn I (S603) were used on the frontal cortex lysates of uninfected, SIV+, and SIV+ ART animals (Fig. 3). We found an overall decreasing trend in Syn I phosphorylation at p-Syn I (S62/67) (one-way ANOVA, F(2,11) = 3.2108, p = 0.08) and no significant change in p-Syn I (S549). Phosphorylation at Syn I (S603) residue exhibited an increasing trend with a significant higher phosphorylation in SIV+ ART animals compared with uninfected lysates (Fig. 3B; one-way ANOVA, F(2,11) = 4.2399, p = 0.04).

Effect of SIV infection on Syn I phosphorylation at S62/S67, S549, and S603 sites in the frontal cortex of rhesus macaques. A, Protein lysates of the frontal cortex from uninfected, SIV+, and SIV+ ART animals were analyzed by Western blot using specific antibodies against Syn I (S62/S67, S549, and S603) sites. B, Band intensities were quantified and normalized against GAPDH and graphed as mean. No significant changes were observed among groups in the phosphorylation levels of S62/S67 and S549. However, a significant increase was detected in Syn I (S603) when frontal cortex lysates from uninfected were compared with SIV+ ART animals. *p < 0.05 (one-way ANOVA with Tukey HSD). Error bars indicate SD of mean; ± SEM.
Phosphorylation of Syn I (S9) in synaptosomes of SIV+ animals
To assess Syn I phosphorylation in preparations of the nerve terminals, we prepared synaptosomes from the frontal cortex of uninfected, SIV+, and SIV+ ART animals and assessed Syn I (S9) phosphorylation by Western blots. We found significant differences among groups (one-way ANOVA, F(2,12) = 14.5737, p = 0.0006). Significantly higher levels of Syn I (S9) phosphorylation were detected in the synaptosome preparations of frontal cortex of SIV+ animals compared with synaptosomes from uninfected or SIV+ ART animals (Fig. 4A,B; post hoc, p < 0.005). We also found that S62/S67 phosphorylations were not different among groups (Fig. 4B). A significant increase in S549 was also observed among three groups in the synaptosome preparations (Fig. 4B; one-way ANOVA, F(2,11) = 4.3469, p = 0.04), with a significant increase between with SIV+ ART compared with SIV+ animals (Fig. 4B; post hoc, p < 0.05). The phosphorylation of S603 site was not significantly different among the groups (Fig. 4B).

A significant increase in Syn I (S9) phosphorylation of synaptosome preparations from SIV+ rhesus macaques. A, Synaptosome preparations of uninfected, SIV+, and SIV+ ART were subjected to immunoblot analysis using antibodies recognizing the indicated phosphorylation sites of Syn I. Antibodies against GAPDH and total Syn I were used to assess protein loads. B, Band intensities from every sample were quantified and normalized against their corresponding GAPDH band, and means were graphed. A significant increase was observed among groups in Syn I (S9) (ANOVA, p < 0.005). Although no significant differences were detected among groups in S62/67, S549, and S603 phosphorylation, there was a significant increase in S549 phosphorylation between synaptosomes of SIV+ and SIV+ ART animals. *p < 0.05 (one-way ANOVA with Tukey HSD). **p < 0.005 (one-way ANOVA with Tukey HSD). Error bars indicate SD of mean; ± SEM.
We hypothesized that the higher levels of phosphorylated Syn I (S9) in SIV+ animals may indicate either a dysregulation of PKA and/or CaMK I/IV kinases responsible for S9 phosphorylation or a lower activity of PP2A, which is responsible for Syn I (S9) dephosphorylation. To verify the pathway(s) responsible for a higher Syn I (S9) phosphorylation in SIV+ animals, we examined the activities of kinases responsible for S9 phosphorylation. The PKA activity was assessed by quantifying the phosphorylated cAMP response element-binding protein (p-CREB) in the frontal cortex synaptosomes (Delghandi et al., 2005). No significant differences were observed in the overall p-CREB levels (Fig. 5A). Furthermore, the expression of PKA catalytic subunit was assessed by Western blot and the PKA activity directly assayed in SIV infected and compared with synaptosomes from uninfected or SIV+ ART animals. There were no significant differences in the expression of PKA catalytic subunit or PKA activity among the groups (Fig. 5B,C).

Assessment of the activities of CREB, PKA, CaMK I, and CaMK IV in synaptosomes of the frontal cortex of SIV+ rhesus macaques. A, Western blots of synaptosome preparations, which were probed with an antibody against p-CREB. No differences among groups were observed. B, Expression of PKA catalytic subunit in synaptosomes of uninfected, SIV+, and SIV+ ART animals. No differences among the groups were observed. C, Assessment of PKA activity in synaptosome preparations of the frontal cortex of rhesus macaques. D, Assessment of CaMKI and CaMKIV activities using phosphospecific antibodies in synaptosome preparations of uninfected, SIV+, and SIV+ ART rhesus macaque frontal cortex. E, No differences among the groups were observed (one-way ANOVA with Tukey HSD).
Next, we examined the activities of both CaMKI and IV kinases known to phosphorylate Syn I (S9) (Huttner et al., 1981). An antibody against the phosphorylated CaMKIV (Tyr196/Tyr200) and one against the phosphorylated CaMKI (Tyr177) were used to test the synaptosome preparations for the CaMKIV and CaMK I activities, respectively (Wayman et al., 2006; Yadav et al., 2009). These experiments indicated no difference in the level of active forms of phosphorylated CaMKI or IV among SIV+, SIV+ ART, or uninfected animals (Fig. 5D,E).
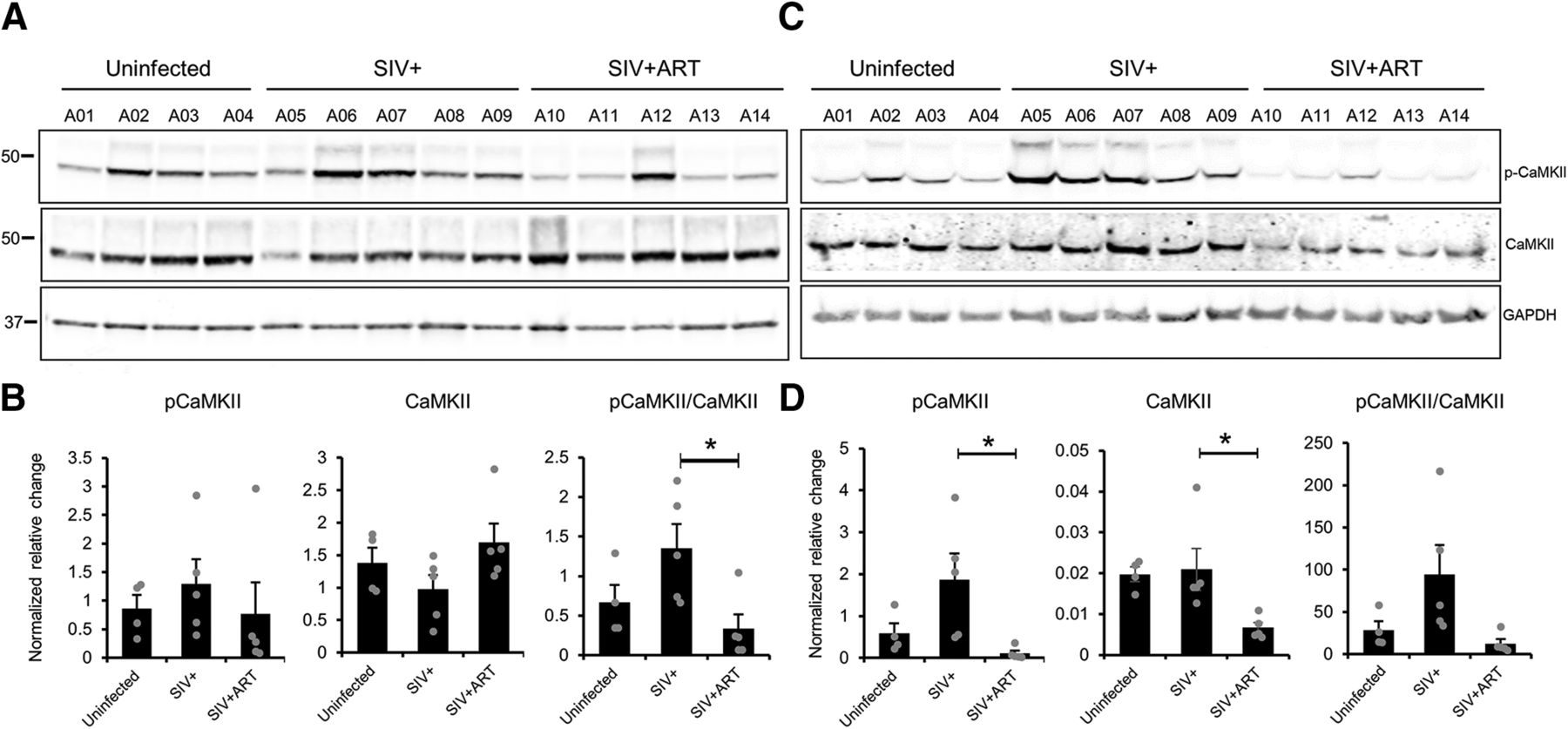
In addition, we verified the CaMKII activity, which promotes the phosphorylation of Syn I (S603) (Onofri et al., 1997). We used an antibody against Thr286, which detects an active form of CaMKII. In synaptosomes, we found no significant differences among groups in the p-CaMKII levels when the loadings were normalized against GAPDH (Fig. 6A,B; one-way ANOVA, F(2,11) = 0.4002, p = 0.68). However, when we compared the p-CaMKII levels with the total CaMKII protein, a significant difference was found among the groups (Fig. 6B; one-way ANOVA, F(2,11) = 4.7433, p = 0.03), specifically a significant reduction of the CaMKII activity in synaptosomes of SIV+ ART compared with SIV+ animals (post hoc, p < 0.05). In total lysates (Fig. 6C,D), the CaMKII activity was found significantly different among groups (one-way ANOVA, F(2,11) = 5.3209, p = 0.02) with SIV+ ART significantly decreased from SIV+ (p < 0.05). We also found significant differences among groups when the lysates were probed with anti-CaMKII (one-way ANOVA, F(2,11) = 5.6849, p = 0.02) with a significant decrease in the CaMKII expression in lysates from SIV+ ART animals (post hoc, p < 0.05) compared with SIV+ animals. When the lysates were normalized with the total CaMKII protein (pCaMKII/CaMKII), a trend for a difference among groups was observed (one-way ANOVA, F(2,11) = 3.9723, p = 0.05) (Fig. 6D).

Analysis of CaMKII activity compared with CaMKII total protein in synaptosomes of SIV+ animals. Synaptosome (A, B) and total lysates (C, D) preparations of uninfected, SIV+, and SIV+ ART rhesus macaques frontal cortex were analyzed using Western blot. The intensities of pCaMKII and CaMKII bands were quantified and graphed alone or as pCaMKII/CaMKII ratio. *p < 0.05 (one-way ANOVA with Tukey HSD).
Alterations in the PP2A A subunit expression, cellular distribution, and PP2A activity in the SIV+ brain
SV trafficking at the presynaptic terminals is regulated by dephosphorylation of Syn I by PP2A and PP2B (calcineurin), which modulate SVs clustering and their associations with actin cytoskeleton (Jovanovic et al., 1996, 2001). Syn I (S9) is selectively dephosphorylated by PP2A in a tonic and Ca2+ independent fashion (Jovanovic et al., 2001). We verified the expression levels of PP2A A subunit and found significant changes among the groups (one-way ANOVA, F(2,11) = 6.8413, p = 0.012) with a significant decrease when synaptosomes from SIV+ compared with SIV+ ART animals (Fig. 7A,B; post hoc, p < 0.05). Indeed, upon examining the cellular distribution of PP2A A subunit using immunolabeling of SIV+ animals frontal cortex, we found that both Syn I and PP2A A subunit were coexpressed in the somata of neurons (Fig. 7C) with significantly lower PP2A A detected compared with uninfected or SIV+ ART frontal cortex (Fig. 7D; one-way ANOVA, F(2,28) = 12.9317, p = 0.0001). Syn I expression, predominantly as perinuclear aggregates, was found in the SIV+ cortex; whereas in uninfected animals, a dispersed distribution of Syn I-positive puncta was detected (Fig. 7C, arrows vs arrowheads). Because of the lower protein levels and impaired distributions of PP2A A subunit, we measured the PP2A activity in SIV+ synaptosomes by immunoprecipitating PP2A A subunit from the synaptosome preparations of uninfected and SIV+ animals and assaying the activity of bound PP2A holoenzyme. We observed a significantly lower PP2A activity in synaptosomes from SIV+ animals compared with uninfected animals (Fig. 7E; t test, p < 0.05). In addition, the application of fostriecin at 1 μm, which specifically inhibits PP2A (Swingle et al., 2007), decreased the PP2A activity in uninfected synaptosomes to that of SIV+ synaptosomes. These experiments strongly suggest that the lower activity of PP2A is responsible for the higher Syn I (S9) phosphorylation in SIV+ brains.

Impairments in the expression, cellular distribution, and activity of PP2A A subunit in SIV+ frontal cortex. A, Synaptosome preparations of the frontal cortex from uninfected, SIV+, and SIV+ ART animals were subjected to Western blot analysis with an antibody recognizing PP2A A subunit. The same blot was reprobed with an antibody against GAPDH to assess and quantify protein loads. B, To graph the means, band densities were measured and normalized against GAPDH. There was a significant difference of PP2A A subunit expression among groups. C, Representative images of the frontal cortex of uninfected, SIV+, and SIV+ ART animals, which were immunoprobed with Syn I (red) and PP2A A (green), revealed lower PP2A A-positive signals in neurons somata. Neurons coexpressing Syn I and PP2A A in sections of SIV+ animals exhibited accumulation of perinuclear staining (arrowhead); whereas in uninfected and SIV+ ART sections, the stainings were more dispersed (arrows). D, Intensities of PP2A A signals were measured and graphed. A significant decrease was observed in the signals from SIV+ sections. E, Equal amounts of lysates from uninfected and SIV+ animals were subjected to immunoprecipitation using PP2A A subunit antibody and phosphatase assay using pNPP as a substrate. A significant reduction of PP2A activity in synaptosomes of SIV+ animals is plotted. fostriecin (1 μm) specifically inhibited PP2A activity. #p < 0.05 (Student's t test). *p < 0.05 (one-way ANOVA with Tukey HSD). ***p < 0.001 (one-way ANOVA with Tukey HSD). Error bars indicate SD of mean; ± SEM; A.U., Fluorescence arbitrary units.
SV endocytosis assay of SIV+ synaptic terminals
To functionally test the efficacy of SVs release in the synaptic terminals of rhesus macaque brains, we used an endocytosis assay using frontal cortex synaptosomes. In the presence of KCl, synaptosomes can undergo depolarization and induce SV exocytosis at the synapse (Coffey et al., 1993). Following depolarization, endocytosis events will recycle the released vesicles (Lou, 2018). We assayed synaptosome preparations from the frontal cortex of uninfected, SIV+, and SIV+ ART animals using FM4-64 by adhering the synaptosomes onto glass microplates (Daniel et al., 2012). Synaptosomes from the frontal cortex of animals were then depolarized using KCl and subsequently imaged. In uninfected animals, we observed a significant increase in the fluorescence signals following KCl treatments indicating an active endocytosis/exocytosis in KCl-treated synaptosomes (Fig. 8A,B; p < 0.005). We found elevated fluorescence signals, even in the absence of KCl in synaptosomes from SIV+ animals (Fig. 8A,B; p < 0.005). The fluorescence signals in synaptosomes from SIV+ ART brains had a similar increase as uninfected synaptosomes in the presence of KCl (p < 0.005). Hydroxy dynasore inhibits dynamin-mediated SV endocytosis (Macia et al., 2006). Application of hydroxy dynasore to the synaptosomes mitigated the fluorescence signals in the presence of KCl, thus inhibiting depolarization (Fig. 8A,B). These experiments further support the existence of more unbound SVs available for fusion at the synaptic terminals in SIV+ than uninfected or SIV+ ART animals, indicating a higher phosphorylation of Syn I (S9).

Endocytosis assay of SVs using synaptosomes from the frontal cortex of rhesus macaques. A, Frontal cortex synaptosomes were assayed on glass microplates in the presence of 40 mm KCl and FM4–64. Brighter fluorescence signals are an indication of increased SV endocytosis, and the dye and lipid interactions thus increase in depolarization of synaptosomes. Significantly higher baseline fluorescence signals were detected in synaptosomes of SIV+ animals, even in the absence of KCl. Synaptosomes from SIV+ animals pretreated with an inhibitor of dynamin-mediated endocytosis exhibited low signals in both the presence and absence of KCl. B, Quantification of fluorescence signals using the total fluorescence signals recorded in different conditions are graphed. H.d., Hydroxy dynasore. *p < 0.05 (one-way ANOVA with Tukey HSD). **p < 0.005 (one-way ANOVA with Tukey HSD). Error bars indicate SD of mean; ± SEM.
A phosphatase-activating drug mitigated SV endocytosis/exocytosis cycle
PP2A plays a key role in cellular pathways and is a therapeutic target for a number of human conditions, such as multiple sclerosis, cancer, and inflammatory chronic respiratory disease (Perrotti and Neviani, 2013). To test the ability of a phosphatase-activating drug FTY720 (a sphingosine analog) to mitigate SV release from synaptosomes from SIV+, FTY720 was added to the synaptosomes in both resting and depolarizing states. FTY720 strongly lowered the fluorescence intensity in a concentration-dependent fashion, indicating slower endocytosis/exocytosis events in the presence of the phosphatase-activating drug (Fig. 9A,B). The quenching effect of FTY720 was significantly slower in synaptosomes from SIV+ than in synaptosomes from uninfected animals. At 10 and 50 μm, FTY720 exhibited stronger effect on uninfected than SIV+ synaptosomes (t test: 10 μm, p < 0.005; 50 μm, p < 0.05). Thus, the activation of PP2A causes a slower depolarization in synaptosomes from SIV+ animals because of the dephosphorylated Syn I and a greater SV in tethered state.

Treatment of synaptosomes with a phosphatase-activating drug (FTY720) mitigates SV endocytosis in frontal cortex synaptosomes of rhesus macaques. A, FTY720 was applied in different concentrations on frontal cortex synaptosomes in SVE assay in the presence of 40 mm KCl and FM4–64. FTY720 produced lower fluorescence signals in a concentration-dependent fashion. In SIV+ synaptosomes, higher concentrations of FTY720 were required to quench the signals. B, Quantification of fluorescence signals detected in A using the ratio of the total fluorescence recorded after depolarization by 40 mm KCl in different conditions. *p < 0.05 (Student's t test). **p < 0.005 (Student's t test). Error bars indicate mean ± SEM.
Discussion
The phosphorylation of synaptic proteins plays an important role in quickly modifying their functions and regulates synaptic efficacy. Among them, Syn I (S9) phosphorylation modulates Syn I association with SVs, and actin cytoskeleton thus regulates the dynamics of neurotransmitter release to the synaptic clefts. Basal levels of Syn I (S9) phosphorylation are low and highly conserved in both vertebrates and invertebrates (Hosaka and Südhof, 1999; Menegon et al., 2000, 2006; Chi et al., 2001, 2003; Bonanomi et al., 2005). Here, we investigated changes in Syn I phosphorylations in the frontal cortex of SIV-infected rhesus macaques as a model of NeuroHIV (Fig. 10). Our results revealed a specific increase in Syn I (S9) phosphorylation in SIV+ but not in uninfected or SIV+ animals treated with ART. Furthermore, we found a lower expression and perinuclear accumulation of PP2A A (scaffold) subunit, which may indicate an impaired cellular distribution. Interestingly, Syn I was coexpressed with these perinuclear accumulations. Therefore, both pathological events might be a secondary effect, which triggered, with the lack of available PP2A A subunit, to form an active PP2A holoenzyme at the nerve terminals. It was suggested that the A subunit is responsible for the catalytic activity of PP2A by inducing a conformational change in catalytic subunit (Zhou et al., 2003). The interaction of PP2A and viral pathways was also reported (Guergnon et al., 2011). PP2A activity reversely regulated HIV-1 transcription through LIS1, a microtubule-associated protein, which binds to PP2A and displaces its regulatory B subunit (Epie et al., 2006). However, a positive regulatory role of PP2A on HIV-1 transcription has also been reported in which higher nonactive core enzyme form of PP2A-inhibited Tat-induced HIV-1 transcription and PP2A indirectly enhanced HIV-1 transcription (Ruediger et al., 1997; Faulkner et al., 2003). HIV-1 viral infectivity factor was able to degrade PP2A regulatory subunits in vitro (Evans et al., 2018). Recently, Schott et al. (2018) have demonstrated that PP2A dephosphorylates SAM domain and HD domain-containing protein 1, a protein that inhibits HIV-1 replication in dendritic and myeloid cells, and identified PP2A as a key regulator of antiviral activity of SAM domain and HD domain-containing protein 1. These studies strongly suggest the role of PP2A in the HIV-1 infection and replication in infected cells, and imply pathological interactions of HIV-expressed proteins with PP2A subunits. Our current findings support a lower activity of PP2A in neurons and provide a strong evidence that changes in PP2A activity might contribute to HIV neuropathogenesis by interfering with normal interaction of PP2A with its substrates.

Dynamics of the impact of SIV infection and ART regimen on Syn I phosphorylations. A, Amino acid residues studied in this work and their approximate positions in the Syn I domains (A–E). B, Observed changes in phosphorylations at multiple sites of Syn I in total lysates versus synaptosome preparations and functional impact of phosphorylation of these sites in normal physiological conditions based on Cesca et al. (2010). Thin arrows indicate insignificant trends. Thick arrows indicate significant changes. NC, No changes; AA, amino acids; NT, neurotransmitter.
HIV-1 Tat and gp120 proteins have been identified as the main candidates to elicit neurotoxic effects on the synaptic density, even before triggering chronic events, such as neuronal cell death (Sá et al., 2004; Bruce-Keller et al., 2008; Kim et al., 2008; Lu et al., 2011; Shin and Thayer, 2013; Bertrand et al., 2014; Puccini et al., 2015). Studies of HIV-1-associated neuronal impairments in HIV-1 transgenic rats and Tat transgenic mice have revealed significant changes in synaptodendritic integrity and behavioral deficits (Fitting et al., 2013; Roscoe et al., 2014; Hahn et al., 2015). For instance, both proteins have been implicated in impairing glutamate recycling by increasing glutamine release and reducing its uptake at the synaptic cleft (Vesce et al., 1997; Musante et al., 2010). The effect of Tat in inducing changes in the synaptic integrity and the inhibition of hippocampal synapses and network have been reported (Hargus and Thayer, 2013; Mohseni Ahooyi et al., 2018). A Tat-induced excitatory state was also proposed in the brain of Tat transgenic mice (Zucchini et al., 2013).
In addition, persistent immune response to HIV-1 infection also affects neuronal function by overproducing cytokines and chemokines. HIV-1-associated activation of macrophage and microglia, the production of proinflammatory cytokines, and subsequent detection in the CSF of PWH were correlated with HIV neurocognitive impairments (Bandaru et al., 2007). Both IL1β and TNFα downregulate the expression of excitatory amino acid transporter 2, which plays an important role in up taking of excess glutamate from the synaptic cleft (Vartak-Sharma et al., 2014).
Our findings demonstrated that PP2A activity is impaired in the brain of SIV-infected animals, which indirectly dysregulates post-translational modification of PP2A targets. This impaired activity may be associated with the presence of SIV proteins, active infection, activation of resident immune brain cells, and/or increased proinflammatory cytokines. These experiments are currently under investigation. In an experimental autoimmune encephalomyelitis model, a mixture of proinflammatory cytokines, including Th1, TNFα, IFNγ, and IL-1β, induced Syn I phosphorylations by ERK1/2 pathway (Guarnieri et al., 2018). Thus, extracellular signals may modulate PP2A activity toward its substrates leading to multiple physiological impairments.
Neuronal markers, such as microtubule-associated protein 2 and the presynaptic protein, synaptophysin, are downregulated in PWH (Desplats et al., 2013). The unchanged Syn I protein expression observed in the synaptosome and lysates preparations of frontal cortex of uninfected and SIV+ using Western blot analysis and previous findings in HIV-1+ human brains showing lower optical density of Syn I immunolabeling in the brain sections in the neocortical with high HIV-1 RNA load indicate a dysregulation in the Syn I protein distributions at the cellular levels. A reduction in the Syn I expression was reported in Tat transgenic mice and in vitro in a neuroblastoma cell line (Gelman and Nguyen, 2010; Hahn et al., 2015; Guha et al., 2018). These findings support the impact of several factors in the Syn I expression and post-translational modifications, which should be considered for future work. Thus, the presence of HIV-1 proteins may contribute to abnormal neuronal transmission by modulating neuronally expressed proteins.
We found a decreasing trend of Syn I (S603) phosphorylation in synaptosomes, a site dephosphorylated by PP2A, which supports a lower PP2A activity in synaptosome of SIV+ animals (Jovanovic et al., 2001). However, we also reported a significant increase in CaMKII activity in the total lysates of frontal cortex from SIV+ animals, which supports higher phosphorylation of Syn I (S603) in the lysates. Recent reports of changes in the CaMKII expression indicated a lower expression of total CaMKII protein when normalized with MAP2 in the frontal cortex of PWH with neurocognitive impairments and in differentiated human SH-5YSY neuroblastoma cells exposed to supernatants of HIV-1-infected macrophages (Guha et al., 2018). However, the CaMKII activity was rapidly induced in mice by intrathecal injection of HIV-1 gp120 (Li et al., 2013). In doxycycline-inducible HIV-1 Tat mice, the CaMKII activity and expression were reduced in male mice (Nookala et al., 2018). In an SIV infection model, an increase in the CaMKII expression was found in the hippocampus and frontal cortex of SIV+ animals (Gupta et al., 2010). Our findings on the CaMKII expression and activity in total lysates versus synaptosome preparations suggest a dysregulation in subcellular distributions of neuronal and non-neuronally expressed CaMKII protein and its activity.
For the first time, we performed the endocytosis assay on nonhuman primate frozen brain samples using FM4–64. This allowed us to functionally test the excitability of SIV+ synaptosomes and assess the differences with uninfected animals. Furthermore, we tested the potential application of an FDA-approved drug, FTY720 (fingolimod), to mitigate the synaptosome depolarization in SIV+ animals by activating PP2A. We found that the application of FTY720 was sufficient to mitigate SV depolarization, which was delayed in synaptosomes of SIV+ animals. FTY720 was also shown to inhibit the cytokine production in vitro (Yu et al., 2015). Interestingly, HIV-1 transgenic rats exhibit altered inflammatory pathways and develop pulmonary hypertension (Lund et al., 2011). FTY720 also significantly represses proinflammatory cytokines (Rahman et al., 2016a,b). These findings further support the role of PP2A deficiency in SV release and provide a proof of principle for clinical intervention in PWH with neurological impairment. Together, our findings suggest a significant dysregulation in the dynamics of SV release during depolarization because of altered Syn I (S9) phosphorylation in SIV+ animals conferring key pathology at the synaptic levels.
Footnotes
This work was supported by National Institutes of Health Grant R01NS082116 to T.H.B. and Comprehensive NeuroAIDS Center Pilot Grant P30MH092 I 777 to M.S.
The authors declare no competing financial interests.
- Correspondence should be addressed to Tricia H. Burdo at tug82705{at}temple.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}