

Abstract
Transient receptor potential melastatin 3 (TRPM3) is a nonselective cation channel that is inhibited by Gβγ subunits liberated following activation of Gαi/o protein-coupled receptors. Here, we demonstrate that TRPM3 channels are also inhibited by Gβγ released from Gαs and Gαq. Activation of the Gs-coupled adenosine 2B receptor and the Gq-coupled muscarinic acetylcholine M1 receptor inhibited the activity of TRPM3 heterologously expressed in HEK293 cells. This inhibition was prevented when the Gβγ sink βARK1-ct (C terminus of β-adrenergic receptor kinase-1) was coexpressed with TRPM3. In neurons isolated from mouse dorsal root ganglion (DRG), native TRPM3 channels were inhibited by activating Gs-coupled prostaglandin-EP2 and Gq-coupled bradykinin B2 (BK2) receptors. The Gi/o inhibitor pertussis toxin and inhibitors of PKA and PKC had no effect on EP2- and BK2-mediated inhibition of TRPM3, demonstrating that the receptors did not act through Gαi/o or through the major protein kinases activated downstream of G-protein-coupled receptor (GPCR) activation. When DRG neurons were dialyzed with GRK2i, which sequesters free Gβγ protein, TRPM3 inhibition by EP2 and BK2 was significantly reduced. Intraplantar injections of EP2 or BK2 agonists inhibited both the nocifensive response evoked by TRPM3 agonists, and the heat hypersensitivity produced by Freund's Complete Adjuvant (FCA). Furthermore, FCA-induced heat hypersensitivity was completely reversed by the selective TRPM3 antagonist ononetin in WT mice and did not develop in Trpm3−/− mice. Our results demonstrate that TRPM3 is subject to promiscuous inhibition by Gβγ protein in heterologous expression systems, primary neurons and in vivo, and suggest a critical role for this ion channel in inflammatory heat hypersensitivity.
SIGNIFICANCE STATEMENT The ion channel TRPM3 is widely expressed in the nervous system. Recent studies showed that Gαi/o-coupled GPCRs inhibit TRPM3 through a direct interaction between Gβγ subunits and TRPM3. Since Gβγ proteins can be liberated from other Gα subunits than Gαi/o, we examined whether activation of Gs- and Gq-coupled receptors also influence TRPM3 via Gβγ. Our results demonstrate that activation of Gs- and Gq-coupled GPCRs in recombinant cells and sensory neurons inhibits TRPM3 via Gβγ liberation. We also demonstrated that Gs- and Gq-coupled receptors inhibit TRPM3 in vivo, thereby reducing pain produced by activation of TRPM3, and inflammatory heat hypersensitivity. Our results identify Gβγ inhibition of TRPM3 as an effector mechanism shared by the major Gα subunits.
Introduction
Transient receptor potential melastatin 3 (TRPM3) is a nonselective cation channel that is widely expressed in mammalian tissues. TRPM3 is present in peripheral sensory neurons where it may act as a heat sensor and its activation in vivo evokes nociceptive behaviors in mice (Vriens et al., 2011). TRPM3 can be activated by the endogenous neurosteroid pregnenolone sulfate (PS), which has been used as a pharmacological tool to study the channel (Wagner et al., 2008). In studies of isolated dorsal root ganglion (DRG) neurons, PS application evoked increases in intracellular Ca2+ concentrations ([Ca2+]i) in ∼58% of cells (Vriens et al., 2011), and a high proportion of TRPM3-expressing DRG neurons also expressed TRPV1 (Vriens et al., 2011), identifying these neurons as nociceptors. Trpm3−/− mice exhibit compromised behavioral responses to noxious heat, altered temperature preferences, and fail to develop heat hyperalgesia associated with inflammation (Vriens et al., 2011).
Like most TRP channels, the activity of TRPM3 can be inhibited by phospholipase C-mediated PtdIns(4,5)P2 hydrolysis (Rohacs, 2014). We recently demonstrated, along with two other groups, that activation of Gi/o-coupled GPCRs inhibits TRPM3 both in vitro and in vivo (Badheka et al., 2017; Dembla et al., 2017; Quallo et al., 2017). TRPM3 can also be inhibited by activation of heterologously expressed Gq-coupled GPCRs in recombinant systems (Badheka et al., 2017). This inhibition is independent of Gαi/o and Gαq subunits and is due to the direct interaction of Gβγ subunits with TRPM3 because direct application of Gβγ to the intracellular face of excised inside-out patches inhibits the channel and TRPM3 can be coimmunoprecipitated together with Gβγ (Badheka et al., 2017; Dembla et al., 2017; Quallo et al., 2017). Earlier studies of some voltage-gated calcium channels (VGCCs) and G protein-coupled inwardly-rectifying potassium channels (GIRKs) have shown that these can be promiscuously modulated by Gβγ released from different Gα subunits (for review, see Dascal, 1997; Yamada et al., 1998; Dolphin, 2003). We therefore examined whether activation of Gs-coupled GPCRs can similarly modulate TRPM3 through liberated Gβγ subunits. Additionally, we assessed whether Gq-coupled GPCR activation modulates TRPM3 activity in native sensory neurons and in vivo.
We have used endogenously and heterologously expressed Gs-coupled adenosine 2B (A2B) receptors in HEK293 cells heterologously expressing TRPM3. We also studied the influence of natively expressed Gs-coupled prostaglandin EP2 and Gq-coupled bradykinin B2 receptors on TRPM3 in mouse isolated DRG neurons. Our results demonstrate that activation of Gs- and Gq-coupled GPCRs inhibits TRPM3 through a direct interaction with Gβγ subunits. The inhibition of TRPM3 by Gs- and Gq-coupled GPCRs also operated in DRG neurons and in vivo, where GPCR agonists reduced pain-related behaviors evoked by activation of TRPM3, as well as the heat hyperalgesia associated with inflammation.
Materials and Methods
Mice.
Male and female wild type (WT) C57BL/6J, WT C57BL/6N, and TRPM3-KO (Trpm3−/−; C57BL/6N background) mice were used.The TRPM3-deficient mouse line was generated as described in detail in Extended Data 1. Mice were kept in a climatically controlled environment with ad libitum access to food and water and were acclimatized in the procedure room for 1 h before the experiments. All behavioral experiments were approved by the King's College London Animal Welfare and Ethical Review Board and conducted under the UK Home Office Project License (PF0C9D185).
Cell culture.
DRG neurons were prepared from adult male and female C57BL/6J mice using methods described previously (Bevan and Winter, 1995). Isolated neurons were plated on poly-d-lysine-coated coverslips and maintained at 37°C in an atmosphere of 95% air and 5% CO2 in MEM AQ (Sigma-Aldrich) supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin, and 50 ng/ml NGF (Promega) for up to 24 h before experimentation. Untransfected HEK293 cells (RRID:CVCL_U427, Thermo Fisher Scientific) were grown in DMEM AQ supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml), and FBS (10%). HEK293 cells stably expressing TRPM3α2 plasmid (pcDNA3.1) DNA were grown in DMEM AQ supplemented with penicillin (100 U/ml), streptomycin (100 μg/ml), FBS (10%), and hygromycin (200 μg/ml). For some experiments, TRPM3 HEK293 cells were transiently transfected with plasmids encoding pEYFP-N1-A2BR (a gift from Robert Tarran, Addgene plasmid #37202), pRK5 BARK1 minigene (a gift from Robert Lefkowitz, Addgene plasmid #14695; http://n2t.net/addgene:14695; RRID:Addgene_14695) and muscarinic M1 receptor (a gift from David Julius), using Lipofectamine 2000 according to the supplier's protocol. All cells used were mycoplasma free.
Imaging intracellular calcium concentrations.
DRG neurons and TRPM3 HEK293 cells were loaded with 2.5 μm fura-2 AM (Invitrogen) in the presence of 1 mm probenecid (Tocris Bioscience) for 1–1.5 h. Dye loading and all experiments were performed in a physiological saline solution containing the following (in mm): 140 NaCl, 5 KCl, 10 glucose, 10 HEPES, 2 CaCl2, and 1 MgCl2, buffered to pH 7.4 (NaOH). Drug solutions were applied to cells by local microsuperfusion of solution through a fine tube placed very close to the cells being studied. The temperature of the superfused solution (25°C) was regulated by a temperature controller (Marlow Industries) attached to a Peltier device with the temperature measured at the outlet orifice of the microperfusion tube. Images of a group of cells were captured every 2 s at 340 and 380 nm excitation wavelengths with emission measured at 520 nm with a microscope-based imaging system (PTI). Analyses of emission intensity ratios at 340 nm/380 nm excitation were performed with the ImageMaster suite of software.
Multiwell readings of calcium and cAMP levels.
To monitor intracellular cAMP levels, we used the pGLOSensor-22F plasmid, which is a biosensor for cAMP that is able to respond rapidly and reversibly to changes in the intracellular concentration of cAMP (Binkowski et al., 2009). Untransfected and pGLOSensor-22F HEK293 cells were plated in poly-d-lysine-coated 96-well black-walled plates (Corning Costar) 1–2 d before experimentation. Cells were loaded with fura-2 using the protocol described above. When measuring changes in intracellular cAMP levels, pGLOSensor-22F-expressing HEK293 cells were loaded with GloSensor (Promega) diluted in physiological saline solution for 45 min before experimentation. In both assays, wells were injected with compounds and responses read using the FlexStation 3 Multi-Mode Microplate Reader (Molecular Devices) using appropriate excitation and emission wavelengths (340/380 excitation, 520 emission for fura-2 and luminescence for Glosensor).
Electrophysiology.
DRG neurons and TRPM3-expressing HEK293 cells were studied under voltage-clamp conditions using an Axopatch 200B amplifier and pClamp 10.0 software (RRID:SCR_011323, Molecular Devices). Recordings were performed at +60 mV using borosilicate electrodes (2.5–5 mΩ) filled with a solution containing the following (in mm): 140 CsCl, 10 EGTA, 2 MgATP, and 2 Na2ATP, buffered to pH 7.4 (CsOH). In some experiments, 500 μm GDPβS or 300 μm GTPγS was also included in the intracellular solution. The extracellular solution was the same used for the [Ca2+]i measurements (see above).
Behavioral assessment of pain responses.
Male C57BL/6J mice (6–10 per group) were administered by intraplantar injection with a combination of PS (5 nmol) and CIM0216 (0.5 nmol in 25 μl in PBS) or capsaicin (5 nmol in 25 μl in PBS) into one of the hindpaws using Luer syringe (Hamilton) fitted with a 26-guage × 3/8 inch needle. Bradykinin was injected 5 min and PGE2 and butaprost (0.3 nmol each in 20 μl, intraplantar, each) were injected 10 min before PS/CIM0216 or capsaicin. Mice were habituated to the experimental Perspex chambers before the experiment and placed in the chambers immediately after injection of the compounds. The duration of pain-related behaviors (licking, biting, flinching, shaking, and elevating the injected paw) were recorded using a digital stopwatch. Total pain response times over the first 2 min were used for analysis as the pain behaviors were largely restricted to this period.
Male and female WT C57BL/6J, C57BL/6N, and Trpm3−/− C57BL/6N mice (6–8 per group) were injected with Freund's Complete Adjuvant (FCA; 15 μl) in one of the hindpaws. Paw withdrawal latencies from the hotplate were assessed before and 72 h after FCA injection. Thermal nociception was examined by lightly restraining the animal and placing one of the hindpaws onto a hotplate maintained at 50°C (Andersson et al., 2011). The paw withdrawal latency was measured using 30 s as a cutoff to avoid tissue damage. To assess the effects of ononetin, PGE2, or bradykinin, paw withdrawal latencies in FCA-injected mice were also measured 1 h after administration of ononetin (10 mg/kg, i.p.), and 10 min and 5 min after intraplantar administration of PGE2 and bradykinin.
Materials.
Stock solutions of PS, capsaicin, forskolin (Sigma-Aldrich), bisindolylmaleimide VIII, butaprost (Cayman Chemical), BAY 60–6583, CIM0216, PSB 603, KT 5720 (Tocris Bioscience), and SSR240612 (Sanofi Aventis) were prepared in DMSO. Stock solutions of GRK2i, carbachol, 2-furoyl-LIGRLO-amide, adenosine, bradykinin, HOE 140 (Tocris Bioscience), and morphine sulfate (Sigma-Aldrich) were made in H2O, and PGE2 (Sigma-Aldrich) and CAY10598 (Cayman Chemical) in ethanol. Pertussis toxin (PTX, 0.2 mg/ml; Sigma-Aldrich) was diluted in cell culture AQMEM media. GTPγS and GDPβS (Sigma-Aldrich) were added to the intracellular solution. FCA was obtained from Sigma-Aldrich.
Experimental design and statistical analyses.
Data are presented as box plots showing the mean (square symbol), median (horizontal line), and SEM (box). For imaging and electrophysiology experiments, n indicates the number of PS responding neurons or cells; for multiwell experiments, n indicates the number of independent experiments performed (each in triplicate wells); and in behavioral experiments, n values indicate the number of animals in each group. A priori power calculations were not performed, but our sample sizes are similar to, or greater than, those generally used in the field. Normality of data was tested using the Shapiro–Wilk Test. Normally distributed data were analyzed using an independent-samples t test. Differences in normally distributed data means between three groups or more were analyzed using two-way and repeated-measures ANOVA, followed by Tukey's or Dunnett's multiple-comparisons post hoc test. Differences in non-normally distributed data means between two groups were analyzed using a Mann–Whitney U test. Differences in non-normally distributed data means between three groups or more were analyzed using a Kruskal–Wallis test, followed by a Dunn's multiple-comparisons post hoc test. All statistical analyses were made using Prism version 7 (GraphPad).
Relative amplitudes in [Ca2+]i imaging experiments were calculated by comparing the minimum response amplitude during the coapplication of PS and GPCR agonists (3–6 min time point) to the maximum amplitude during PS application (0–3 min time point). All PS-responsive cells were included in the analysis.
The effect of GPCR agonist application on PS-evoked currents in patch-clamp electrophysiology were calculated in a similar manner and represented as the percentage inhibition of the maximal PS response.
Results
Non–Gi/o-mediated inhibition of TRPM3
Because Gi/o-coupled GPCRs inhibit TRPM3 by an interaction with Gβγ (Badheka et al., 2017; Dembla et al., 2017; Quallo et al., 2017), we examined whether Gβγ subunits liberated by other Gα proteins can exert a similar influence on TRPM3. To assess the effects of the irreversible Gα activator GTPγS and inhibitor GDPβS, we studied HEK293 cells stably expressing TRPM3 by voltage clamp. In the absence of either GTP analog, currents evoked by repeated application of PS (100 μm) showed a time-dependent, but modest, decline in amplitude (Fig. 1A,D). Inclusion of GTPγS (300 μm) in the pipette solution significantly accelerated the desensitization of TRPM3 after ∼3 min (third PS application) (Fig. 1B,D). In contrast, dialyzing the cell with GDPβS (500 μm) led to a progressive and significant increase in the PS-evoked current amplitude, which reached a maximum after ∼4 min (fourth PS application) (Fig. 1C,D). To test whether the effects of GTPγS were partly mediated by non-Gi/o signaling, we incubated the cells with PTX (200 ng/ml, for 24 h), which specifically locks the αi/o subunits into an inactive, GDP-bound state and inhibits Gαi/o subunits coupling to their cognate GPCRs. GTPγS was still able to inhibit PS-evoked currents in PTX-treated cells, albeit more slowly than in the absence of PTX (10th PS application) (Fig. 1E–G), which is consistent with a mechanism independent of Gi/o signaling.

Non–Gi/o-mediated inhibition of TRPM3. A–C, Example traces of multiple applications of PS (100 μm) in whole-cell voltage-clamp recordings (+60 mV) of TRPM3 HEK293 cells with control, GTPγS (300 μm), or GDPβS (500 μm) intracellular solutions. D, Bar chart of the average responses ± SEM of (A) (n = 7), (B) (n = 6), and (C) (n = 8) (F(2,18) = 11.8, p = 0.0005). E, F, Example traces of multiple applications of PS (100 μm) in whole-cell voltage-clamp recordings (60 mV) of TRPM3 HEK293 cells treated with PTX (200 ng/ml) with control or GTPγS (300 μm) intracellular solutions. G, Bar chart of the average responses ± SEM of (E) (n = 7 or 8) and (F) (n = 9 or 10) (t(14) = 3.62, p = 0.028). *p < 0.05; **p < 0.01; compared with control, repeated-measures two-way ANOVA with Dunnett's multiple-comparisons test (D) and multiple t test (G).
Gs-mediated inhibition of TRPM3
Because our experiments with GTPγS demonstrate that G-proteins other than Gi/o can inhibit TRPM3 currents, we examined the effects of agonists of GPCRs coupled to Gαs on TRPM3 activity. The adenosine A2B receptor is expressed at high levels in HEK293 cells (Cooper et al., 1997) and couples to Gαs, thereby generating substantially increased intracellular cAMP upon stimulation. To confirm that the receptor is expressed in our cell line, we used HEK293 cells stably expressing the pGLOSensor (Binkowski et al., 2009) to monitor the effect of A2B agonists on cAMP levels. Application of the endogenous full agonist adenosine or the selective partial A2B agonist BAY 60–6583 (Hinz et al., 2014) produced robust intracellular cAMP responses (Fig. 2A). The response profile and pEC50 values of adenosine (4.75 ± 0.05 SEM, n = 4) and BAY 60–6583 (6.04 ± 0.18 SEM, n = 4) agree with previous studies (Goulding et al., 2018). To confirm that both agonists produced cAMP by activating the A2B receptor, we generated inhibitory concentration response curves using the selective A2B antagonist PSB603 and submaximally effective (EC80) concentrations of adenosine (50 μm) and BAY 60–6583 (5 μm). PSB603 inhibited the responses with a similar pIC50 for both adenosine (7.18 ± 0.15 SEM, n = 4) and BAY 60–6583 (6.71 ± 0.3 SEM, n = 4) and almost completely inhibited intracellular cAMP responses at the highest concentrations tested (Fig. 2B). To assess whether A2B can also couple to Gαq, we applied adenosine and used fura-2 to measure [Ca2+]i (Fig. 2C). As a positive control for Gq-mediated [Ca2+]i increases, we used the selective PAR2 agonist, 2-furyl LIGRLO-NH2 (2F-LIGRLO), as the receptor has been shown to couple to Gαq and produce calcium responses in HEK293 cells (Kawabata et al., 1999). Stimulation of A2B evoked substantial intracellular cAMP production, but no detectable increase in [Ca2+]i (Fig. 2A,C), and we therefore used activation of the A2B receptor to explore the influence of Gs-mediated activity on TRPM3.

Gs-coupled adenosine 2B receptor activation inhibits TRPM3-mediated responses in HEK293 cells. A, Concentration response curves of intracellular cAMP production in response to adenosine or BAY 60–6583 application mean ± SEM in pGLOSensor HEK293 cells. B, Inhibitory concentration response curves using the selective A2B antagonist, PSB603, on intracellular cAMP production evoked by adenosine (50 μm) or BAY 60–6583 (5 μm) mean ± SEM. C, Concentration response curve of [Ca2+]i responses in HEK293 cells following application of 2F-LIGRLO or adenosine mean ± SEM (n = 4, both). D, [Ca2+]i responses in TRPM3 HEK293 cells with PS application (20 μm). E, F, [Ca2+]i responses in TRPM3 HEK293 cells treated with PS (20 μm) and adenosine (100 μm) or BAY 60–6583 (20 μm). G, Scatter plot representing the mean, median, and SEM of the relative amplitude from (D) (n = 731), (E) (n = 588), and (F) (n = 1145). The minimum response amplitude during coapplication of PS and A2B agonists was compared with the maximum amplitude during PS application. H, I, Example traces of adenosine (100 μm) and BAY 60–6583 (20 μm) mediated inhibition of PS (100 μm) evoked currents in whole-cell voltage-clamp recordings (+60 mV) in TRPM3 HEK293 cells. J, Scatter plot representing TRPM3-evoked current inhibition in (H) (n = 12) and (I) (n = 9) (t(19) = 0.17, p = 0.867, two-tailed unpaired t-test). K, L, Example traces of adenosine (100 μm) and BAY 60–6583 (20 μm) mediated inhibition of PS (100 μm) evoked currents in whole-cell voltage-clamp recordings (60 mV) in TRPM3 and A2B transfected HEK293 cells. M, Scatter plot representing TRPM3-evoked current inhibition in K and L (n = 4, both) (t(6) = 0.902, p = 0.402, two-tailed unpaired t-test). ***p < 0.001 compared with control; ###p < 0.001 compared with adenosine treatment; Kruskal–Wallis with Dunn's multiple-comparisons test.
The TRPM3 agonist PS (20 μm) evoked [Ca2+]i increases in TRPM3-expressing HEK293 cells, which were only subject to a minor degree of desensitization during the 9 min application period (22.4 ± 0.81% of reduction; Fig. 2D,G). A2B receptor stimulation with adenosine (100 μm) or BAY 60–6583 (20 μm) significantly and reversibly reduced the amplitudes of the TRPM3-mediated responses by 65.8% ± 1.1% and 35.9% ± 0.5% (Fig. 2E–G). The smaller degree of inhibition produced by BAY 60–6583 is consistent with its properties as a partial agonist at A2B receptors (Hinz et al., 2014) (Fig. 2A,G). Next, we used patch-clamp recordings to confirm that the observed A2B-mediated inhibition of PS-evoked [Ca2+]i responses was associated with a corresponding inhibition of TRPM3 currents. Adenosine (100 μm) and BAY 60–6583 (20 μm) significantly inhibited PS-evoked (100 μm) currents to a similar extent (by 41.6 ± 6.4% and 43.1 ± 5.8%) (Fig. 2H–J). Since the current inhibition achieved by A2B stimulation was variable, we repeated the experiments using TRPM3 HEK293 cells overexpressing the A2B receptor. Overexpression of A2B generated more uniform and robust inhibition of PS-evoked currents (by 87.5 ± 3.8% for adenosine and 80.6 ± 6.7% for BAY 60–6583) (Fig. 2K–M). Together, these observations demonstrate that Gs-coupled GPCR activation can inhibit TRPM3.
Gs-mediated inhibition of TRPM3 in sensory neurons
We next measured [Ca2+]i responses in isolated mouse DRG neurons to confirm that the Gs-mediated inhibition of TRPM3 operates in native cells. PS (20 μm) was applied to evoke a response and agonists of different GPCRs were coadministered as described for HEK293 cell experiments above. Continuous application of PS evoked a sustained increase in [Ca2+]i in DRG neurons (Fig. 3A). In control experiments, the amplitude of PS-evoked responses after 9 min of application was reduced by 17.8 ± 1.2% of the maximal response amplitude, consistent with a small degree of desensitization (Fig. 3C). Applications of the inflammatory mediator PGE2 (1 μm), which has been widely used to sensitize nociceptive afferents (Meves, 2006), strongly inhibited PS-induced [Ca2+]i responses in a subpopulation of DRG neurons (>50% inhibition in 97 of 187 cells; Fig. 3B). Treatment with PGE2 significantly reduced the amplitude of PS responses measured in all neurons by 48.4 ± 2.6% (Fig. 3C). Scatter plots of response amplitudes (Fig. 3C,I) revealed a large inhibition in some neurons and little or no inhibition in other neurons. The effect of PGE2 was unaffected by the overnight incubation of neurons with PTX (200 ng/ml, 18 h) (Fig. 3C). Conversely, PTX (200 ng/ml, 18 h) treatment fully reversed morphine-mediated Gi/o inhibition of PS-evoked responses in DRG neurons (Fig. 3D–F) (Badheka et al., 2017; Dembla et al., 2017; Quallo et al., 2017). These findings demonstrate that PGE2 inhibits PS responses independently of Gi/o.

Prostaglandin EP2 receptor activation inhibits TRPM3-mediated responses in sensory neurons. A, [Ca2+]i responses in isolated mouse DRG neurons challenged by PS (20 μm) followed by KCl (50 mm) application. B, [Ca2+]i responses in mouse isolated DRG neurons treated with PS (20 μm) and PGE2 (1 μm). C, Scatter plots representing the relative response amplitudes for control (n = 207), 1 μm PGE2 (n = 175), and PGE2 + PTX (200 ng/ml; n = 151) treated DRG neurons. Relative responses represent the minimum amplitudes recorded 3–6 min during PS application (corresponding to the time of drug application) expressed as a percentage of the maximum amplitude recorded in the 0–3 min period. Some reduction in response amplitude was typically seen in control conditions. D, [Ca2+]i responses in isolated DRG neurons treated with PS (20 μm) and morphine (10 μm). E, [Ca2+]i responses in PTX-treated (200 ng/ml) isolated DRG neurons challenged with PS (20 μm) and morphine (10 μm). F, Scatter plot representing the relative amplitudes of control (94.3 ± 1.6% SEM, n = 164), (D) (48.4 ± 2.7% SEM, n = 109), and (E) (86 ± 2.3% SEM, n = 135) neurons. G, [Ca2+]i responses in isolated mouse DRG neurons treated with PS (20 μm) and butaprost (1 μm). H, Scatter plot representing the relative amplitude in control (n = 212), butaprost (n = 250), and butaprost + PTX (n = 140) treated DRG neurons. I, Scatter plot representing the relative amplitude in control (n = 409), PGE2 (n = 185), butaprost (n = 294), and CAY10595 (30 nm; n = 207) treated DRG neurons. J, Ononetin (10 μm) mediated inhibition of PS (50 μm) evoked currents in whole-cell voltage-clamp recordings (60 mV) in isolated DRG neurons (n = 6). K, L, Example traces of PGE2 and butaprost (both 1 μm) mediated inhibition of PS (50 μm) evoked currents in whole-cell voltage-clamp recordings (60 mV) in isolated DRG neurons. M, Scatter plot representing PGE2 and butaprost current inhibition of TRPM3-evoked currents inhibition in (K) (n = 6) and (L) (n = 7) (t(11) = 1.11, p = 0.289, two-tailed unpaired t-test). ***p < 0.001 compared with control; ###p < 0.001 compared with morphine-treated cells; Kruskal–Wallis with Dunn's multiple-comparisons test.
PGE2 acts on two cognate Gs-coupled E prostanoid receptors, EP2 and EP4, both of which are found in DRG neurons (Cruz Duarte et al., 2012). To determine the relative contribution of these receptors to the PGE2-mediated inhibition of TRPM3, we used the selective EP2 agonist butaprost and the selective EP4 agonist CAY10598. Butaprost (1 μm) significantly inhibited PS-induced [Ca2+]i responses in a subpopulation of neurons (>50% inhibition in 90 of 294 cells) (Fig. 3G), reducing the overall amplitude of PS-evoked responses by 28.1 ± 1.8% compared with control experiments (12.8 ± 1.4%) (Fig. 3H). Butaprost (1 μm) was still able to inhibit PS-evoked responses in neurons treated with PTX (200 ng/ml, 18 h) (Fig. 3H). In contrast to the effect of the EP2 receptor activator, the selective EP4 receptor agonist CAY10598 (30 nm) had no effect on PS responses in DRG neurons compared with untreated controls (Fig. 3I).
TRPM3 is widely expressed in DRG neurons and mediates PS-evoked [Ca2+]i responses (Vriens et al., 2011). Here we found that 64.2% (52 of 81) of DRG neurons studied by voltage-clamp responded to stimulation with PS, which agrees well with previously reported results (Vriens et al., 2011). To confirm that PS-evoked currents in DRG neurons were mediated by TRPM3, the selective TRPM3 antagonist ononetin (10 μm) (Straub et al., 2013) was applied during PS application. Ononetin inhibited PS evoked currents completely (by 98 ± 2%) and reversibly in all treated cells (Fig. 3J). We therefore conclude that TRPM3 mediates PS-evoked current responses in DRG neurons.
Voltage-clamp recordings confirmed that PGE2 and butaprost inhibited PS-evoked TRPM3 currents in isolated DRG neurons. Application of PGE2 (1 μm) or butaprost (1 μm) reversibly inhibited PS-evoked TRPM3 currents to a similar extent (by 65.7 ± 10.6% and 50.5 ± 8.9%) (Fig. 3K–M). These results demonstrate that activation of Gs-coupled EP2 receptor inhibits TRPM3 in DRG neurons.
Gq-mediated inhibition of TRPM3 in sensory neurons
It has previously been shown that stimulation of Gq-coupled muscarinic acetylcholine M1 and BK2 receptors leads to the inhibition of TRPM3-evoked currents in recombinant cells (Badheka et al., 2017). Consistent with these results, we found that activation of muscarinic M1 receptors inhibited TRPM3-mediated [Ca2+]i responses in HEK293 cells (Fig. 4A,B). As M1 receptor activation can increase [Ca2+]i, we therefore used a low concentration of carbachol (0.1 μm) that did not evoke a [Ca2+]i response in control experiments (Fig. 4C).

Gq-coupled muscarinic M1 receptor activation inhibits TRPM3-mediated responses in HEK293 cells. A, [Ca2+]i responses in TRPM3/muscarinic M1 HEK293 cells treated with PS (20 μm) and carbachol (0.1 μm) followed by application of a high concentration of carbachol (1 μm) to confirm M1 transfection. B, Scatter plot representing the mean, median, and SEM of the relative amplitude from control cells (60 ± 0.9% SEM, n = 431) and (A) (51.7 ± 1.0% SEM, n = 329), (Mann–Whitney U = 52821, p < 10−4). ***p < 0.001. C, [Ca2+]i responses in TRPM3/muscarinic M1 HEK293 cells with application of different carbachol concentrations.
We next examined whether Gq-coupled GPCRs can inhibit the activity of TRPM3 in its native environment by stimulating isolated DRG neurons with bradykinin, a pronociceptive mediator often used to sensitize nociceptors (Petho and Reeh, 2012). Application of bradykinin (100 nm) strongly, and rapidly inhibited PS-induced [Ca2+]i responses in a subpopulation of DRG neurons (>50% inhibition in 86 of 293 cells) (Fig. 5A). Bradykinin treatment significantly reduced the amplitude of PS responses by 34.4 ± 1.4% compared with control experiments (22 ± 1.8%; Fig. 5B). The effect of bradykinin was unaltered by PTX pretreatment (200 ng/ml, 18 h; Fig. 5C). Furthermore, pretreatment with the selective BK2 receptor antagonist HOE 140 (50 nm) completely prevented the effect of bradykinin (control, reduced by 30.9 ± 1.6% from the maximal amplitude; bradykinin: 41.5 ± 1.7%; bradykinin + HOE 140: 31.3 ± 1.6%) (Fig. 5D). In contrast, the BK1 receptor antagonist SSR240612 (50 nm) was without effect on bradykinin-mediated inhibition of TRPM3 (control: reduced by 17.9 ± 1.7%; bradykinin: 33.0 ± 2.6%; bradykinin + SSR240612: 31.6 ± 1.8%; Fig. 5E).

Bradykinin BK2 receptor activation inhibits TRPM3-mediated responses in sensory neurons. A, [Ca2+]i responses in isolated mouse DRG neurons treated with PS (20 μm) and bradykinin (100 nm). B, Scatter plot representing the relative amplitudes in control (n = 293) and bradykinin (n = 293) treated DRG neurons (Mann–Whitney U = 29892, p < 10−4). ***p < 0.001. C, Scatter plot representing the relative amplitudes in control (n = 190), bradykinin (n = 157), and bradykinin + PTX (200 ng/ml; n = 165) treated DRG neurons. D, Scatter plot representing the relative amplitudes in control (n = 157), bradykinin (n = 197), and bradykinin + HOE 140 (50 nm; n = 182) treated DRG neurons. E, Scatter plot representing the relative amplitudes in control (n = 118), bradykinin (n = 106), and bradykinin + SSR240612 (50 nm; n = 144)-treated DRG neurons. ***p < 0.001 compared with control; ###p < 0.001 compared with bradykinin-treated cells, Kruskal–Wallis with Dunn's multiple-comparisons test C–E. F, Example trace of bradykinin (100 nm) mediated inhibition of PS (50 μm) evoked currents in whole-cell voltage-clamp recordings (60 mV) in isolated DRG neurons.
We also examined the effects of bradykinin on DRG neurons electrophysiologically. Application of bradykinin (100 nm) to DRG neurons in the voltage-clamp configuration reversibly inhibited PS-evoked TRPM3 currents (by 53.7 ± 10.8% SEM, n = 5; Fig. 5F). Together, these results show that bradykinin inhibits TRPM3 in DRG neurons by activating Gq-coupled BK2 receptors.
Gs- and Gq-mediated inhibition of TRPM3 is independent of PKA and PKC activity
Canonical Gs- and Gq-coupled receptor signaling leads to the downstream activation of PKA and PKC. This has been previously shown to increase nociceptor excitability, by sensitizing voltage-gated sodium channels and T-type VGCCs (England et al., 1996; Gold et al., 1998; Kim et al., 2006; Chemin et al., 2007) and the noxious heat transduction channel TRPV1 (Bevan et al., 2014). We used the selective PKA inhibitor KT5720 to determine whether PKA contributed to the EP2-mediated inhibition of TRPM3. The butaprost-induced inhibition of PS-evoked [Ca2+]i responses in DRG neurons was unaffected by KT5720 (1 μm) (control: 19.3 ± 1.8%; butaprost: 29.2 ± 2.6%; butaprost + KT5720: 33.3 ± 2%; Fig. 6A,B). Conversely, we used the adenylyl cyclase activator forskolin to raise intracellular cAMP and activate PKA. Forskolin (10 μm) was applied 6 min before and during PS and butaprost application. Forskolin treatment did not alter PS-induced [Ca2+]i responses in neurons and had no effect on butaprost-mediated inhibition of PS responses (control: 16.3 ± 1.40%; butaprost: 36.2 ± 2.15%; forskolin: 13.8 ± 2.2%; butaprost + forskolin: 35.8 ± 2.2%; Fig. 6C,D).

Gs- and Gq-coupled mediated inhibition of TRPM3 is independent of PKA and PKC activity. A, [Ca2+]i responses in isolated mouse DRG neurons treated with PS (20 μm), butaprost (1 μm), and KT5720 (1 μm). B, Scatter plot representing the relative amplitudes in control (n = 112), butaprost-treated (n = 151), and butaprost + KT5720-treated (n = 151) cells. C, [Ca2+]i responses in isolated mouse DRG neurons treated with PS (20 μm), butaprost (1 μm), and forskolin (10 μm). D, Scatter plot representing the relative amplitudes in control (n = 185), butaprost-treated (n = 183), forskolin-treated (n = 148), and butaprost + forskolin-treated (n = 155) cells. E, [Ca2+]i responses in isolated mouse DRG neurons treated with PS (20 μm), bradykinin (100 nm), and BIM VIII (1 μm). F, Scatter plot representing the relative amplitudes in control (n = 236), bradykinin-treated (n = 257), and bradykinin + BIM VIII-treated (n = 248) cells. **p = 0.0014; ***p < 0.001 compared with control; ###p < 0.001 compared with bradykinin-treated cells; Kruskal–Wallis with Dunn's multiple-comparisons test.
We examined whether PKC may be responsible for the inhibition of TRPM3 by BK2 receptor activation by applying the selective PKC inhibitor BIM VIII (1 μm) before and during bradykinin application. BIM VIII treatment did not prevent bradykinin-mediated inhibition of PS-induced [Ca2+]i responses in neurons (control: 21.9 ± 1.2%; bradykinin: 40.9 ± 1.5%; bradykinin + BIM VIII: 55.9 ± 1.4%; Fig. 6E,F). Together, these observations strongly suggest that Gs- and Gq-coupled GPCRs inhibit TRPM3 independently of cAMP, PKA, and PKC.
Gβγ subunits mediate inhibition of TRPM3 in heterologous and endogenous systems
Because the major canonical signaling events activated by Gαs and Gαq did not affect TRPM3 inhibition, we next examined the potential involvement of Gβγ protein. TRPM3 HEK293 cells were transiently transfected with the Gβγ sink βARK1-ct (C terminus of the β-adrenergic receptor kinase-1) (Koch et al., 1994) and the effects of A2B activation on TRPM3 examined using [Ca2+]i measurements. βARK1-ct transfection abolished the inhibitory effect of adenosine (100 μm) and BAY 60–6583 (20 μm) application on TRPM3-mediated responses compared with untransfected cells (control: 83.7 ± 1.40%; adenosine: 34.2 ± 1.1%; adenosine + βARK1-ct: 75.3 ± 0.8%; BAY 60–6583: 64.1 ± 0.5%; and BAY 60–6583 + βARK1-ct: 80.5 ± 0.6%). Similarly, TRPM3 inhibition by muscarinic M1 receptor activation was completely prevented by transfection with βARK1-ct (control: 59.9 ± 0.86%; carbachol: 51.7 ± 0.95%; and carbachol + βARK1-ct: 60.3 ± 0.84%; Fig. 7A–F). To confirm the involvement of G beta-gamma, we also used GRK2i, a smaller 28 amino acid polypeptide that corresponds to the Gβγ binding region of βARK1 (β-adrenoreceptor kinase 1 also known as GRK2, G-protein receptor kinase 2). GRK2i (10 μm) dialysis of TRPM3 HEK293 cells in the whole-cell recording configuration for >5 min significantly reduced adenosine-mediated inhibition of TRPM3-evoked currents (control: 67.4 ± 6.4%; GRK2i: 36.6 ± 6.3%). We next examined the involvement of Gβγ in EP2- and BK2-mediated inhibition of TRPM3 by dialyzing GRK2i in isolated DRG neurons. GRK2i (10 μm) dialysis significantly reduced the butaprost- and bradykinin-mediated inhibition of TRPM3-evoked currents (butaprost: 50.5 ± 8.9%; GRK2i and butaprost: 17.6 ± 1.9%; bradykinin: 53.7 ± 10.8%; and GRK2i and bradykinin: 26.2 ± 3.2%; Fig. 7G–L). These results demonstrate that Gβγ subunits are responsible for TRPM3 inhibition produced by activation of GPCRs coupled to Gαs and Gαq in heterologous and native cell systems.
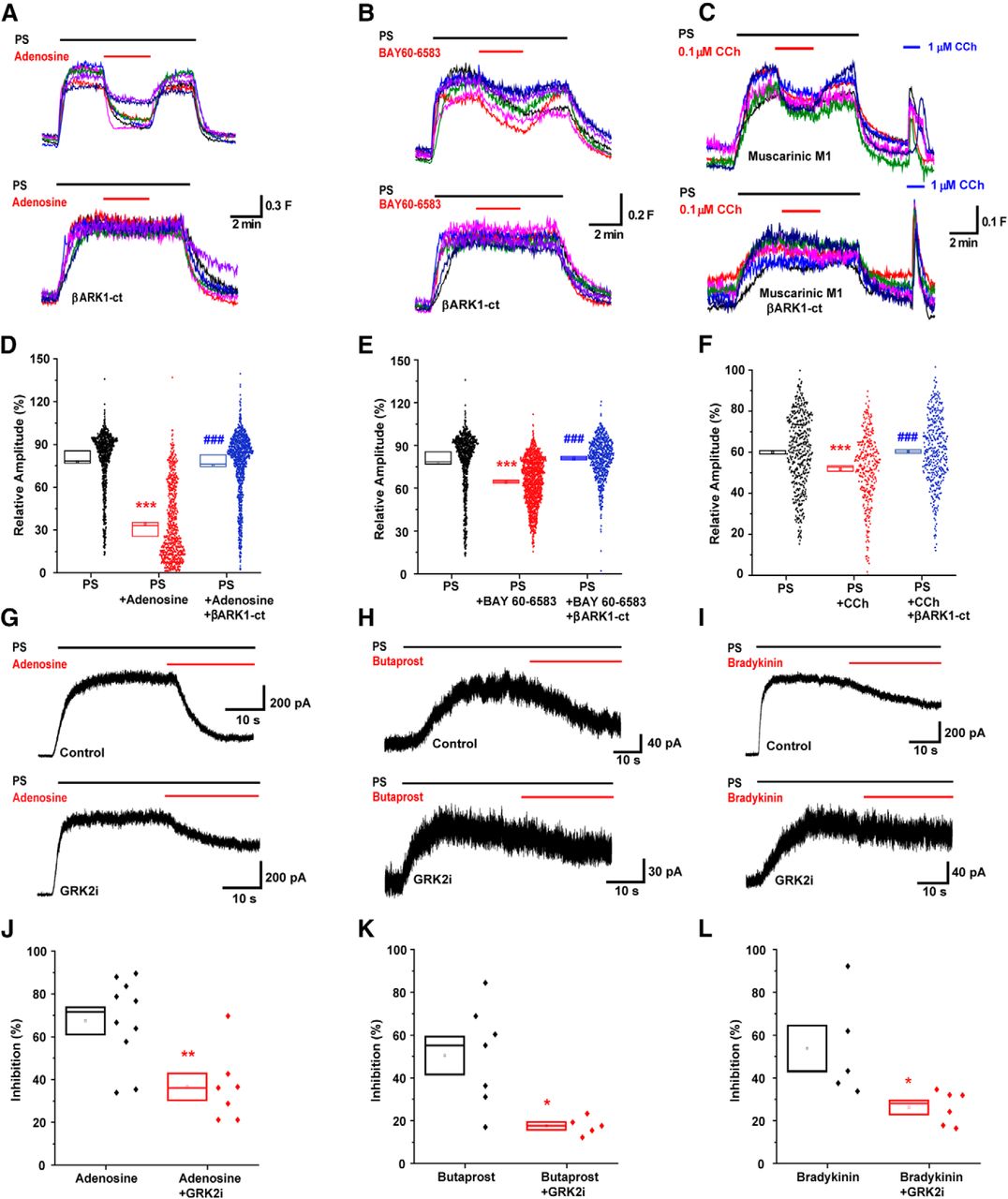
Gs- and Gq-coupled GPCR inhibition of TRPM3 is reliant on Gβγ protein. A–C, [Ca2+]i responses in control (top) and βARK1-ct transfected (bottom) TRPM3 HEK293 cells treated with PS (20 μm) and adenosine (100 μm, n = 852), BAY 60–6583 (20 μm, n = 553), or carbachol (0.1 μm, n = 386). D–F, Scatter plots representing the relative amplitudes of A–C with βARK1-ct untransfected control and treated TRPM3 HEK293 cells. ***p < 0.001 compared with control; ###p < 0.001 compared with treated cells; Kruskal–Wallis with Dunn's multiple-comparisons test. G–I, Example traces of adenosine (100 μm), butaprost (1 μm), and bradykinin (100 nm) mediated inhibition of PS (100 μm for HEK cells and 50 μm for DRG neurons)-evoked currents in whole-cell voltage-clamp recordings (+60 mV) in HEK293 cells (for adenosine) and isolated DRG neurons (for butaprost and bradykinin) in control (top) and GRK2i (10 μm, bottom) intracellular solutions. J–L, Scatter plots representing TRPM3-evoked current inhibition in (G) (t(15) = 3.32, p = 0.0047, control n = 10, GRK2i n = 7, two-tailed unpaired t-test), (H) (t(10) = 3.06, p = 0.012, control n = 7, GRK2i n = 5, two-tailed unpaired t-test), and (L) (t(9) = 2.67, p = 0.026, control: n = 5, GRK2i: n = 6, two-tailed unpaired t-test). *p < 0.05. **p < 0.01.
Gs- and Gq-coupled GPCRs inhibit TRPM3-mediated nociception
As activation of EP2 and BK2 receptors inhibits TRPM3 in isolated DRG neurons, we next examined whether activation of EP2 and BK2 receptors can inhibit pain evoked by TRPM3 agonists in vivo. Previous studies in Trpm3−/− mice showed that the behavioral nocifensive responses to intraplantar injections of PS and a second TRPM3 agonist CIM0216 are dependent on TRPM3 (Held et al., 2015). Intraplantar administration of a combination of 5 nmol PS and 0.5 nmol CIM0216 evoked a robust paw licking/flinching behavior in WT mice. Intraplantar administration of 0.3 nmol of PGE2 (10 min before), butaprost (10 min before), or bradykinin (5 min before) before PS/CIM0216 administration, significantly reduced the pain-related paw licking and flicking behavioral responses evoked by the TRPM3 agonists (Fig. 8A–C). The doses of PGE2, butaprost, and bradykinin were chosen not to directly evoke pain-related behavioral responses.

Gs- and Gq-coupled GPCR activation prevents mouse nociceptive behavior in response to TRPM3 agonists and reverses heat hyperalgesia in FCA-treated mice. A–C, Scatter plots representing the duration of pain responses in mice with intraplantar hindpaw injections of PGE2 (t(18) = 2.86, p = 0.0103, n = 10, two-tailed unpaired t-test), butaprost (Mann–Whitney U = 10.5; p = 0.0258, n = 8, two-tailed), and bradykinin (Mann–Whitney U = 15.5, p = 0.0062, n = 10, two tailed) (all at a dose of 0.3 nmol) or with vehicle before PS (5 nmol)/CIM0216 (0.5 nmol) administration. *p < 0.05; **p < 0.01. D–F, Scatter plots representing the duration of pain responses in mice administered with intraplantar hindpaw injections of PGE2 (t(10) = 1.11, p = 0.29, two-tailed unpaired t-test), butaprost (Man–Whitney U = 16, p = 0.82, two-tailed), and bradykinin (t(10) = 0.69, p = 0.5, two-tailed unpaired t-test) (0.3 nmol, n = 6, all) or with vehicle before capsaicin (5 nmol) administration. G, Bar chart comparing heat withdrawal latencies of mice from 50°C hotplate before and 72 h after intraplantar FCA (15 μl) injection in Trpm3+/+ (t(14) = 5.49, p < 0.0001, two-tailed unpaired t-test) and Trpm3−/− mice (t(14) = 1.31, p = 0.21, two-tailed unpaired t-test) (n = 8, both) ***p < 0.001. H, Bar chart comparing heat withdrawal latencies of mice from 50°C hotplate before and 72 h after injection with FCA (15 μl), vehicle or ononetin (10 mg/kg; F(2,30) = 7.2, p = 0.0028) application (n = 6, both). I, Bar chart comparing heat withdrawal latencies of mice from 50°C hotplate before and 72 h after injection with FCA (15 μl) or vehicle, PGE2 (0.3 nmol; F(2,30) = 4.7, p = 0.0168), or bradykinin (0.3 nmol; F(2,30) = 6.21, p = 0.0055) (n = 6, each). *p < 0.05; **p < 0.01, compared with baseline; #p < 0.05; ##p < 0.01, compared with 72 h after FCA; two-way ANOVA followed by Tukey's multiple-comparisons test.
To test whether this effect is specific for TRPM3-dependent nociception, we examined whether activation of EP2 and BK2 receptors could also inhibit TRPV1-mediated behavioral responses because many TRPM3 expressing neurons also express TRPV1 (Vriens et al., 2011). However, neither PGE2 (vehicle: 68.5 ± 3.7 s; PGE2: 62.7 ± 3.7 s), nor butaprost (vehicle: 77 ± 7.3 s; butaprost: 72 ± 7.9 s), nor bradykinin (vehicle: 57 ± 4.2 s; bradykinin: 65 ± 10.7 s) inhibited capsaicin-evoked behaviors (Fig. 8D–F), in marked contrast to their inhibitory effect on responses elicited by PS/CIM0216.
TRPM3 is required for the development and maintenance of inflammatory heat hyperalgesia, which consequently has been found to be absent from Trpm3−/− mice (Vriens et al., 2011). We compared the heat sensitivity of Trpm3+/+ and Trpm3−/− mice 3 d after intraplantar injections of Freund's complete adjuvant (FCA, 15 μl). FCA reduced the paw withdrawal latency in WT mice in the hotplate test (50°C) but was without effect in Trpm3−/− mice, in good agreement with earlier observations (Vriens et al., 2011), indicating that TRPM3 is of critical importance for inflammatory heat hypersensitivity (Fig. 8G). Administration of the selective TRPM3 inhibitor ononetin (10 mg/kg, i.p.) completely reversed established FCA-induced heat hypersensitivity (Fig. 8H), demonstrating that the loss of hypersensitivity in Trpm3−/− mice was unlikely to be caused by developmental or compensatory mechanisms and suggests that TRPM3 may be a tractable target for inflammatory pain.
FCA has long been used as a model for inflammatory hypersensitivity and pain, but the precise mechanisms by which it produces pain and hypersensitivity are not known. To determine the influence of local, intraplantar injections of PGE2 and bradykinin on established FCA-induced heat hyperalgesia, we administered the same doses that inhibited the behavioral response to TRPM3 agonists (Fig. 8A,C) but did not evoke a behavioral response on their own (5 or 10 min before hotplate test). Perhaps counterintuitively, intraplantar administration of either PGE2 or bradykinin fully reversed heat hyperalgesia in FCA-treated mice (Fig. 8I). These observations are consistent with our observations that EP2 and BK2 receptor activation inhibit TRPM3 in vitro and mimic the effects of pharmacological blockade or genetic inactivation of TRPM3 on heat hypersensitivity.
Discussion
Our results demonstrate, for the first time, that TRPM3 is inhibited by activation of GPCRs coupled to any of the three major classes of Gα in heterologous and native cellular environments. Based on our findings with GDPβS-treated cells, we show that TRPM3 is tonically inhibited by G-proteins in HEK293 cells, in good agreement with the tonic inhibition of TRPM3 previously observed in DRG neurons and in vivo (Quallo et al., 2017). Here we show that stimulation of Gs-coupled GPCRs exerts an inhibitory effect on TRPM3 in HEK293 cells and DRG neurons. This inhibition is PTX-insensitive, demonstrating that Gi/o is not responsible. Furthermore, the inhibition is not mediated by the canonical signaling pathways engaged by Gs activation because it is unaffected by PKA inhibition or by stimulation of cAMP production. In contrast, Gs-coupled GPCR inhibition of TRPM3 is fully prevented by expression of βARK1-ct in HEK293 cells, demonstrating that Gβγ protein is responsible for this inhibition.
Previous investigations demonstrated that Gαq does not coimmunoprecipitate with TRPM3 (Dembla et al., 2017) and that inhibition of TRPM3 by heterologous expression of Gq-coupled muscarinic M1 and BK2 receptors is unaffected by PtdIns(4,5)P2 supplementation but is reduced by βARK1-ct (Badheka et al., 2017). Here, we confirmed these findings by showing that TRPM3-mediated [Ca2+]i responses in HEK293 cells is inhibited by muscarinic M1 receptor activation and that this inhibition is prevented by expression of βARK1-ct, and we have further shown that BK2 receptor activation inhibits TRPM3 in vivo and in isolated DRG neurons. This inhibition is mediated by Gβγ and is independent of PKC. Along with our observations that βARK1-ct and GRK2i reversed TRPM3 inhibition by A2B and EP2 activation, these results clearly demonstrate that Gβγ mediates Gi/o-, Gs-, and Gq-induced inhibition of TRPM3.
Gβγ proteins directly modulate N- and P/Q-type VGCCs and GIRK channels independently of Gα and regardless of whether they are liberated from Gαi/o, Gαs, or Gαq (for review, see Dascal, 1997; Yamada et al., 1998; Dolphin, 2003). Our results with GTPγS in control and PTX-treated TRPM3 HEK293 cells show that, although PS-evoked currents were gradually inhibited after PTX treatment, the inhibition developed more quickly in the absence of PTX. A more effective inhibition of TRPM3 by Gi/o-coupled GPCRs is supported by the stronger inhibition observed following activation of μ-opioid, NPY, and GABAB receptors (Quallo et al., 2017) than with Gs- or Gq-coupled receptors. This finding agrees with the dominant modulation of VGCCs and GIRK channels by activating Gi/o-coupled GPCRs (Dolphin, 2003). Promiscuous GPCR activation of GIRK channels by Gβγ has been observed, but primarily in heterologous overexpression systems. In sinoatrial node pacemaker cells, GIRK is preferentially activated by the Gi/o-coupled M2 muscarinic receptor rather than by the Gs-coupled β2-adrenergic receptor (Hein et al., 2006; Digby et al., 2008; Touhara and MacKinnon, 2018). Reports that Gβγ proteins dissociate more readily from Gαo than from Gαs (Digby et al., 2008) lend further support to the notion that Gi/o-coupled Gβγ is more effective at inhibiting TRPM3.
The proinflammatory mediators PGE2 and bradykinin are both thought to produce pain and hypersensitivity at least in part by sensitization of sensory neuron TRP channels, such as TRPA1 and TRPV1, downstream of EP2 and BK2 receptors (for review, see Bautista et al., 2013; Veldhuis et al., 2015). Here, low doses of PGE2 and bradykinin that did not evoke a behavioral response on their own significantly reduced the pain-related behavioral responses evoked by topical injections of a combination of the TRPM3 agonists PS and CIM0216, in a manner similar to that observed with application of agonists of the Gi/o-coupled μ opioid receptor (Badheka et al., 2017; Dembla et al., 2017; Quallo et al., 2017). This effect appears to be specific for TRPM3 because pain-related behaviors produced by the TRPV1 agonist capsaicin were not inhibited by application of PGE2, butaprost, and bradykinin. Thus, activation of EP2 and BK2 receptors is unlikely to inhibit pain produced by TRPM3 agonists by interfering with action potential generation or membrane excitability because many TRPM3 expressing neurons also express TRPV1 (Vriens et al., 2011).
We evaluated the effects of PGE2 and bradykinin on heat hypersensitivity produced by intraplantar FCA. The roles of PGE2 and bradykinin in adjuvant-induced hypersensitivity are not clear and the behavioral effects produced by FCA are, for example, unchanged in mice lacking both BK1 and BK2 bradykinin receptors (Cayla et al., 2012). Intra-articular injections of FCA in the rat produced mechanical hyperalgesia, which was unaffected by inhibition of BK2 receptors 3 d after FCA induction, whereas coadministration of a BK2 antagonist together with FCA prevented the development of hypersensitivity, suggesting a role for BK2 in the development, rather than maintenance of hypersensitivity (Perkins et al., 1993). An evaluation of the efficacy of analgesic drugs in rats treated with intraplantar FCA demonstrated that the nonselective NSAIDs indomethacin and diclofenac, at doses that would completely prevent cyclooxygenase-mediated PGE2 formation, produced no, or only a minor reduction of heat hypersensitivity (Nagakura et al., 2003). Here, we found that FCA failed to produce heat hypersensitivity in Trpm3−/− mice, and that a selective TRPM3 antagonist, ononetin, produced a complete reversal of the behavioral sensitization produced by FCA in WT mice. Surprisingly, local intraplantar injections of either of the proinflammatory mediators PGE2 or bradykinin also produced a complete reversal of the established FCA-induced heat hypersensitivity in WT mice. Together, our behavioral analysis thus confirms that TRPM3 is critically important for inflammatory heat hyperalgesia (see Vriens et al., 2011), and strongly indicates that activation of GPCRs may produce analgesia by inhibiting TRPM3. Somewhat surprisingly, this was the case following local administration of PGE2 and bradykinin. These latter findings are consistent with the absence of high concentrations of PGE2 and bradykinin in the inflamed paw at this post-FCA time point.
Our results demonstrate that TRPM3 is promiscuously inhibited by Gβγ after activation of receptors coupled to any of the major classes of G-proteins and thus may act as a pan-GPCR effector molecule. We show that Gs- and Gq-coupled GPCRs inhibit TRPM3 in cell lines and in isolated sensory neurons in vitro and that they can produce antinociception and analgesia by inhibiting TRPM3 in vivo.
Footnotes
This work was supported in part by Medical Research Council UK Grant MR/L010747/1, Saudi Arabian Custodian of the Two Holy Mosques Scholarship to O.A., Coordenação de Aperfeiçoamento de Pessoal de Nível Superior Brazil (CAPES, finance code 001), the Royal Society UK Newton International Fellowship to R.d.C., the DZHK (German Centre for Cardiovascular Research), the BMBF (German Ministry of Education and Research), the DFG (German Research Foundation) Project-ID 239283807-TRR 152 and the FOR 2289 to M.F. and by the DFG (SFB 894) and Saarland University [HOMFOR] to S.E.P.
The authors declare no competing financial interests.
- Correspondence should be addressed to David A. Andersson at david.andersson{at}kcl.ac.uk or Omar Alkhatib at omar.alkhatib{at}kcl.ac.uk





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}