

Abstract
Transient periods of childhood hearing loss can induce deficits in aural communication that persist long after auditory thresholds have returned to normal, reflecting long-lasting impairments to the auditory CNS. Here, we asked whether these behavioral deficits could be reversed by treating one of the central impairments: reduction of inhibitory strength. Male and female gerbils received bilateral earplugs to induce a mild, reversible hearing loss during the critical period of auditory cortex development. After earplug removal and the return of normal auditory thresholds, we trained and tested animals on an amplitude modulation detection task. Transient developmental hearing loss induced both learning and perceptual deficits, which were entirely corrected by treatment with a selective GABA reuptake inhibitor (SGRI). To explore the mechanistic basis for these behavioral findings, we recorded the amplitudes of GABAA and GABAB receptor-mediated IPSPs in auditory cortical and thalamic brain slices. In hearing loss-reared animals, cortical IPSP amplitudes were significantly reduced within a few days of hearing loss onset, and this reduction persisted into adulthood. SGRI treatment during the critical period prevented the hearing loss-induced reduction of IPSP amplitudes; but when administered after the critical period, it only restored GABAB receptor-mediated IPSP amplitudes. These effects were driven, in part, by the ability of SGRI to upregulate α1 subunit-dependent GABAA responses. Similarly, SGRI prevented the hearing loss-induced reduction of GABAA and GABAB IPSPs in the ventral nucleus of the medial geniculate body. Thus, by maintaining, or subsequently rescuing, GABAergic transmission in the central auditory thalamocortical pathway, some perceptual and cognitive deficits induced by developmental hearing loss can be prevented.
SIGNIFICANCE STATEMENT Even a temporary period of childhood hearing loss can induce communication deficits that persist long after auditory thresholds return to normal. These deficits may arise from long-lasting central impairments, including the loss of synaptic inhibition. Here, we asked whether hearing loss-induced behavioral deficits could be reversed by reinstating normal inhibitory strength. Gerbils reared with transient hearing loss displayed both learning and perceptual deficits. However, when animals were treated with a selective GABA reuptake inhibitor during or after hearing loss, behavioral deficits were entirely corrected. This behavioral recovery was correlated with the return of normal thalamic and cortical inhibitory function. Thus, some perceptual and cognitive deficits induced by developmental hearing loss were prevented with a treatment that rescues a central synaptic property.
Introduction
Developmental hearing loss (HL) is the most prevalent childhood sensory impairment, posing a risk for deficits in both perceptual and cognitive skills, including delayed language acquisition (Svirsky et al., 2004; Nicholas and Geers, 2006; Moeller et al., 2007; Niparko et al., 2010; Tobey et al., 2013; Tomblin et al., 2014; Kishon-Rabin et al., 2015; Davidson et al., 2019). Indeed, auditory behavioral deficits can persist even after auditory thresholds return to normal following a period of transient HL, such as that caused by middle ear infections (Pillsbury et al., 1991; Hall and Grose, 1994; Hall et al., 1995, 1998; Hogan et al., 1996; Asbjørnsen et al., 2000, 2005; Hogan and Moore, 2003; Whitton and Polley, 2011; Sanes, 2016). In contrast, brief periods of mild HL in adults lead to a change in loudness perception that resolves within ∼24 h (Formby et al., 2003; Munro and Blount, 2009; Munro et al., 2014). One hypothesis that explains the persistence of these behavioral deficits when occurring during childhood is that HL during a developmental critical period induces persistent changes to inhibitory synapse function that degrade central auditory processing (Sanes, 2013). Indeed, transient childhood HL is associated with altered central auditory physiology, both in the brainstem and cortex (Folsom et al., 1983; Gunnarson and Finitzo, 1991; Hall and Grose, 1993; Haapala et al., 2014, 2016). Here, we ask whether developmental HL-induced auditory behavioral deficits can be rescued by maintaining or restoring normal inhibitory synaptic function.
A broad range of neurodevelopmental disorders, including HL, are associated with a decline in the strength of synaptic inhibition (Turrigiano and Nelson, 2004; Chao et al., 2010; Richardson et al., 2012; Braat and Kooy, 2015). For example, synapses between interneurons and pyramidal cells are weakened in the visual cortex following monocular deprivation (Maffei et al., 2004), the auditory cortex (ACx) of animals raised with HL (Takesian et al., 2012; Mowery et al., 2015), and the somatosensory cortex of animals subjected to whisker trimming (Jiao et al., 2006). These effects result from the downregulation of GABAA receptors, or the loss of GABA-containing presynaptic terminals (Fuchs and Salazar, 1998; Kilman et al., 2002; Sarro et al., 2008; Braat et al., 2015). This led us to target inhibitory synapses as a candidate for ameliorating behavioral deficits. Support for this idea emerges from research showing that better performance is correlated with stronger GABAergic transmission (Gleich et al., 2003; Leventhal et al., 2003; Edden et al., 2009). However, these behavioral benefits are only present while the GABA-enhancing drug is in the system, whereas our goal is to permanently rescue normal function. If developmental HL-induced inhibitory deficits cause perceptual impairments, then preventing or restoring normal cortical GABAergic inhibition should rescue normal behavioral performance.
We evaluated the relationship between weakened cortical inhibition and auditory perceptual deficits following developmental HL in juvenile gerbils reared with bilateral earplugs. Developmental HL impairs ACx synaptic inhibition, and also degrades an associated perceptual skill, amplitude modulation (AM) detection (Caras and Sanes, 2015; Mowery et al., 2015, 2017). Here, we report that HL also reduced inhibition in auditory thalamus, the ventral nucleus of the medial geniculate body (MGv). Daily injections with a drug that enhances GABAergic inhibition prevented the reduction of MGv and ACx inhibition, and normalized auditory behavioral skills. Together, these results demonstrate that inhibitory synapse dysfunction can account for perceptual deficits that attend childhood HL. More generally, our results suggest that central impairments may explain some of the educational barriers that persist following a transient period of HL.
Materials and Methods
Experimental animals.
For brain slice experiments, we recorded from 299 pyramidal neurons in layer 2/3 of ACx, using a total of 50 male and female gerbils (Meriones unguiculatus). We also recorded from 74 MGv neurons, using a total of 12 male and female gerbils. Depending on the experiment, the age of recording varied from postnatal day (P) 16 to 91. For behavioral testing, 61 male and female gerbils were used. All animals were obtained from commercially obtained breeding pairs (Charles River Laboratories). Animal care and maintenance were in accordance with the guidelines and rules of the institutional care and use committee, New York University approved by the Office of Laboratory Animal Welfare, Office of Extramural Research, U.S. National Institutes of Health.
Reversible auditory deprivation.
Mild auditory deprivation was induced by inserting a malleable plug (BlueStik Adhesive Putty, RPM International) into the opening of each ear canal at P11 (Caras and Sanes, 2015; Mowery et al., 2015). Animals were checked daily, and earplugs were adjusted to accommodate growth. Earplugs were removed at P23. Postmortem examination confirmed that the tympanic membranes were intact and patent. Earplugs attenuate auditory brainstem responses and perceptual thresholds by ∼15–50 dB, depending on frequency, and the attenuation is completely reversible (Caras and Sanes, 2015; Mowery et al., 2015).
Pharmacological manipulation.
Some animals received subcutaneous injections of a selective GABA reuptake inhibitor (SGRI, 5 mg/ml, 10 mg/kg; NO-711 hydrochloride, Sigma-Aldrich) once daily while the earplugs were in place (P11-P23, HL+SGRI) or after the earplugs were removed (P23-P35, HL+late SGRI). Other animals received subcutaneous injections of the GABAA α1 receptor agonist zolpidem (1 mg/ml, 10 mg/kg) once daily while the earplugs were in place (P11-P23, HL+zolpidem). Another group of animals received subcutaneous injections of saline during earplugging (P11-P23, HL+saline). Injections were typically delivered in the morning. A final group consisted of uninjected, normal-hearing control animals. All behavioral and most neural measurements were obtained several days after the final injection (see Fig. 1: 12–14 d; see Fig. 2: 15–19 d; see Fig. 3: 63–70 d; see Fig. 4: 15 d; see Fig. 7: 6–18 d), and the pharmacokinetics of each drug suggest that none would have remained in the system at this latency. For one experiment (see Figs. 5, 6), the final drug injection occurred ∼24 h before the day of recording.
SGRI is an anticonvulsant that crosses the blood–brain barrier, and is a selective antagonist for the GABA transporter, GAT-1(Suzdak et al., 1992; Borden et al., 1994; Kubová, 1999). After injection, animals typically exhibited a decline in motor activity for ∼1–2 h but displayed no other behavioral signs thereafter.
Behavioral training.
AM depth detection thresholds were assessed with an aversive conditioning procedure used in our laboratory (Sarro and Sanes, 2010, 2011; Rosen et al., 2012; Caras and Sanes, 2015, 2017, 2019). The procedure was controlled by custom Python (Dr. Bradley Buran, Oregon Health and Sciences University) or MATLAB scripts (Dr. Daniel Stolzberg, University of Maryland), interfaced with a digital signal processor (TDT). AM stimuli (3–20 kHz noise, 5 Hz rate, 45 dB SPL) varied from 0 to −24 dB re: 100% depth in 3 dB steps. Stimuli were delivered via a calibrated tweeter (Vifa) 1 m above the test cage within an attenuation booth. Behavioral training and testing were typically performed from late morning to early afternoon. Procedural training: After placement on controlled water access, gerbils rapidly learned to drink from a water spout in the presence of continuous, unmodulated noise (the “safe” stimulus). Animals were trained to withdraw from the spout when the sound changed to 5 Hz AM noise (the “warn” stimulus) by pairing the AM cue with a mild shock. Breaking contact with the water spout was scored as a correct response (hit) on warn trials and an incorrect response (false alarm) on safe trials. The signal detection metric was calculated as d′ = z(hit rate) − z(false alarm rate). Warn trials were interspersed with 3–5 safe trials to avoid temporal conditioning. Animals continued training until they reach criterion performance (d′ ≥ 1.5). Perceptual training: Psychometric performance was assessed for 5 consecutive days using AM depths that bracket detection thresholds, and psychometric functions were fit to the data (Green and Swets, 1966; Wichmann and Hill, 2001a, b; Schütt et al., 2016).
Thalamocortical brain slice preparation.
The surgery and details for thalamocortical brain slice preparation have been previously described (Kotak et al., 2005; Mowery et al., 2015). Animals were deeply anesthetized (chloral hydrate, 400 mg/kg, i.p.) and brains dissected into 4°C oxygenated ACSF as follows (in mm): 125 NaCl, 4 KCl, 1.2 KH2PO4, 1.3 MgSO4, 26 NaHCO3, 15 glucose, 2.4 CaCl2, and 0.4 l-ascorbic acid; and bubbled with 95% O2/5% CO2, pH 7.4. Brains were vibratome-sectioned to obtain 300–400 μm perihorizontal auditory thalamocortical slices. To validate thalamorecipient ACx, a bipolar stimulating electrode (FHC) was placed at rostral border of the medial geniculate (MG), and MG-evoked field responses were recorded in the ACx.
Whole-cell current-clamp recordings were obtained (Warner, PC-501A) from ACx layer 2/3 pyramidal neurons at 32°C in oxygenated ACSF. Recording electrodes were fabricated from borosilicate glass (1.5 mm OD; Sutter, P-97). The internal recording solution contained the following (in mm): 5 KCl, 127.5 K-gluconate, 10 HEPES, 2 MgCl2, 0.6 EGTA, 2 ATP, 0.3 GTP, and 5 phosphocreatine, pH 7.2 with KOH. The resistance of patch electrodes filled with internal solution was between 5 and 10 mΩ. Access resistance was 15–30 mΩ, and was compensated by ∼70%.
Recordings were digitized at 10 kHz and analyzed offline using custom Igor-based macros (IGOR, WaveMetrics). All recorded neurons had a resting potential ≤−50 mV and overshooting action potentials. Frequency-current curves were constructed from the responses to 1500 ms current pulses, in steps of 100 pA (Mowery et al., 2015).
In juvenile animals, IPSPs were evoked via biphasic stimulation of layer 4 (1–10 mV, 10 s interstimulus interval) in the presence of ionotropic glutamate receptor antagonists (20 μm DNQX; 50 μm AP-5). The drugs were applied for a minimum of 8 min before recording IPSPs. Peak amplitudes of the short-latency hyperpolarization (putative GABAA component) and long-latency hyperpolarization (putative GABAB component) were measured from each response at a holding potential (Vhold) of −50 mV.
In adult animals, IPSPs were evoked by optogenetic activation of ACx interneurons (Dimidschstein et al., 2016). Targeted expression of channelrhodopsin (ChR2) to GABAergic interneurons was achieved via cortical injections of a recombinant adeno-associated viral vector (rAAV-mDlx-ChR2-mCherry) (Dimidschstein et al., 2016). All viral injections were conducted in sterile conditions under isoflurane. Craniotomies were made at stereotaxic coordinates to target ACx (Radtke-Schuller et al., 2016), the pipette was inserted ∼200 μm, the virus injected (∼50 nl, 13 nl/s), and the pipette left in place for 20 min. The craniotomy was covered with sterile bone wax, the scalp was sutured, and the animal was allowed to recover for several weeks before in vitro recordings. Peak GABAA and GABAB IPSP amplitudes were obtained at a Vhold of −50 mV at 10 mW light intensity (470 nm, 1 ms pulse). These recordings were performed with a 40× objective that was focused on the recorded neuron. In pilot experiments, we found that moving the objective ∼15 μm away from the recorded neuron led to a significant reduction in maximum-evoked IPSP amplitudes. Therefore, the stimulus likely activated interneuron terminals, rather than cell bodies. In a subset of experiments, we verified that the short- and long-latency IPSP components were selectively blocked by GABAA-specific (20 μm bicuculline) or GABAB-specific antagonists (10 μm SCH-50911), respectively.
Thalamic brain slice preparation.
The surgery for obtaining MGv brain slices was similar to that described for ACx slices. However, brains were vibratome-sectioned to obtain 300–400 μm coronal slices through the medial geniculate body. A bipolar stimulating electrode was placed at the dorsomedial border of the MGv, and focally evoked IPSPs were recorded from MGv neurons.
Statistical analyses.
Statistical tests for distribution and significance were performed using the SAS-based package JMP (SAS Institute). When data were normally distributed (as assessed by the Shapiro–Wilk W Test), values are given as mean ± SEM and ANOVA tests were performed, followed by a Dunnett's test to control for the family-wise error rate. The HL+saline group was specified as the control group for all post hoc tests. When multiple measures were obtained from the same neuron or animal, a two-way mixed-model ANOVA (linear regression analysis with repeated measures) was used to verify a main effect of treatment group. When the assumption of sphericity was violated (as assessed using Mauchley's test), the degrees of freedom and p values were adjusted using the Greenhouse–Geisser correction. When data were not normally distributed, values are given as medians and ranges, and Kruskal–Wallis Rank Sum Tests were performed, followed by Steel's method for nonparametric comparisons with a control. For in vitro analyses, animals were assigned project numbers that blinded the analyzer to experimental group. Exact n values used to calculate the statistics are provided in the Results.
Results
SGRI treatment prevented HL-induced deficits in learning and memory consolidation
Transient HL during a developmental critical period leads to auditory perceptual deficits (Caras and Sanes, 2015). If HL-induced perceptual deficits are caused by weak auditory cortical inhibition, then preventing the loss of IPSP strength should maintain performance on an auditory psychometric task. A previous study demonstrated that a specific GABA reuptake inhibitor (SGRI) could rescue ACx inhibitory strength following developmental HL (Kotak et al., 2013). To test whether enhancing inhibition also rescues perceptual deficits, AM depth detection thresholds were assessed using a yes-no aversive conditioning paradigm (Sarro and Sanes, 2010, 2011, 2014; Sarro et al., 2011; Rosen et al., 2012; Buran et al., 2014; Kang et al., 2014; Caras and Sanes, 2015, 2017, 2019). Animals were first trained to drink from a water spout when a safe stimulus (unmodulated noise) was present, and to avoid the water spout during a warn stimulus (0 dB AM re: 100% depth; Fig. 1a). Hits and false alarms were acquired, and sensitivity was scored as the signal detection metric, d′ (see Materials and Methods). As shown in Figure 1b, the treatment groups initially included uninjected, normal-hearing controls (n = 15), and three groups of animals raised with HL from P11-P23 and simultaneously treated with saline (n = 12), SGRI (n = 11), or zolpidem (n = 12). In addition, after discovering a positive effect of SGRI treatment on HL-induced deficits (see below), we added an additional group (n = 11) to determine whether late SGRI treatment from P23-P35 (i.e., after hearing was restored at P23) yielded similar effects even after the ACx critical period closed. Five HL-reared animals (3 saline-treated, 1 SGRI-treated, 1 late SGRI-treated) did not reach or maintain a d′ ≥ 1.5 during procedural training and were removed from the study. One control subject was excluded from the analyses in Figure 1 because data for one of the procedural training sessions were lost due to a computer malfunction.

SGRI treatment rescues HL-induced deficits in procedural learning and consolidation. a, Behavioral task schematic. Animals learned to withdraw from the water spout during amplitude modulated noise (Warn) to avoid a brief shock. Correctly avoiding the shock is scored a hit, and failing to withdraw from the spout is scored a miss. During unmodulated noise (Safe), animals can drink freely from the water spout. Correctly drinking from spout is scored a correct reject, and incorrectly withdrawing from the spout is scored a false alarm. b, Experimental timeline shows five groups: Group 1: The control group was unmanipulated. Group 2: The HL+saline group received bilateral earplugs from P11–P23 and daily injections of 0.9% saline during this period. Group 3: The HL+SGRI group received bilateral earplugs from P11–P23 and daily injections of SGRI during this period. Group 4: The HL+late SGRI group received bilateral earplugs from P11–P23 and daily injections of SGRI after earplug removal, from P23–P35. Group 5: The HL+zolpidem group received bilateral earplugs from P11–P23 and daily injections of zolpidem during this period. After earplugs were removed, animals experienced normal hearing (NH). Animals trained on the AM detection task from P35–P37 (procedural training), and psychometric functions were obtained daily from P38–P42 (perceptual training). c, HL-reared animals required more trials than controls to reach a d′ ≥ 1.5, the procedural training criterion. However, none of the drug treatment groups was significantly different from the HL+saline group. Plots represent median, 25th and 75th percentile (box), and 1.5 × the interquartile range (whiskers) (for statistics, see Results). d, The HL-induced learning delay in saline-treated animals was explained by slower task acquisition during the initial training session. HL+saline animals also displayed diminished memory consolidation, illustrated by the drop in performance at the beginning of Sessions 2 and 3. SGRI treatment improved the rate of learning in Session 1 and abolished the HL-induced consolidation deficit. Plots represent mean ± SEM calculated with a 15-trial-wide sliding window. e, Administration of SGRI after the period of HL was not sufficient to rescue learning and memory consolidation. f, Zolpidem treatment did not resolve the initial learning delay in Session 1, nor the consolidation deficit in Session 2. However, zolpidem-treated animals performed no differently from controls by the beginning of Session 3.
The effect of HL and drug treatment on auditory procedural learning was assessed from P35-P37 by determining how many trials were required to reach our performance criterion for learning the task (d′ ≥ 1.5). Figure 1c shows that transient HL caused a significant increase in the number of trials to criterion, compared with controls (Kruskal–Wallis Rank Sum Test, H(4) = 15.33, p = 0.0041; post hoc multiple comparisons with the HL+saline control group using Steel's method, control vs HL+saline, Z = 3.54, p = 0.0015). However, the number of trials to criterion for both groups of SGRI-treated HL animals and zolpidem-treated animals did not differ from the HL+saline group (HL+SGRI vs HL+saline: Z = −1.55, p = 0.3225; HL+late SGRI vs HL+saline: Z = −2.29, p = 0.0710; HL+zolpidem vs HL+saline: Z = −1.06, p = 0.6432). Thus, HL delayed procedural learning, and the drug treatments did not restore it.
To determine the behavioral basis for this HL-induced learning delay, we examined performance within each of the three daily training sessions by calculating d′ with a 15-trial-wide sliding window. Figure 1d shows that control animals displayed rapid learning during training Session 1. Control animals began Session 2 at a performance level superior to where they ended Session 1, suggesting overnight memory consolidation, and reached asymptotic performance by the end of Session 2. In contrast, animals reared with transient HL and treated with saline displayed slower task acquisition (mixed-model ANOVA significant effect of group, F(4,34) = 3.27, p = 0.0227), barely reaching a d′ of 1 during training Session 1 (Fig. 1d). Furthermore, HL+saline animals did not display control-like performance at the beginning of Session 2 in that there was an effect of group on d′ calculated over the first 25 trials performed (Kruskal–Wallis Rank Sum Test, H(4) = 13.04, p = 0.0111; post hoc comparison with the HL+saline group using Steel's method, control vs HL+saline: Z = 3.07, p = 0.0076), but reached control levels by the final 25 trials of Session 2 (Kruskal–Wallis Rank Sum Test: H(4) = 4.457, p = 0.3476). However, the HL+saline group again performed more poorly than controls during the first 25 trials of Session 3, suggesting impaired memory consolidation (Kruskal–Wallis Rank Sum Test: H(4) = 13.22, p = 0.0102; post hoc comparison with the HL+saline control group using Steel's method, control vs HL+saline: Z = 2.74, p = 0.0210). Early SGRI treatment rescued both learning and consolidation as evidenced by the fact that the performance of HL+SGRI (Fig. 1d) animals improved substantially during Session 1, and differed significantly from the HL+saline group at the start of Session 2 (HL+SGRI vs HL+saline: Z = 2.50, p = 0.0412) and the start of Session 3 (HL+SGRI vs HL+saline: Z = 2.78, p = 0.0187). In contrast, learning was not rescued in either late SGRI-treated HL animals (HL+late SGRI vs HL+saline; start of Session 2: Z = 1.96, p = 0.1488; start of Session 3: Z = 1.76, p = 0.2209; Fig. 1e) or zolpidem-treated HL animals (HL+zolpidem vs HL+saline; start of Session 2: Z = 1.35, p = 0.4434; start of Session 3: Z = 1.89, p = 0.1713; Fig. 1f). All animals ultimately achieved similar maximum d′ values during procedural training (Kruskal–Wallis Rank Sum Test, no significant effect of group, H(4) = 6.82, p = 0.1458).
Improvements in procedural learning across sessions were driven by a substantial increase in hit rates (mixed-model ANOVA, significant effect of trial window, F(68,3400) = 78.75, p < 0.0001). False alarm rates were low (median = 5%, 90th percentile = 9%, range 5%–24%) with a small, but significant decrease as training progressed (mixed-model ANOVA, significant effect of trial window, F(67,3350) = 3.72, p < 0.0001). These analyses were limited to the first 69 (hit rates) or 68 (false alarm rates) trial windows, for which we had data from all animals. The poor performance by HL+saline animals during the initial training session was not explained by insufficient practice, as this group performed a similar number of trials (mean ± SEM: 33 ± 2) as controls (27 ± 2; post hoc comparison with the HL+saline group using Dunnett's test, control vs HL+saline: p = 0.2988), HL+SGRI animals (38 ± 3, HL+SGRI vs HL+saline: p = 0.7144), HL+late SGRI animals (25 ± 4, HL+late SGRI vs HL+saline: p = 0.1372), and HL+zolpidem animals (32 ± 2, HL+zolpidem vs HL_saline: p = 0.9921). Similarly, the poor performance of the HL+saline animals was not explained by body size because HL-reared animals did not differ from one another, regardless of drug treatment (control = 27.3 ± 0.7 g, HL+saline = 20.3 ± 1.6 g, HL+SGRI = 22.0 ± 1.0 g; HL+late SGRI = 20.0 ± 0.4; HL+zolpidem = 22.4 ± 1.4 one-way ANOVA, F(4,51) = 8.75, p < 0.0001; post hoc multiple comparisons using Dunnett's method: control vs HL+saline: p < 0.0001; HL+SGRI vs HL+saline: p = 0.6868; HL+late SGRI vs HL+saline: p = 0.9993; HL+zolpidem vs HL+saline: p = 0.4732).
SGRI treatment prevented HL-induced perceptual deficits
Once animals in each treatment group reached performance criterion on the AM detection task, we presented animals with a range of AM depths to generate psychometric functions and threshold estimates. We obtained an initial threshold on P38, and then continued to train animals for four additional sessions (see Fig. 1b). Representative psychometric functions from the fifth day of perceptual testing are shown in Figure 2a–c with AM detection threshold defined as the depth at which d′ = 1 (gray horizontal lines). While HL-rearing impaired AM depth detection in saline-treated animals, both early (Fig. 2d) and late SGRI treatment (Fig. 2e) maintained thresholds at control-like levels (mixed-model ANOVA, significant effect of group, F(4,51) = 3.05, p = 0.0250). In contrast, zolpidem treatment did not rescue control-like performance (Fig. 2f). Perceptual training improved AM depth thresholds in all groups (mixed-model ANOVA, significant effect of day, F(3.15,160.77) = 24.12, p < 0.0001). However, the HL+saline and HL+zolpidem groups maintained elevated thresholds compared with the control and HL+SGRI groups throughout training (mixed-model ANOVA, no day × group interaction, F(12.61,160.77) = 1.28, p = 0.2312).

SGRI treatment rescues HL-induced deficits in AM depth detection. a–c, Representative psychometric functions from 3 animals obtained on the fifth day of perceptual testing. Threshold was defined as the AM depth at which d′ = 1 (gray horizontal line). The same representative control and HL+saline psychometric functions are plotted in a–c. a, An SGRI-treated HL-reared animal performs similarly to the control animal. b, An animal treated with SGRI after HL displays control-like performance. c, A zolpidem-treated animal displays HL-like performance. d, Perceptual training improved AM depth thresholds in all groups. However, HL+saline animals continued to display elevated thresholds compared with the control and HL+SGRI animals. e, The HL+late SGRI group displayed control-like thresholds throughout perceptual training. f, The performance of HL+zolpidem animals was significantly poorer than controls (for statistics, see Results). Data are mean ± SEM.
These findings were not explained by proxies for motivation or experience. While there was a significant effect of group on the number of trials completed during the first day of perceptual training (one-way ANOVA, F(4,51) = 7.55, p < 0.0001), saline-treated HL animals performed a similar number of trials (325 ± 17) as controls (384 ± 22, comparison with the HL+saline group using Dunnett's test, control vs HL+saline: p = 0.2067), HL+SGRI animals (406 ± 18, HL+SGRI vs HL+saline: p = 0.0795), HL+late SGRI animals (237 ± 24, HL+late SGRI vs HL+saline: p = 0.0559), and HL+zolpidem animals (351 ± 28, HL+zolpidem vs HL+saline: p = 0.8386). Similarly, while there was also a significant effect of group on false alarm rates during the first day of perceptual training (Welch's ANOVA, F(4,22.55) = 2.96, p = 0.0417), saline-treated HL animals made a similar number of false alarms (3 ± 1%) as controls (3 ± 1%, comparison with the HL+saline control group using Dunnett's test, control vs HL+saline: p = 0.9515), HL+SGRI animals (3 ± 1%, HL+SGRI vs HL+saline: p = 0.9808), HL+late SGRI animals (6 ± 1%, HL+late SGRI vs HL+saline: p = 0.0744), and HL+zolpidem animals (2 ± 0%, HL+zolpidem vs HL+saline: p = 0.5680).
SGRI treatment permanently rescued HL-induced loss of cortical inhibition
Given the close association between synaptic inhibition and developmental disorders (Turrigiano and Nelson, 2004; Chao et al., 2010; Richardson et al., 2012; Braat and Kooy, 2015), we next evaluated whether HL disrupted ACx inhibition, and whether this effect was rescued by the drug treatments that preserved learning and perception (Figs. 1, 2). To evaluate this possibility, animals were reared with HL and administered saline, SGRI, or zolpidem from P11-P23 (Fig. 3a). ACx inhibitory neurons were subsequently targeted to express ChR2 at P56, using a recombinant adeno-associated viral vector (Fig. 3b) (Dimidschstein et al., 2016). When animals reached sexual maturity (>P90), ACx brain slices were prepared and IPSPs were evoked by optogenetic activation of ACx interneuron terminals. In a separate set of control recordings, we verified that SGRI and zolpidem do enhance stimulus-evoked IPSP amplitude at low concentrations (Fig. 3c). Light-evoked IPSPs displayed short- and long-latency components, and control recordings indicated that they represented GABAA and GABAB responses, respectively. First, by applying either the GABAA receptor antagonist (20 μm bicuculline), or the GABAB receptor antagonist (10 μm SCH-50911, SCH), the short- and long-latency responses were blocked, respectively (Fig. 3d, left). Second, by holding the neuron at successively more negative potentials, the short-latency response was found to reverse at ≈−70 mV, near the chloride equilibrium potential, whereas the long-latency component reversed at a more hyperpolarized potential (Fig. 3d, right). Therefore, the amplitude of short- and long-latency components of the IPSP is referred to as the GABAA and GABAB components, respectively.

SGRI treatment during developmental HL leads to long-term prevention of cortical inhibitory deficits. a, Experimental timeline shows four treatment groups: controls, HL+saline, HL+SGRI, and HL+zolpidem. Earplug and drug injection details are identical to those described in Figure 1. At P56, all animals were injected with a vector (rAAV-mDlx-ChR2-mCherry) that expresses ChR2 and mCherry (mCh) in GABAergic interneurons under the control of a Dlx promotor (Dimidschstein et al., 2016). IPSPs were then measured using an optogenetic approach at ≥P86. b, Schematics show injection of the vector into ACx (top), and the perihorizontal brain slice preparation (bottom) containing ChR2-expressing GABAergic interneurons (red). Light stimulation (470 nm) was used to evoked IPSPs in recorded layer 2/3 pyramidal neurons. c, To verify that SGRI and zolpidem both served to enhance IPSPs, control recordings were obtained at −50 mV, and the slice was bathed in a low concentration of either SGRI (left) or zolpidem (right). All recordings displayed an increase in IPSP amplitude. d, Recordings of light-evoked IPSPs displayed both a short- and a long-latency component. To assess whether these components represented GABAA and GABAB responses, respectively, we performed two set of control recordings. Application of the GABAA receptor antagonist (20 μm bicuculline [BIC]), or the GABAB receptor antagonist (10 μm SCH-50911, SCH), blocked the short- and long-latency components, respectively (left). In addition, holding the neuron at increasingly negative potentials revealed that the short-latency response reversed at ≈−70 mV, near the chloride equilibrium potential, whereas the long-latency component reversed at a more depolarized potential (right). Therefore, the amplitude of short- and long-latency components is presented as GABAA and GABAB IPSPs in e. e, Representative current-evoked IPSPs are shown for neurons from each treatment group. These IPSPs displayed short- and long-latency responses, referred to as the GABAA (A) and GABAB (B) components. Plots represent that optogenetically evoked (f) GABAA and (g) GABAB receptor-mediated IPSP amplitudes remained significantly smaller in adult neurons, long after a transient period of developmental HL (HL+saline) at P11-P23, but SGRI treatment during that same period (HL+SGRI) permanently prevented this deficit (for statistics, see Results). For both plots, the number of recorded neurons was control = 21, HL+saline = 17, HL+SGRI = 21, and HL+zolpidem = 16. Plots represent mean ± SEM. *Significant difference from the HL+saline group. n.s. indicates no significant difference.
Maximum current-evoked IPSP amplitudes were recorded in layer 2/3 pyramidal neurons at a holding potential of −50 mV. The exemplar IPSPs in Figure 3e illustrate that the brief period of HL resulted in the persistent reduction of both GABAA and GABAB IPSP amplitudes, compared with controls. Figure 3f plots individual GABAA IPSP values, and Figure 3g plots individual GABAB IPSP values. For both measures, HL resulted in a significant reduction of IPSP amplitude (mixed-model ANOVA to test significant effect of group, F(3,71) = 5.75, p = 0.0014; followed by a Dunnett's Test to control for multiple comparisons; GABAA IPSP amplitude: control vs HL+saline: p = 0.0011; GABAB IPSP amplitude: control vs HL+saline: p = 0.0030). ACx neurons from animals that were treated with SGRI from P11-P23 displayed control-like GABAA and GABAB IPSP amplitudes (GABAA IPSP: HL+saline vs HL+SGRI: p = 0.0356; GABAB IPSP: HL+saline vs HL+SGRI: p = 0.0004). In contrast, zolpidem treatment did not restore GABAA or GABAB IPSP amplitudes (GABAA IPSP: HL+saline vs HL+zolpidem: p = 0.9602; GABAB IPSP: HL+saline vs HL+zolpidem: p = 0.0664). Together, these results suggested that SGRI was far more effective than zolpidem at rescuing HL-induced cortical deficits.
SGRI treatment was effective when delivered during or after the ACx critical period
We next asked whether there was a critical period for rescuing cortical inhibition following developmental HL. Here, we assessed ACx inhibition at P38, the age at which behavioral testing was initiated. As shown in Figure 4a, the treatment groups included uninjected normal-hearing controls, saline-treated HL animals, and HL animals that received SGRI-treatment from P11-P23, or from P23-P35. Zolpidem was not studied because it was less effective at rescuing behavior or inhibition in HL-reared animals. Maximum current-evoked IPSP amplitudes were recorded from layer 2/3 pyramidal neurons at a holding potential of −50 mV in auditory cortical brain slices at P38 (15 d after earplug removal), which corresponds to the age at which perceptual training began (Fig. 4b). Recordings were performed in the presence ionotropic glutamate receptor antagonists (50 μm AP-5; 20 μm DNQX). As shown in Figure 4c, IPSPs displayed short- (A) and long-latency components (B), which are presented as GABAA and GABAB responses, respectively (for validation, see Fig. 3d).

SGRI treatment prevents HL-induced reduction of cortical inhibition at age of behavioral testing. a, Experimental timeline shows four treatment groups: controls, HL+saline, HL+SGRI, and HL+late SGRI. Earplug and drug injection details are identical to those described in Figure 1. Brain slices were obtained for synaptic physiology at P38. b, Perihorizontal brain slices containing the ACx and MG were obtained, and electrical stimuli (Stim) was used to activate inhibitory interneurons (gray). Evoked IPSPs were recorded from layer 2/3 pyramidal neurons in the presence of DNQX and AP-5. c, Representative current-evoked IPSPs are shown for neurons from each treatment group, and the GABAA (A) and GABAB (B) components are indicated. d, Plot represents that GABAA receptor-mediated IPSP amplitudes were significantly reduced by HL (HL+saline). SGRI treatment from P11-P23 (HL+SGRI) prevented this effect, but late SGRI treatment at P23-P35 did not restore GABAA IPSP amplitude. e, Plot represents that GABAB receptor-mediated IPSP amplitudes were significantly reduced by HL (HL+saline). SGRI treatment prevented this effect when delivered at either age. Small upward arrows represent data values that were larger than maximum y axis value (for statistics, see Results). Number of recorded neurons at P38 was control = 13, HL+saline = 14, HL+SGRI = 12, and HL+late SGRI = 17. Plots represent mean ± SEM. *Significant difference from HL+saline group. n.s. indicates no significant difference.
As shown in Figure 4d, HL induced a significant reduction of GABAA receptor-mediated IPSPs at P38 (mixed-model ANOVA to test significant effect of group, F(3,52) = 11.22, p = 0.0014; followed by a Dunnett's Test to control for multiple comparisons; GABAA IPSP amplitude: control vs HL+saline: p < 0.0001). SGRI treatment prevented the loss of GABAA inhibition when introduced during the period of HL (HL+saline vs HL+SGRI: p < 0.0001). However, the ability of SGRI to restore GABAA inhibition when introduced after the earplugs were removed did not attain significance (HL+saline vs HL+late SGRI: p = 0.0547).
As shown in Figure 4e, HL also led to a decrease in GABAB receptor-mediated IPSP amplitudes (control vs HL+saline: p = 0.0001). SGRI treatment prevented the loss of GABAB inhibition when introduced during the period of HL, and also restored GABAB inhibition when introduced after the earplugs were removed (HL+saline vs HL+SGRI: p < 0.0001; HL+saline vs HL+ late SGRI: p = 0.0068). Therefore, SGRI treatment during the period of HL is sufficient to prevent the loss of auditory cortical GABAA and GABAB inhibition. However, when introduced after the period of HL, SGRI does not effectively rescue GABAA inhibition. This suggests that, like HL itself, there may be a critical period during which SGRI treatment can fully restore normal inhibitory function.
SGRI treatment prevented the HL-induced loss of cortical inhibition
To determine whether early SGRI treatment prevented the HL-induced reduction of ACx inhibitory synapse function, or reversed it after the fact, we recorded maximum current-evoked IPSP amplitudes from animals between P14 and P20 while the earplugs were in place (Fig. 5a). As shown in Figure 5b, HL induced a significant reduction of GABAA receptor-mediated IPSPs by P14-P17, but SGRI treatment prevented this effect (mixed-model ANOVA to test significant effect of group, F(5,126) = 24.58, p < 0.0001; followed by a Dunnett's Test to control for multiple comparisons; GABAA IPSP amplitude recorded at P14-P17: control vs HL+saline: p < 0.0001, HL+saline vs HL+SGRI: p < 0.0001, GABAA IPSP amplitude recorded at P18-P20: control vs HL+saline: p < 0.0001, HL+saline vs HL+SGRI: p < 0.0001).

SGRI treatment prevents HL-induced reduction of cortical inhibition. a, Experimental timeline shows three treatment groups: controls, HL+saline, and HL+SGRI. Earplug and drug injection details are identical to those described in Figure 1. Brain slices were obtained for synaptic physiology at P14-P20, as illustrated in Figure 4b. b, Plot represents that GABAA receptor-mediated IPSP amplitudes were significantly reduced by HL (HL+saline) during both age ranges examined, and SGRI treatment (HL+SGRI) prevented this effect. c, Plot represents that GABAB receptor-mediated IPSP amplitudes were significantly reduced by HL (HL+saline) beginning after P17, and SGRI treatment (HL+SGRI) prevented this effect. Small upward arrows indicate data values that were larger than maximum y axis value (for statistics, see Results). Number of recorded neurons at P14-P17 was control = 27, HL+saline = 30, and HL+SGRI = 26. Number of recorded neurons at P18-P20 was control = 15, HL+saline = 17, and HL+SGRI = 17. Plots represent mean ± SEM. *Significant difference from HL+saline group.
Similarly, HL led to a decrease in GABAB receptor-mediated IPSP amplitudes (Fig. 5c), but this effect was not observed until after P17 (GABAB IPSP amplitude recorded at P14-P17: control vs HL+saline: p = 0.1880, GABAB IPSP amplitude recorded at P18-P20: control vs HL+saline: p = 0.0059). SGRI not only prevented this effect (GABAB IPSP amplitude recorded at P18-P20: HL+saline vs HL+SGRI: p < 0.0001), but even increased GABAB responses above control levels at P18-P20 (Tukey HSD test, control vs HL+SGRI: p < 0.0001). Therefore, SGRI treatment during the period of HL is sufficient to completely prevent the loss of auditory cortical inhibition.
SGRI treatment prevented the HL-induced loss of cortical α1 GABA receptor subunit
HL was induced during an age range when GABAA receptor-mediated synaptic current duration normally decreases in conjunction with an upregulation of α1 subunit expression (Kotak et al., 2008). Therefore, we next asked whether SGRI treatment prevented HL-induced inhibitory dysfunction by facilitating the functional expression of the α1 subunit. Functional expression of the α1 subunit was assessed by measuring the enhancement of current-evoked IPSPs in response to the α1 subunit-specific agonist, zolpidem, which displays a high affinity to the benzodiazepine-binding site (Pritchett and Seeburg, 1990; Wafford et al., 1994; Lüddens et al., 1995; Rudolph and Möhler, 2004; Kotak et al., 2008). The effects of HL and SGRI treatment were assessed at P13-P17 in ACx brain slices (Fig. 6a) by recording maximum current-evoked IPSP amplitudes and durations in layer 2/3 pyramidal neurons at a holding potential of −50 mV in the presence of ionotropic glutamate receptor antagonists (50 μm AP-5; 20 μm DNQX). Figure 6b, c shows that control neurons displayed a significant increase in IPSP amplitude and duration in response to 100 μm zolpidem exposure, whereas HL neurons displayed almost no response, suggesting that α1 subunits were either expressed at lower levels or not trafficked into the membrane. In contrast, neurons from SGRI-treated HL animals displayed control-like amplitudes in response to zolpidem (mixed-model ANOVA to test significant effect of group; amplitude: F(2,33) = 32.2, p < 0.0001; duration: F(2,33) = 20.31, p < 0.0001). This indicated that functional α1 subunit expression was pharmacologically rescued. This finding suggests that the HL-induced reduction of auditory cortical inhibition has a postsynaptic locus that is associated with a decline in the expression or trafficking of GABAA receptors, consistent with our previous findings (Sarro et al., 2008), and SGRI treatment prevents this reduction.
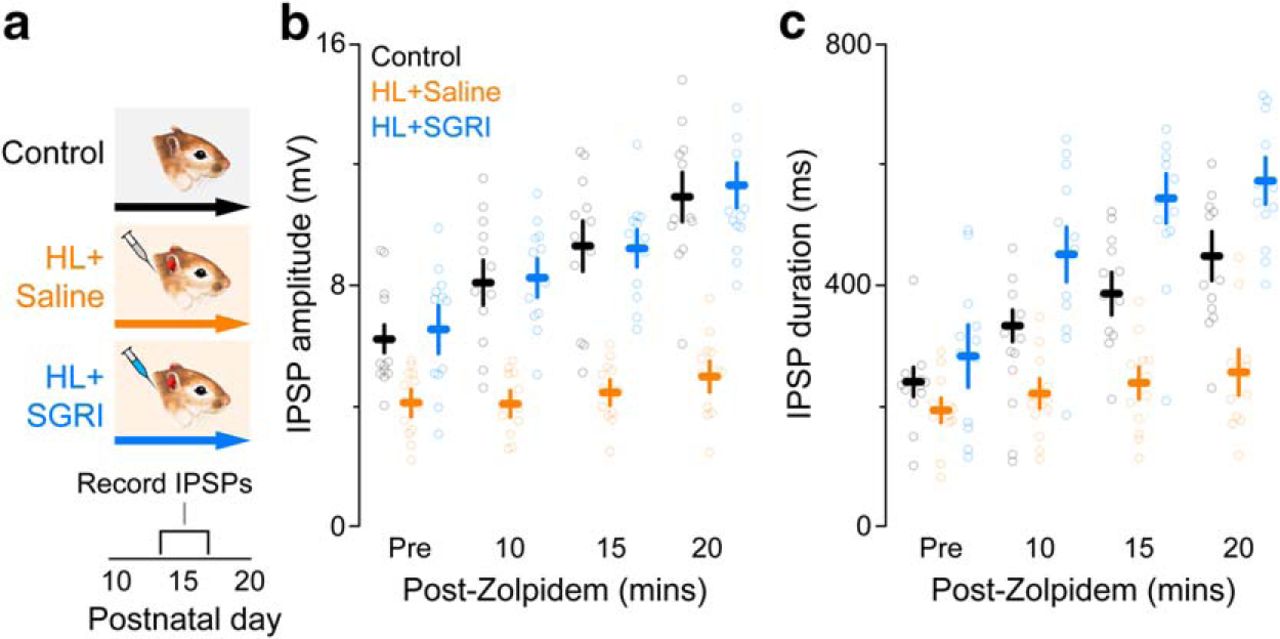
SGRI treatment during developmental HL prevents the loss of cortical α1 GABAR subunits. a, Experimental timeline shows three treatment groups: controls, HL+saline, and HL+SGRI. Earplug and drug injection details are identical to those described in Figure 1. Brain slices were obtained for synaptic physiology at P14-P20, as illustrated in Figure 4b. b, c, To assess the functional expression of α1 subunits, we measured the magnitude of zolpidem-induced increases in evoked IPSPs. Plot represents that neurons in the HL+saline group remained insensitive to zolpidem over the 20 min exposure period, as assessed with IPSP amplitude (b) or duration (c) compared with neurons in either the control or the HL+SGRI groups (for statistics, see Results). Plots represent mean ± SEM. For both plots, the number of recorded neurons was control = 12, HL+saline = 12, and HL+SGRI = 12.
SGRI treatment prevented the HL-induced loss of thalamic inhibition
To determine whether developmental HL caused a reduction in auditory thalamic inhibition, we assessed inhibition in MGv neurons following earplugging from P11-P23. As shown in Figure 7a, the treatment groups included uninjected normal-hearing controls and saline-treated animals that received earplugs from P11-P23 (HL+saline), as well as SGRI-treated animals that received earplugs from P11-P23 (HL+SGRI). Maximum current-evoked IPSP amplitudes were recorded from MGv neurons between P29 and P41 (Fig. 7b), encompassing the age range during which perceptual testing occurred. As shown in Figure 7c, IPSPs displayed short- (A) and long-latency components (B), which are presented as GABAA and GABAB responses, respectively. As shown in Figure 7d, HL caused a significant reduction of GABAA receptor-mediated IPSPs (mixed-model ANOVA to test significant effect of group, F(2,71) = 46.51, p < 0.0001; followed by a Dunnett's Test to control for multiple comparisons; GABAA IPSP amplitude: control vs HL+saline: p < 0.0001). To determine whether early SGRI treatment prevented the HL-induced reduction of MGv inhibitory synapse function, animals were earplugged and treated with SGRI from P11 to P23 (Fig. 7a). SGRI treatment prevented the loss of GABAA inhibition when introduced during the period of HL (HL+saline vs HL+SGRI: p < 0.0001).

SGRI treatment prevents HL-induced reduction of thalamic inhibition at age of behavioral testing. a, Experimental timeline shows three treatment groups: controls, HL+saline, and HL+SGRI. Earplug and drug injection details are identical to those described in Figure 4. Brain slices were obtained for synaptic physiology between P29 and P41. b, Coronal brain slices containing the MGv were obtained, and electrical stimuli (Stim) were used to activate inhibitory terminals (gray). Evoked IPSPs were recorded from MGv neurons in the presence of DNQX and AP-5. c, Representative current-evoked IPSPs are shown for neurons from each treatment group, and the GABAA (A) and GABAB (B) components are indicated. d, Plot represents that GABAA receptor-mediated IPSP amplitudes in MGv were significantly reduced by HL (HL+saline). SGRI treatment from P11 to P23 (HL+SGRI) prevented this effect. e, Plot represents that GABAB receptor-mediated IPSP amplitudes in MGv were significantly reduced by HL (HL+saline). SGRI treatment prevented this effect. Number of recorded neurons was control = 22, HL+saline = 24, and HL+SGRI = 28. Plots represent mean ± SEM. *Significant difference from HL+saline group.
As shown in Figure 4e, HL also led to a decrease in GABAB receptor-mediated IPSP amplitudes (control vs HL+saline: p = 0.0001). SGRI treatment prevented the loss of GABAB inhibition when introduced during the period of HL (HL+saline vs HL+SGRI: p < 0.0001). Therefore, HL induces the loss of inhibition in MGv and SGRI treatment during the period of HL is sufficient to completely prevent this.
Discussion
HL is the most common sensorineural impairment (Fortnum et al., 2001; Kennedy and McCann, 2004; Morton and Nance, 2006; Hilgert et al., 2009; Aithal et al., 2012; Smith et al., 2014), posing a risk for deficits in both perceptual and cognitive skills, including language acquisition (Svirsky et al., 2004; Nicholas and Geers, 2006; Moeller et al., 2007; Niparko et al., 2010; Tobey et al., 2013; Tomblin et al., 2014; Kishon-Rabin et al., 2015). Although HL research typically focuses on cochlear dysfunction for which there is a clear relationship between cellular and perceptual deficits, behavioral problems can persist long after audibility returns to normal (Whitton and Polley, 2011; Sanes, 2016). For example, transient periods of HL, such as those found during otitis media with effusion, have been associated with persistent auditory processing and language impairments (Pillsbury et al., 1991; Hall and Grose, 1994; Hall et al., 1995, 1998; Hogan et al., 1996; Hogan and Moore, 2003; McKenna Benoit et al., 2019). Furthermore, children with a history of otitis media with effusion, but normal hearing at the time of testing, display longer latencies between evoked potentials in the brainstem (Folsom et al., 1983; Gunnarson and Finitzo, 1991; Hall and Grose, 1993), and abnormal neural responses to speech syllable variants (Haapala et al., 2014). To study a model in which behavioral deficits could not be attributed to a damaged cochlea, we induced a perceptual deficit by transiently attenuating sound with bilateral earplugs (Caras and Sanes, 2015).
The behavioral consequences of many developmental disorders, including congenital deafness and blindness, have been linked to diminished GABAA receptor-mediated inhibition (Chao et al., 2010; Richardson et al., 2012; Braat and Kooy, 2015). Inhibitory synapses between cortical interneurons and pyramidal cells are weakened following auditory, visual, or somatosensory deprivation (Maffei et al., 2004; Kotak et al., 2005; Jiao et al., 2006; Takesian et al., 2012). These effects are correlated with a downregulation of GABAA receptors or a loss of GABAergic terminals (Fuchs and Salazar, 1998; Kilman et al., 2002; Sarro et al., 2008; Braat et al., 2015). Moreover, a HL-induced reduction of glycinergic or GABAergic inhibitory synaptic responses has previously been reported in auditory brainstem structures (for review, see Takesian et al., 2009). Here, we have shown that a similar reduction of IPSP amplitude was observed in the MGv following a transient period of HL (Fig. 7).
A transient period of developmental HL in gerbils induces perceptual deficits that are associated with reduced ACx inhibitory synaptic strength (Caras and Sanes, 2015; Mowery et al., 2015). If the observed HL-induced inhibitory deficits are causally linked to perceptual impairments, then these impairments should resolve when inhibition is preserved. This strategy draws support from research showing that better performance is correlated with stronger GABAergic transmission (Gleich et al., 2003; Leventhal et al., 2003; Edden et al., 2009; Han et al., 2012). Here, we found that treating HL-reared animals with a specific GABA reuptake inhibitor could prevent learning and auditory perceptual deficits in juveniles, and this could be explained by the ability of this treatment to prevent or restore the loss of cortical and thalamic GABAA and GABAB receptor-mediated inhibition, although this effect was diminished when SGRI was delivered after the ACx critical period.
Developmental HL: preventing learning and perceptual deficits in juveniles
We have found that developmental HL impairs several auditory perceptual skills, as well as task learning. Adults reared with permanent conductive HL display poorer performance on frequency modulation detection, AM detection, AM discrimination, and modulation masking release tasks (Rosen et al., 2012; Buran et al., 2014; Ihlefeld et al., 2016; von Trapp et al., 2017; Yao and Sanes, 2018). Furthermore, adult animals reared with conductive HL display slower task learning and perceptual learning (von Trapp et al., 2017). Similar results were found for juvenile animals following a brief period of HL that occurs during a well-defined critical period (Caras and Sanes, 2015; Mowery et al., 2015, 2016). These behavioral findings are consistent with auditory critical periods in several species during which stimulus encoding can be altered by environmental manipulations, including HL (Knudsen et al., 1984a, b; Moore et al., 1999; de Villers-Sidani et al., 2007; Razak et al., 2008; Insanally et al., 2009; Popescu and Polley, 2010; Barkat et al., 2011; Keating and King, 2013; Keating et al., 2013; Polley et al., 2013).
Our current findings confirm that a brief period of HL during the ACx critical period leads to behavioral deficits in juvenile animals, and newly demonstrate that perceptual deficits are completely ameliorated by administration of SGRI during, but not after, the period of HL. Whereas HL+saline animals displayed slower task learning, and an apparent decline in memory consolidation between training sessions, HL+SGRI animals exhibited control-like consolidation and learning (Fig. 1d). Treatment with SGRI after the period of HL or a GABAA receptor enhancer (zolpidem) were less effective, such that procedural learning was still delayed, relative to controls.
SGRI treatment also overcame the HL-induced deficit in AM depth detection. This deficit, which manifests as poorer AM detection thresholds in animals raised with HL, persists throughout perceptual training, and is also observed in children with a history of otitis media with effusion (McKenna Benoit et al., 2019). Here, we found that AM detection thresholds were completely ameliorated in HL+SGRI and HL+late SGRI animals (Fig. 2d,e). In contrast, zolpidem had no effect (Fig. 2f). Because SGRI facilitates both GABAA and GABAB receptor-mediated inhibition, it is possible both receptor types must be properly activated to promote normal maturation.
Developmental HL: preventing the loss of ACx inhibition
Transient HL during a well-described critical period (Mowery et al., 2015) not only disrupted cognitive and perceptual abilities, but also led to a significant reduction of both GABAA and GABAB receptor-mediated inhibition, an effect that persisted to adulthood (Fig. 3e). Daily injection of SGRI during the period of deprivation completely prevented the loss of both GABAA and GABAB inhibition, whereas zolpidem was less effective, a finding that parallels our behavioral results. As expected, the HL-induced reduction of inhibition was present at the time of behavioral testing (Fig. 4). To determine whether there was a critical period during which SGRI could rescue inhibition, we compared the effect of drug delivery during the HL, and immediately after the earplugs were removed. Although SGRI treatment was more effective when initiated during HL, it continued to improve GABAB receptor-mediated inhibition when delivered from P23 to P35 (Fig. 4). Indeed, recordings obtained during the period of HL demonstrate that inhibitory strength declines within days of earplug insertion and SGRI can prevent this reduction (Fig. 5). A final set of experiments determined whether SGRI treatment rescued the functional expression of the adult α1 GABAA receptor subunit, as assessed by the sensitivity of IPSPs to the α1 subunit-specific GABAA receptor enhancer, zolpidem (Pritchett and Seeburg, 1990; Kralic et al., 2002; Rudolph and Möhler, 2004). Figure 6 shows that bath application of zolpidem had a significantly smaller effect on IPSPs from HL+saline neurons, compared with controls, but a control-like response was maintained in neurons from HL+SGRI animals. Together, these results suggest that SGRI can prevent the HL-induced loss of inhibition when delivered during the ACx critical period. Furthermore, the nervous system remains somewhat sensitive to SGRI exposure even after the critical period closes (Fig. 4e).
Although GABAB receptors have an uncertain relationship to developmental disorders, sensory deprivation also influences this inhibitory signaling system (Takesian et al., 2010; Balmer and Pallas, 2015). In addition, there is evidence for selective downregulation of the postsynaptic GABAB1b subunit in adult primate somatosensory cortex following peripheral nerve injury (Mowery et al., 2013). Here, we found that postsynaptic GABAB receptor function was also profoundly reduced by a transient period of HL. Like GABAA receptor-mediated IPSP amplitude, the loss of GABAB-mediated IPSPs was prevented by SGRI treatment, and the effect of both HL and SGRI treatment was apparent long after the transient period of developmental HL (Figs. 3–5). Since neurogliaform interneurons are a known source of GABAB receptor-dependent IPSPs (Tamás et al., 2003; Oláh et al., 2007; Wozny and Williams, 2011; Chittajallu et al., 2013; Jiang et al., 2013), it is possible that synapses from this class of interneurons onto ACx layer 2/3 pyramidal neurons are weakened by developmental deprivation, similar to weakening of inhibitory synapses from fast-spiking (FS) (parvalbumin-positive) and low threshold-spiking (somatostatin-positive) interneurons (Takesian et al., 2010).
Mechanism of pharmacological action
Because SGRI increases GABA receptor activation (Fig. 3c) but is no longer present during behavioral and neural measurements, it must have exerted a sustained effect. Such a mechanism would be consistent with a broad literature demonstrating that both GABAA and GABAB receptor-dependent signaling can have a broad trophic influence during development (Owens and Kriegstein, 2002; Represa and Ben-Ari, 2005; Ben-Ari et al., 2007; Sernagor et al., 2010; Gaiarsa and Porcher, 2013; Le Magueresse and Monyer, 2013). Our core hypothesis is that normal environmental stimulation ordinarily promotes GABAergic neurotransmission and facilitates the maturation of GABAergic synapses. Indeed, enhancing environmental stimulation can hasten GABAergic synapse maturation (He et al., 2010). At the level of gene expression, GABAergic activity has been shown to increase mRNA levels of a chloride transporter that is required for hyperpolarizing IPSPs (Ganguly et al., 2001). One intracellular pathway that could mediate such an effect is the GABAA receptor-mediated release of Mg2+ from mitochondria, which stimulates both the CREB and mTOR signaling pathways (Yamanaka et al., 2018). Activation of metabotropic GABAB receptors can also induce long-lasting or permanent changes to the developing CNS. For example, GABAB receptor activation triggers secretion of BDNF, thereby inducing the addition of perisomatic GABAergic synapses (Fiorentino et al., 2009). Therefore, the ability of SGRI to induce permanent changes in the developing CNS is consistent with a trophic GABAergic signal.
Relationship between synaptic and behavioral findings
Fast ionotropic synaptic inhibition, mediated by glycine and GABAA receptors, plays a fundamental role in auditory processing (Davis and Young, 2000; Grothe, 2003; Xie et al., 2007; Wu et al., 2008). While metabotropic inhibition is less studied, GABAB IPSPs are observed in ACx neurons (Metherate and Ashe, 1994; Buonomano and Merzenich, 1998; Hefti and Smith, 2000; Cruikshank et al., 2002; Wehr and Zador, 2005; Oswald et al., 2009), and their in vivo blockade suggests a contribution to stimulus selectivity in other systems (Lee et al., 1994; Allison et al., 1996; Mann et al., 2009; Palmer et al., 2012; Craig et al., 2013). Support for a relationship between synaptic inhibition and AM encoding draws from in vivo experiments in which GABAA receptors are pharmacologically blocked with bicuculline. The observations include reduced synchronization to AM stimuli in cochlear nucleus and ACx (Backoff et al., 1999; Kurt et al., 2006). Furthermore, inhibition influences response properties, which could each have an indirect impact on AM encoding. GABAA receptor blockade results in cortical neuron responses at lower sound levels, to a broader range of sound levels, to a broader range of modulation rates, and to a broader range of stimulus durations (Chen and Jen, 2000; Wang et al., 2002, 2016; Razak and Fuzessery, 2009). Specific manipulations of FS parvalbumin-positive interneurons or low threshold spiking somatostatin-positive interneurons, suggest that FS cells provide temporally precise feedforward inhibition to auditory cortical pyramidal neurons (Hamilton et al., 2013; Li et al., 2014, 2015; Natan et al., 2017; Cai et al., 2018; Keller et al., 2018; Liu et al., 2019). Together, these observations suggest that the strong FS inhibition contributes to the temporal following ability of pyramidal neurons. Therefore, the reduction of synaptic strength at FS to pyramidal connections would be expected to have the greatest impact on AM processing. Indeed, previous observations indicate that FS-evoked inhibitory responses are significantly reduced by developmental HL (Takesian et al., 2010). Thus, it is plausible that the HL-induced reduction of GABAA and GABAB receptor-mediated inhibition could disrupt auditory processing. This idea is also consistent with the observation that blockade of ACx activity with muscimol can significantly and reversibly diminish AM depth detection (Caras and Sanes, 2017).
An alternative hypothesis that could explain our results is that stress is an intervening variable, such that handling and/or HL induces stress, thereby degrading behavioral performance. If so, then SGRI could have exerted its effect by mitigating stress. This idea is plausible because childhood HL is correlated with elevated salivary cortisol levels at awakening, suggesting that dysregulation of the hypothalamic-pituitary-adrenal axis could be induced by the secondary consequences of HL, such as fatigue due to increased vigilance (Bess et al., 2016). Furthermore, maternal separation and restraint can lead to impaired behavioral performance in gerbils (Hardy et al., 2019; Ye et al., 2019). In the present study, both the HL+saline and the HL+SGRI animals did not gain weight as quickly as the control group. Therefore, if weight is a proxy measure for stress, then SGRI must have restored behavioral performance without alleviating stress. However, to address these questions empirically, it will be necessary to obtain unambiguous measures of both stress and effort in HL-reared animals, and to subsequently selectively manipulate these variables. A related issue is whether the sleep-inducing effect of SGRI caused synapse maturation, as opposed to a direct effect on ACx or thalamic inhibitory synapses. This is also a plausible hypothesis in that manipulations of sleep can influence CNS development (Miyamoto and Hensch, 2003; Peirano and Algarín, 2007; Frank, 2015; Del Rio-Bermudez and Blumberg, 2018). However, the literature showing that GABAergic signaling can have a direct effect on intracellular signaling and gene expression, discussed above, suggests that SGRI likely had a direct effect on GABAergic synapse maturation.
Several studies have asked whether behavioral deficits can be ameliorated by manipulating GABA receptor activity. For example, mutation of an autism-associated sodium channel subunit (SCN1A) reduces GABAergic interneuron spiking and disrupts social interactions, and treating Scn1a+/− mice with a benzodiazepine restores normal social behaviors (Han et al., 2012). In the auditory system, a drug that elicits tinnitus (sodium salicylate) impairs cochlear function yet increases sound-evoked ACx activity and acoustic startle responses. ACx activity and startle are each normalized by a GABAB agonist, baclofen (Lu et al., 2011), suggesting that rescuing GABAergic inhibition can ameliorate behavioral deficits. Our results in MGv suggest a more global effect of SGRI treatment. Therefore, we suggest that the SGRI treatment mitigated the behavioral deficits that attend developmental HL, at least in part, by preserving normal CNS inhibition along the auditory neuraxis.
Footnotes
The work was supported by National Institute on Deafness and Other Communication Disorders R01DC011284 to D.H.S. and T.M.M.
The authors declare no competing financial interests.
- Correspondence should be addressed to Todd M. Mowery at tm106{at}nyu.edu





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}