

Abstract
N-methyl-D-aspartate receptor (NMDAR) hypofunction has been implicated in several neurodevelopmental disorders. NMDAR function can be augmented by positive allosteric modulators, including endogenous compounds, such as cholesterol and neurosteroid pregnenolone sulfate (PES). Here we report that PES accesses the receptor via the membrane, and its binding site is different from that of cholesterol. Alanine mutagenesis has identified residues that disrupt the steroid potentiating effect at the rat GluN1 (G638; I642) and GluN2B (W559; M562; Y823; M824) subunit. Molecular dynamics simulation indicates that, in the absence of PES, the GluN2B M1 helix residue W559 interacts with the M4 helix residue M824. In the presence of PES, the M1 and M4 helices of agonist-activated receptor rearrange, forming a tighter interaction with the GluN1 M3 helix residues G638 and I642. This stabilizes the open-state position of the GluN1 M3 helices. Together, our data identify a likely binding site for the NMDAR-positive allosteric modulator PES and describe a novel molecular mechanism by which NMDAR activity can be augmented.
SIGNIFICANCE STATEMENT There is considerable interest in drugs that enhance NMDAR function and could compensate for receptor hypofunction associated with certain neuropsychiatric disorders. Positive allosteric modulators of NMDARs include an endogenous neurosteroid pregnenolone sulfate (PES), but the binding site of PES on the NMDAR and the molecular mechanism of potentiation are unknown. We use patch-clamp electrophysiology in combination with mutagenesis and in silico modeling to describe the interaction of PES with the NMDAR. Our data indicate that PES binds to the transmembrane domain of the receptor at a discrete group of residues at the GluN2B membrane helices M1 and M4 and the GluN1 helix M3, and that PES potentiates NMDAR function by stabilizing the open-state position of the GluN1 M3 helices.
Introduction
The N-methyl-D-aspartate receptor (NMDAR) is a heteromeric ligand-gated ion channel permeable to Ca2+. During synaptic transmission, postsynaptic NMDARs are transiently activated by released glutamate, giving rise to the NMDAR component of the EPSC. Virtually all CNS circuits use NMDAR EPSCs to regulate physiological functions; in addition, NMDAR activity has been implicated in various forms of synaptic plasticity (Dingledine et al., 1999; Lynch, 2004; Traynelis et al., 2010; Huganir and Nicoll, 2013). Recent studies indicate that dysregulation of NMDARs is involved in the pathophysiology of several neurologic and psychiatric disorders. Hypofunction of NMDARs induced pharmacologically by acute administration of ion channel blockers, such as ketamine and phencyclidine, produces psychotic symptoms and cognitive deficits (Luby et al., 1959) mimicking schizophrenia in humans (Tamminga, 1998; Coyle, 2006). In addition, de novo mutations in genes encoding NMDAR subunits have been implicated in the etiology of autism, intellectual disability, epilepsy, and schizophrenia (Burnashev and Szepetowski, 2015; Yuan et al., 2015; Hu et al., 2016; Swanger et al., 2016; Platzer et al., 2017; Fedele et al., 2018; Vyklicky et al., 2018).
There is considerable interest in discovering drugs that augment NMDAR function and could be used to treat neuropsychiatric diseases associated with receptor hypofunction. Among powerful allosteric modulators of NMDARs are natural substances, including neuroactive steroids and newly synthesized steroid-like compounds (Krausova et al., 2018). Neurosteroids represent a broad class of compounds playing multiple biological roles both during development and in various physiological states (Dubrovsky, 2005). In addition, metabolomic studies found differences in the steroid spectrum in control and schizophrenia patients with a predictive model that stated the diagnosis with a high sensitivity (Bicikova et al., 2013).
Despite structural similarities among endogenous neurosteroids affecting NMDAR activity, they have diverse effects apparently mediated by distinct molecular mechanisms of action. Positive allosteric modulators of NMDARs include pregnenolone sulfate (PES; 20-oxo-pregn-5-en-3β-yl sulfate) (Wu et al., 1991), oxysterols (Paul et al., 2013), and cholesterol (Korinek et al., 2015), whereas pregnanolone sulfate (PAS; 20-oxo-5β-pregnan-3α-yl sulfate) is a negative allosteric modulator (Park-Chung et al., 1994). Administration of neurosteroids in rodents has biological consequences: PES enhances memory and reverses memory impairment induced by NMDAR blockers (Flood et al., 1992; Mathis et al., 1994); 24(S)-hydroxycholesterol (24(S)-HC) derivatives restore behavioral and cognitive deficits in rodents treated with NMDAR ion channel blockers (Paul et al., 2013); and structural analogs of PAS have neuroprotective effects surprisingly devoid of psychomimetic symptoms typical for other classes of NMDAR inhibitors (Rambousek et al., 2011). Given the potential of PES analogs in the treatment of neuropsychiatric disorders, the molecular mechanism by which they exert their effect at the NMDAR is of considerable interest.
We used patch-clamp electrophysiology in combination with mutagenesis and in silico modeling to improve our understanding of the molecular mechanism by which NMDAR function may be enhanced. Our data suggest that PES binds to the transmembrane domain (TMD) of the receptor at a discrete group of residues at the GluN2B membrane helices M1 and M4, and at the GluN1 M3 helix. This site is distinct from sites described for other steroids and may be a potential target for the development of therapeutics.
Materials and Methods
Molecular biology and transfection of cells
Human embryonic kidney-293T (HEK) cells were transfected with cDNAs encoding rat NMDAR subunits: GluN1-1a (GluN1; GenBank accession number U08261) (Hollmann et al., 1993); GluN1-4a (GenBank accession number U08267) and GluN1-4a(F484A), generous gifts from J. J. Woodward (Smothers and Woodward, 2009); GluN2B (GenBank accession number M91562) (Monyer et al., 1992); GluN3A-eGFP (GluN3A) (GenBank accession number NM_138546), generous gift from S. A. Lipton (Sucher et al., 1995); and for the GFP in pQBI 25 vector (Takara). HEK cells were transfected using Matra-A reagent (IBA) as described previously (Borovska et al., 2012). Mutations in the M1-M4 regions of both GluN subunits were made using QuikChange site-directed mutagenesis kit (Agilent Technologies) according to the manufacturer's instructions with primers manually designed and purchased from Sigma Millipore. The amino acids are numbered on the basis of the full-length protein, with the initiating methionine marked as 1. Mutated cDNA was transformed into ultra-competent XL1Gold Escherichia coli cells, multiplied, positive clones were selected, and isolated cDNA was verified by sequencing (GATC Biotech). Transfected HEK cells were identified by GFP epifluorescence.
Electrophysiology
Electrophysiological experiments on cultured HEK cells transfected with cDNA encoding NMDAR subunits were performed 24-48 h after the end of transfection. Whole-cell currents from HEK cells were recorded in the voltage-clamp mode using a patch-clamp amplifier (Axopatch 200B; Molecular Devices) after the compensation of capacitance and series resistance (<10 mΩ) by 80%–90%. Data were collected (sampled at 10 kHz and low-pass filtered at 2 kHz) and analyzed using pClamp 10 software (Molecular Devices). Electrophysiological experiments were made at room temperature (21°C–25°C). Borosilicate glass pipettes (3-5 mΩ) were filled with intracellular solution (ICS) containing the following (in mm): 120 gluconic acid δ lactone, 15 CsCl, 10 HEPES, 10 BAPTA, 1 CaCl2, 3 MgCl2, and 2 ATP-Mg salt, pH adjusted to 7.2 with CsOH. Extracellular solution (ECS) contained the following (in mm): 160 NaCl, 2.5 KCl, 10 glucose, 10 HEPES, 0.7 CaCl2, and 0.2 EDTA, pH adjusted to 7.3 with NaOH. Glycine (10 μm), a coagonist of NMDARs, was present in all control and test solutions, except when noted. Steroids were dissolved in DMSO and added to the ECS at the indicated concentrations, with the final DMSO concentration of 1%. An equivalent amount of DMSO (1%) was present in all tested and control solutions. The final dilution of steroids in the ECS was made at 50°C, and the solution was sonicated for 1 min (Sonorex Digitec DT 100/H, Badelin Electronic). Application of solutions was made using a microprocessor-controlled multibarrel fast perfusion system. The solution exchange rate was estimated to be τ ∼ 12.0 ms for dish-attached cells (Vyklicky et al., 2016). Unless otherwise stated, all compounds were purchased from Sigma Millipore.
Cholesterol depletion
HEK cells were bathed in a culture medium (Opti-MEM; 1% FBS, both from Invitrogen), 20 mm MgCl2, 3 mm kynurenic acid, and 1 mm DL-2-amino-5-phosphonovaleric acid, supplemented with 10 mm methyl-β-cyclodextrin (βCDX; Sigma Millipore) at 37°C for 60 min. Assessment of cholesterol content using liquid chromatography/mass spectrometry showed 50%-60% depletion.
Analysis of the steroid effect
The degree of PES modulation (E; potentiation or inhibition) was determined using the following formula:
 where Ie is the value of the current amplitude during glutamate and steroid coapplication and Ia is the current amplitude value for glutamate application.
where Ie is the value of the current amplitude during glutamate and steroid coapplication and Ia is the current amplitude value for glutamate application.
The relative degree of steroid-induced potentiation (E) determined for different steroid doses in individual HEK cells was fit to the following equation:
 where Emax is the maximal value of the potentiation, EC50 is the concentration of the steroid that produces half-maximal potentiation of glutamate-induced current, [PES] is steroid concentration, and h is the apparent Hill coefficient.
where Emax is the maximal value of the potentiation, EC50 is the concentration of the steroid that produces half-maximal potentiation of glutamate-induced current, [PES] is steroid concentration, and h is the apparent Hill coefficient.
The EC50 value for glutamate was calculated from a single dose using the following formula:
 where [Glu] is the concentration of glutamate used, Imax is peak current response to the saturating concentration of glutamate (1 mm), I[Glu] is the current response to the corresponding glutamate concentration, and h is the apparent Hill coefficient that was fixed to a value of 1.6 (Laube et al., 1997).
where [Glu] is the concentration of glutamate used, Imax is peak current response to the saturating concentration of glutamate (1 mm), I[Glu] is the current response to the corresponding glutamate concentration, and h is the apparent Hill coefficient that was fixed to a value of 1.6 (Laube et al., 1997).
Experimental design and statistical analysis
Data are presented as mean ± SEM; statistical analysis was performed using paired or unpaired Student's t test for comparisons of two groups and one-way ANOVA for multiple comparisons (p < 0.050 was used to determine the level of significance).
Steroid synthesis
Pregnenolone sulfate (Pyridinium salt) was synthesized as described previously (Stastna et al., 2009). 5α-Androstan-3β-yl-sulfate (Pyridinium salt; ANDS): To a mixture of commercially available 5α-androstan-3β-ol (205 mg, 0.72 mmol) and sulfur trioxide-pyridine complex (480 mg, 1.51 mmol), dried under reduced pressure (30 min, 25°C, 100 Pa), freshly dried chloroform (7.5 ml), and dried pyridine (three drops) were added, and the reaction mixture was stirred under inert atmosphere at room temperature for 4 h. The reaction mixture was then cooled to −5°C for 18 h and then filtered through cotton wool. The filtrate was evaporated under reduced pressure, and the residue was dried for 1 h (25°C, 100 mbar). The residue was reslurried in freshly dried chloroform (minimum volume) and cooled to −5°C for 2 h. The solids were filtered, the filtrate evaporated under reduced pressure, and dried (1 h, 25°C, 100 mbar). Compound ANDS (115 mg, 48%) was obtained as amorphous solids: [α]D20 −1.9 (c 0.268, CHCl3). 1H NMR (400 MHz, CDCl3): δ 0.67 (3H, s, H-18), 0.79 (1H, s, CH-19), 4.42 (1H, m, H-3), 7.95-7.98 (2H, m, H-2′ and H-4′, pyridinium), 8.44 (1H, tt, J1 = 7.8, J2 = 1.6, H-3′, pyridinium), 8.97 (2H, dt, J1 = 5.2, J2 = 1.6, H-1′ and H-5′, pyridinium). 13C NMR (101 MHz, CDCl3): δ 145.36 (C-1′ and C-5′, pyridinium), 142.84 (C-3′, pyridinium), 127.12 (C-2′ and C-4′, pyridinium), 79.11 (C-3), 54.74, 45.11, 40.61, 39.06, 37.29, 36.05, 35.36, 32.60, 28.89, 28.85, 25.68, 21.41, 20.67, 17.71, 12.43. IR spectrum (CHCl3): 1260, 1178, 1050, 970 (OSO3). MS (EI) m/z: 355.5 (100%, M-pyridinium). HR-MS (ESI) m/z: For C19H31O4S calcd: 355.19 485; found: 355.19 509. The structure of synthesized steroids was confirmed by IR, NMR, MS, and HR-MS spectra.
5-(((3S,5S,10S,13S)−10,13-dimethylhexadecahydro-1H-cyclopenta[a]phenanthren-3-yl)oxy)−5-oxopentanoic acid (AND-hGlut; 5α-androstan-3β-yl hemiglutarate):

In a round-bottom flask, 3α5β-androstane (200 mg, 0.72 mmol, 1.0 eq), DMAP (88 mg, 0.72 mmol, 1.0 eq), and glutaric anhydride (578 mg, 5.06 mmol, 7.0 eq) were placed and dried in a thermostat at 50°C for 4 h. Under argon atmosphere, dry pyridine (15 ml) was added to the solids and the mixture was heated at 105°C for 21 h. After cooling to room temperature, the reaction mixture was poured into H2O (50 ml). The aqueous phase was extracted with CHCl3 (4 × 50 ml). The combined chloroform phases were washed with brine (50 ml), dried over Na2SO4, and concentrated in vacuo. The residue was purified by column chromatography (silica gel, CH2Cl2/acetone [10:1-8:1]) followed by preparative HPLC (CHCl3/acetone, 90:10) to afford the title compound 5α-androstan-3β-yl hemiglutarate as white amorphous solid (131 mg, 0.34 mmol, 47%). LC-MS purity = 95%. Mp 83.1°C-85.2°C (EtOAc/heptane (1:2)), [α]D20 −7.8 (c 0.195, CHCl3). 1H NMR (400 MHz, CDCl3): δ 0.62-0.75 (m, 2H), 0.69 (s, 3H), 0.82 (s, 3H), 0.85-1.85 (m, 22H), 1.94 (p, J = 7.3 Hz, 2H), 2.39 (dt, J = 27.6, 7.3 Hz, 4H), 4.64-4.76 (m, 1H). 13C NMR (101 MHz, CDCl3): δ 178.79, 172.58, 74.06, 54.64, 54.62, 44.83, 40.98, 40.56, 39.00, 36.97, 35.99, 35.74, 34.20, 33.74, 33.08, 32.50, 28.78, 27.66, 25.66, 21.37, 20.64, 20.08, 17.68, 12.41. IR spectrum (CHCl3): 3517 (OH), 3114 (NH), 2948, 2936, 2871, 2849 (CH2), 1725 (ester C = O), 1713 (C = O), 1416, 1268 (acid C-O), 1189 (ester C-O). MS (negative-ESI): m/z 390.3 (26%, M); 389.3 (100%, M-H). HR-MS (negative ESI) m/z: For C24H37O4 [M-H] calcd, 389.2697; found, 389.2695. Elemental analysis: theoretical: C = 73.81, H = 9.81; found: C = 73.80, H = 9.68.
Light scattering analysis
Light scattering (Zetasizer Nano ZS, Malvern Instruments) was used to characterize particle size of neurosteroids in ECS. PES was dissolved in DMSO at a concentration of 20 mm and then in the ECS at a final concentration. This solution was sonicated (Ultrasonic Sonorex Super RK 103 H, Bandelin Electronic) at 50°C for 1 min. Light scattering analysis was repeated 3 times for each sample at 25°C.
Computations
Computational methods
The solvation free energy (ΔGsolv) of steroids was calculated as described previously (Vyklicky et al., 2015).
Homology modeling
As described previously (Ladislav et al., 2018), the all-atom model of the rat GluN1/GluN2B receptor containing the extracellular and the TMD parts (GluN1, uniprot P35439, residues 23-847; GluN2B, uniprot Q00960, residues 30-852) (UniProt Consortium, 2017) was built with MODELLER version 9.14 (Webb and Sali, 2014) using the available crystal structures (4pe5, 4tll, 4tlm) as templates (Karakas and Furukawa, 2014; Lee et al., 2014). The residues missing in the template were further refined using the loopmodel function.
Modeling of ion channel open and closed states
Models of the GluN1/GluN2B receptor in open and closed states were obtained from a molecular dynamics (MD) simulation of the homology model with and without glycine and glutamate bound within the ligand-binding domains. The parameters of implicit solvation/lipid membrane model (EEF1/IMM1) (Lazaridis, 2003) were assigned using a web-based graphical user interface CHARMM-GUI (Jo et al., 2008). The MD simulation was then performed using the CHARMM version c41b1 MD package (Brooks et al., 2009). For the liganded open state simulation, we have introduced distance restraints between glycine and the GluN1 (R524) residue and glutamate and the GluN2B (R519) residue to compensate for their relatively low affinity. The geometry was collected every 10 ps for a total simulation time of 150 ns (Cerny et al., 2019).
MD of steroids in the membrane
A PES steroid molecule was positioned to a selected depth and orientation to the preequilibrated lipid bilayer built of 72 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) molecules, replacing one of the DOPC molecules. The atomic charges of the steroid were obtained by a standard quantum chemical procedure by HF/6-31G* electrostatic potential fit at the B3LYP/6-31G* gradient optimized geometry. The General Amber Force Field and General Amber Force Field lipid force fields were used for the steroid and lipid bilayer, and the system was then placed inside a 48 × 50 × 98 Å box, hydrated using a TIP3P water model, and Na+ and Cl– ions to reach a neutral state and a 100 mm salt concentration. The starting positions covered the whole outer leaflet of the membrane by changing the position of the steroid C17 atom 5 times with 5 Å increment for both the “upward” and “downward” orientation of the steroid sulfate group. A series of 50 ns simulations was obtained using the Amber 14 package. The trajectories were combined and for each steroid the resulting 750 ns trajectory was further analyzed.
Steroid interaction with the NMDAR by MD simulation
Docking of PES to the TMD of GluN1/GluN2B receptor models in open and closed states was performed using the AutoDock Vina program. The steroid was allowed to sample docking poses in the space containing the whole TMD part of the receptor. For more efficient sampling, this space was split into eight overlapping boxes of 32 Å × 32 Å × 32 Å. In total, we performed 200 separate Vina runs, which provided 2000 docked poses for each combination of PES and NMDAR state.
We used the preferential steroid interaction sites obtained from the docking analysis for the initial orientation of PES around the TMD of models of open and closed state of the GluN1/GluN2B receptor, forming a steroid ring around the extracellular portion of the TMD part of receptor. The system was completed using CHARMM-GUI by being inserted into a membrane containing DOPC and cholesterol (85/20). The simulation box (98 Å × 98 Å × 123 Å) was filled with TIP3P water and a charge-neutral system with a final salt concentration of 100 mm created adding Na+ and Cl– ions. The simulations were performed using Gromacs 5 with the geometry collected every 10 ps for a total simulation time of 100 ns.
Calculation of the free energy of binding
We have used snapshots stored every 10 ps from the MD simulations of PES and PAS in the model membrane for further calculation of the free energy (ΔG) of interaction. The 100 ns MD simulations were performed with the open or closed state of the TMD in the DOPC membrane, and in the presence of the steroids. The combined system showed that the TMD structure did not deviate significantly from the initial geometry representing the equilibrium geometry of the full-length NMDAR in the open or closed state. Further, the observed changes in the steroid/TMD interaction were mostly local, involving changes of rotameric states of interacting residues. Taking the preequilibrated components and analyzing the trajectory where the components behave similarly to their more complete description was used for the assessment of relative binding energies. Theoretical binding affinity was calculated from the MD trajectories using the Molecular Mechanics Poisson Boltzmann Surface Area (MMPBSA) and Molecular Mechanics Generalized Born Surface Area methods as implemented in the MMPBSA.py script from AmberTools14. The MMPBSA analysis of the Gromacs all-atom MD simulations of the steroid and NMDAR TMD in the DOPC membrane was performed using the g_mmpbsa program.
Results
PES acts at the membrane domain of the NMDAR
Figure 1A shows the dose–response analysis of the PES effect on the responses of HEK cells transfected with cDNA encoding rat GluN1 and GluN2B subunits to 1 μm glutamate, recorded in the presence of 10 μm glycine and no added Mg2+ (efficacy (Emax) = 120 ± 16%; apparent affinity (EC50) = 21 ± 3 μm; h = 1.5 ± 0.2; n = 7). PES in the absence of any agonist or in the presence of only glycine or glutamate does not induce membrane currents (Fig. 1A). The amphipathic character of the PES molecule together with the computational values of its physicochemical properties predict a propensity of this steroid to form micelles in aqueous solutions (ΔGsolv transfer from vacuum to water –21.10 kcal/mol [neutral] and –75.45 kcal/mol [charged], ΔGsolv transfer from n-octanol to water 1.76 kcal/mol [neutral] and –5.67 kcal/mol [charged], with negative values of ΔGsolv signifying free energy gained, and positive values signifying free energy required during the transfer from the first phase to the second phase; logP = 2.02 and logD = 1.26). Consistent with the prediction, the results of light scattering analysis of extracellular solution containing 0.1, 1, 10, or 100 μm PES showed particle sizes of ∼40 and 230 nm. The distribution of both particle sizes changed with changing steroid concentration, with a tendency for an increase in the density of larger particles at higher steroid concentrations and with a delay between solution preparation and its analysis (up to 6 h). We suppose that larger particles represent an agglomeration complex of small particles. These data suggest that the critical micelle concentration (CMC) for PES is < 100 nm.
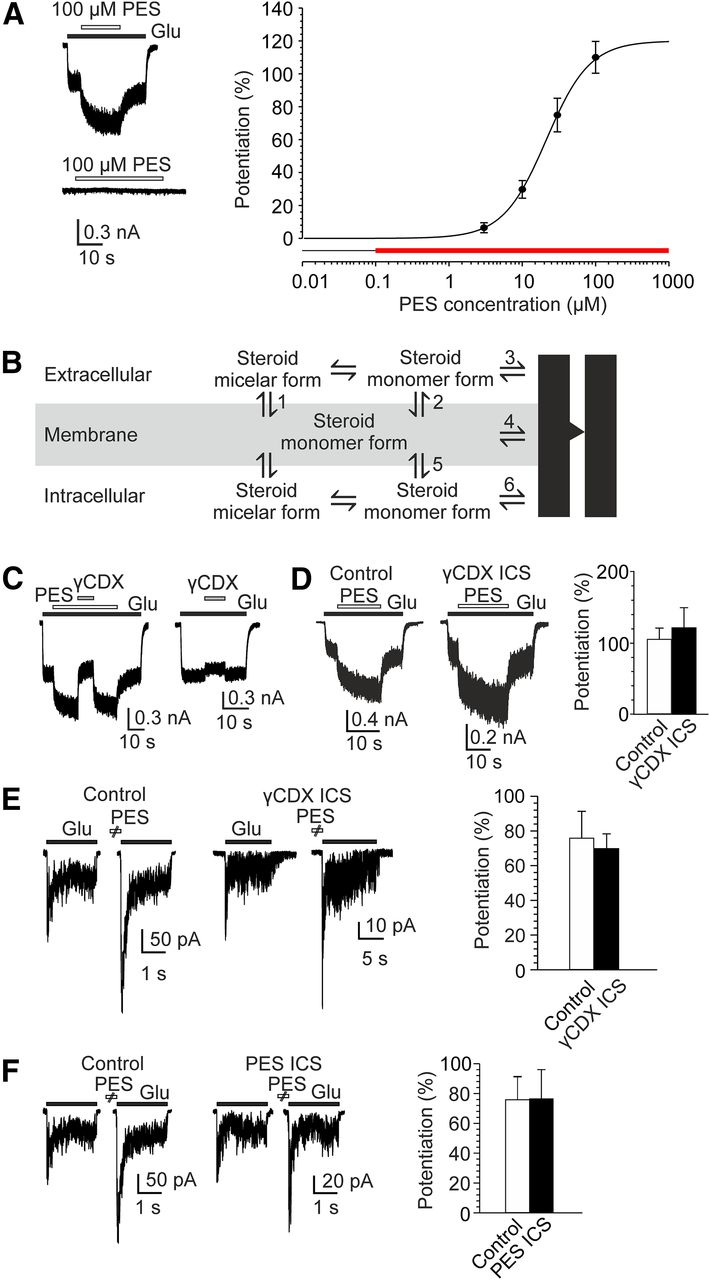
Membrane-limited positive allosteric effect of PES on the NMDAR. A, Example trace obtained from an HEK cell expressing GluN1/GluN2B receptors. PES (100 μm) was coapplied with 1 μm glutamate (above) and in the absence of glutamate (below). Open and filled bars represent duration of steroid and glutamate application, respectively. Concentration-response curve for PES (3-100 μm). Data points are average values of potentiation from 7 cells. Red bar represents the concentration range at which PES is present in the extracellular solution in the micelle form. B, Scheme illustrating routes considered for steroid access to the sites of action at the NMDAR. C, Extracellularly applied γCDX reversibly eliminates PES-induced NMDAR potentiation. Example trace obtained from an HEK cell expressing GluN1/GluN2B receptors. PES (100 μm) was coapplied with 1 μm glutamate as indicated. γCDX (10 mm) was coapplied during the glutamate response made in the presence of PES (gray filled bar). Right, The effect of 10 mm γCDX on currents induced by 1 μm glutamate. D, Comparison of the effect of PES (100 μm) on responses induced by 1 μm glutamate and recorded in control ICS or in ICS containing 10 mm γCDX. Graph represents the results from 4 cells (control ICS) and 5 cells (ICS containing γCDX). No significant differences were observed (p = 0.666, unpaired t test). E, Examples of traces obtained from outside-out patches isolated from HEK cells expressing GluN1/GuN2B receptors. Responses to glutamate (1 mm) and glycine (100 μm), with or without PES (100 μm) preapplication for 10 s are shown. Control ICS (left) or ICS containing 100 μm PES (right) were used. Graph represents the results from 11 (control ICS) and 8 (ICS containing PES) patches. No significant differences were observed (p = 0.977, unpaired t test). F, Examples of traces obtained from outside-out patches isolated from HEK cells expressing GluN1/GuN2B receptors. Responses to glutamate (1 mm) and glycine (100 μm), with or without PES (100 μm) preapplication for 10 s are shown. Control ICS (left) or ICS containing 10 mm γCDX (right) were used. Graph represents the results from 11 (control ICS) and 6 (ICS containing γCDX) patches. No significant differences were observed (p = 0.782, unpaired t test). Data are mean ± SEM.
By definition, at doses lower than the CMC, the monomer concentration of the steroid increases with the steroid added; however, at doses greater than the CMC, micelles form and all additional steroid molecules added to the system go into micelles (McNaught and Wilkinson, 1997). Since the free steroid concentration is constant at doses relevant for the potentiation of GluN1/GluN2B receptor responses, the action of PES is not typical of simple drug-receptor interaction in aqueous solution; therefore, we have considered other mechanisms: (1) steroid in the micelle form dissolves in the membrane from where it directly interacts with the membrane domain of the receptor (Steps 1 and 4 in the scheme in Fig. 1B); and (2) steroid in the micelle form dissolves in the plasma membrane from where it exits in the single-molecule form to bind to the extracellular or the intracellular domain of the receptor (Steps 1-3 described for steroids with inhibitory action on the NMDAR, Vyklicky et al., 2015; or Steps 1, 5, and 6 in Fig. 1B).
Next, we explored the route of steroid access to the receptor in more detail using cyclodextrins, cyclic sugar molecules that can accommodate lipophilic compounds, such as steroids, and manipulate their free content in the membrane (Yancey et al., 1996; Szejtli, 1998; Shu et al., 2004). Figure 1C shows that responses of GluN1/GluN2B receptors to glutamate (1 μm) were potentiated in the presence of PES (100 μm), on average, by 103.0 ± 10.5% (n = 6). Following methyl-γ-cyclodextrin (γCDX; 10 mm) application in the continuous presence of the steroid and glutamate, the potentiation was abolished and the amplitude of the responses was smaller than of control glutamate responses (on average by −7.7 ± 2.2%; n = 7; γCDX significantly reduced the potentiation, p < 0.001, paired t test). The amplitude of the responses recorded in the presence of γCDX and the steroid was similar to glutamate responses recorded in the presence of only γCDX (−12.8 ± 2.0%; n = 9) (unpaired t test p = 0.118) (Fig. 1C). The effect of γCDX assessed in the presence or absence of steroid was fully reversible. To test whether the potentiating effect of PES may be associated with site(s) located at the receptor intracellular domains, we analyzed the steroid effect in HEK cells dialyzed with γCDX. Responses of GluN1/GluN2B receptors to glutamate (1 μm) in HEK cells intracellularly dialyzed for 3-18 min with ICS containing 10 mm γCDX were potentiated by 100 μm PES to a similar extent as in HEK cells dialyzed with control ICS (Fig. 1D). Further, we analyzed the effect of intracellular PES dialysis on the steroid ability to potentiate NMDAR responses when applied extracellularly. Since PES is highly lipophilic and cell interior is rich in membranes, it may take a long time to equilibrate steroid concentration in the intracellular compartment with that in the pipette. To avoid uncertain intracellular steroid concentration, we used outside-out patch-clamp recording rather than whole-cell recording. Responses to glutamate (1 mm) in outside-out patches isolated from HEK cells expressing GluN1/GluN2B receptors and dialyzed with ICS containing 100 μm PES were potentiated following extracellular PES (100 μm) preapplication for 10 s to the same extent as in outside-out patches dialyzed with control ICS (Fig. 1E). Similar results for the effect of intracellular dialysis with PES were found for the effect of intracellular dialysis with γCDX. The degree of PES (100 μm)-induced potentiation of responses to 1 mm glutamate was similar in outside-out patches intracellularly dialyzed with control ICS and that containing 10 mm γCDX (Fig. 1F). Together, these results argue against a mechanism of steroid potentiation mediated by its interaction with the intracellular domain of the receptor and/or a membrane domain in contact with the inner membrane leaflet.
To determine whether steroids with a potentiating action act at the extracellular domain of the receptor, similarly to the inhibitory steroids (Vyklicky et al., 2015), or instead interact with receptor membrane domain in contact with the outer membrane leaflet, we made use of ANDS, a steroid that exhibited signs of dual (positive and negative) action at NMDARs in screening experiments. Responses of GluN1/GluN2B receptors to glutamate (1 mm) after ANDS (10 μm) preapplication for 15 s were potentiated by 17 ± 6% (n = 5) and exhibited complex kinetics indicating a mixed effect of the steroid (Fig. 2). In contrast, responses induced by coapplication of glutamate (1 mm) and γCDX (10 mm) after ANDS preapplication were potentiated significantly more (by 172 ± 18%; n = 5; p = 0.002, paired t test) (Fig. 2). We reasoned that γCDX abolished the inhibitory effect of ANDS at its extracellular binding site and unmasked the potentiating effect associated with steroid action at the TMD. This is presumably due to the fact that the depletion of ANDS from the outer membrane leaflet by γCDX is relatively slow. At a low steroid concentration (3 μm), the potentiating and the inhibitory effects were both markedly reduced to a similar extent, 7.6-fold and 7.0-fold, respectively (Fig. 2). Together, results shown in Figures 1 and 2 suggest that there are distinct sites for steroids mediating positive and negative allosteric effects at NMDARs and argue in favor of the steroid potentiation being mediated by its interaction with the receptor membrane domain in contact with the outer membrane leaflet.

γCDX abolishes the inhibitory effect of steroids with mixed effects at the NMDAR. Representative whole-cell responses of GluN1/GluN2B receptors to glutamate (1 mm) made after preapplication of ANDS for 15 s or to glutamate (1 mm) coapplied with γCDX (10 mm) after ANDS preapplication. ANDS was used at a concentration of 3 μm (top) or 10 μm (bottom). Right, Responses to glutamate (black) and to glutamate and γCDX (red) are overlaid to show the difference in their kinetics. Inset, Chemical structure of ANDS. The graph summarizes the potentiation (empty columns) and the inhibition (black columns) of peak NMDAR responses induced by preapplication of 3, 10, and 30 μm ANDS. Data are mean ± SEM. Potentiation was calculated according to the following formula: (c – a)/a × 100; and inhibition: (1 – (b/c)) × 100, where a is the peak response to 1 mm glutamate, b is the peak response to 1 mm glutamate after ANDS preapplication, and c is the peak response to 1 mm glutamate coapplied with γCDX after ANDS preapplication (see a-c in the raw data).
The mechanisms of PES and sterol action at the NMDAR are different
Multiple mechanisms have been reported for steroid-induced modulation of NMDARs. Steroids with a flat ring structure (pregn-5-ene and 5α-pregnane) exert positive or dual effect with the potentiating effect prevailing. Molecules with a flat-ring structure include PES (Wu et al., 1991), cholesterol (Korinek et al., 2015), and oxysterols (Paul et al., 2013). Therefore, we tested whether PES shares a common mechanism of action with sterols 24(S)-HC (a member of the oxysterol family) and cholesterol; both sterols share similar structure with an –OH residue at the C3 carbon of the ring skeleton. We hypothesized that, if PES and cholesterol exert their function at the NMDAR through the same mechanism, then PES will beable to compensate for the diminution of NMDAR responses induced by cholesterol depletion. The mean amplitude of responses to 1 μm glutamate in HEK cells expressing GluN1/GluN2B receptors was −547 ± 193 pA (n = 5), and the responses were potentiated by 119 ± 32% (n = 5) when recorded in the presence of PES (100 μm) (Fig. 3A). Following cholesterol depletion induced in sister cultures by 60 min incubation of HEK cells with βCDX (10 mm; at 37°C), the mean amplitude of current responses to 1 μm glutamate was only −48 ± 18 pA (n = 6) (Fig. 3B). This diminution agrees well with that observed previously (Korinek et al., 2015). Glutamate responses induced in βCDX-treated HEK cells were potentiated by coapplication of PES (100 μm) by 122 ± 66% (n = 6), to the same extent as in control HEK cells (p = 0.973; unpaired t test) (Fig. 3A-C). This potentiation was not sufficient to compensate for the diminution of glutamate responses in βCDX-treated HEK cells (Fig. 3D).

PES is unable to compensate for the diminution of NMDAR current responses in cholesterol-depleted cells. A, Potentiating effect of PES (100 μm) on NMDAR response to 1 μm glutamate in control HEK cells. Right, NMDAR response to 1 μm glutamate from the same control cell following a 5 min preapplication of 24(S)-HC (10 μm). B, Effect of PES (100 μm) on NMDAR response to 1 μm glutamate in HEK cells treated with βCDX (10 mm) in culture media for 60 min (cholesterol-depleted). Right, NMDAR response to 1 μm glutamate from the same cholesterol-depleted cell following a 5 min preapplication of 24(S)-HC (10 μm). C, Summary of the results from a minimum of 5 cells per bar. Potentiation of NMDAR responses to 1 μm glutamate coapplied with 100 μm PES and to 1 μm glutamate after 5 min preapplication of 10 μm 24(S)-HC recorded from control and cholesterol-depleted HEK cells. D, Summary of raw current amplitudes from the same experiments as shown in C. Glutamate-induced current responses were significantly diminished in cholesterol-depleted cells (*, unpaired t test). In cholesterol-depleted cells, the glutamate responses recorded following 24(S)-HC treatment were neither significantly changed (NS) from those recorded from control HEK cells (p = 0.667) nor from responses recorded from control HEK cells following 24(S)-HC treatment (p = 0.061).
In contrast, 24(S)-HC (10 μm) potentiated the amplitude of glutamate responses in control cells by 221 ± 32% (n = 9) and in βCDX-treated HEK cells by 1009 ± 378% (n = 5) (Fig. 3B,C). The mean amplitude of responses to 1 μm glutamate recorded in βCDX-treated HEK cells following 24(S)-HC preapplication was not significantly different from that recorded in control untreated HEK cells and that recorded from control untreated HEK cells following 24(S)-HC treatment (Fig. 3D). These results indicate that the potentiating mechanism of PES action at the NMDAR is different from the mechanism of NMDAR potentiation by sterols. In addition, since 24(S)-HC compensated for the current amplitude diminution caused by cholesterol depletion, it is likely that the modulatory effect of 24(S)-HC (Paul et al., 2013) and cholesterol (Korinek et al., 2015) is mediated by the same or similar site(s).
Steroid orientation in the membrane
Our results (Fig. 1) suggest that PES accesses the NMDAR via the outer membrane leaflet; therefore, steroid orientation in the plasma membrane is relevant for its effect on the NMDAR. In order to understand the atomic details, we designed a series of MD simulations. Overall, the studied process covers a wide range of conformations with many degrees of freedom. We addressed this highly complex task by performing and combining the results of MD simulations with varying the starting position and the orientation of the steroid moiety with respect to the lipid bilayer, collecting a cumulative trajectory of 750 ns for each steroid. Data show that PES quickly reorients in the membrane to point with the sulfate group in the direction of the phosphates of the corresponding membrane leaflet. Snapshots of the trajectory taken every 10 ps were used for further calculation of the free energy(ΔG) of interaction. We used Molecular Mechanics Generalized Born Surface Area and MMPBSA (see Materials and Methods) to calculate the interaction energy. Both methods provided qualitatively similar energy profiles differing in the absolute value of the interaction energy (as a result of different terms involved in the energy calculation). Energy profiles for PES show a high-affinity interaction in the lower half of the respective leaflet (Fig. 4A). The overall orientation is perpendicular to the membrane plane without significant tilting. We have observed steroid leaving and entering the membrane in a few cases. Steroid transfer between inner and outer leaflets was observed occasionally with similar energy profiles on both sides. MD simulation was also used to analyze membrane orientation for PAS, a steroid with inhibitory effect at the NMDAR (Park-Chung et al., 1994). The data show that PAS, similarly to PES, quickly reorients in the membrane to point with the sulfate group in the direction of the phosphates; however, in contrast to PES, the preferred orientation of PAS is parallel to the membrane plane with the C17 located just below the lipid phosphates, where it reaches a similar ΔG as that estimated for PES with the C17 approaching the middle of the membrane (Fig. 4A,B). PAS was also observed in an orientation perpendicular to the membrane plane; however, this corresponds to a less stable local minimum with lower occurrence. Because androstene derivatives (lacking the C17-bound acetyl moiety) are even more potent positive allosteric modulators than PES (Krausova et al., 2018), it is reasonable to speculate that the position of the sulfate moiety and the C17 of the steroid in the membrane is critical for the potentiating effect of the steroid and outlines residues at the NMDAR TMD that may be part of the binding site.

In silico analysis of neurosteroid molecules in the membrane. A, Left, Profiles of MMPBSA-derived free energy of PES (red) and PAS (blue) within a model membrane. The value on the x axis indicates the predicted ΔG in kcal/mol, where a more negative value corresponds to a higher binding affinity and a more probable steroid position. The y axis scale shows the relative position of the C17 atom of the steroid molecule with respect to membrane thickness. The values of 0 and 1 are defined to overlay with average coordinates of phosphate groups in each membrane leaflet. The values between 0.5 and 1 cover positions within the outer leaflet of the membrane, while the positions between 0 and 0.5 belong to the inner leaflet. Relative positions of Cα atoms of selected amino-acid residues from the membrane-facing edge of the GluN2B M4 helix are shown for reference. Right, Representative positions of PES (red) and PAS (blue) in the model membrane corresponding to the minima in the free energy of binding, extracted from 750 ns MD simulations (shown as sticks). B, Snapshot of the TMD of the GluN1 (gray)/GluN2B (orange) homology model (Ladislav et al., 2018) as a diagram representation in the model membrane (light gray lines) from a 100 ns MD simulation. Sticks represent GluN2B M4 helix residues.
Amino-acid residues influencing the modulatory effect of PES on the NMDAR
To identify the neurosteroid binding site, we sequentially replaced amino-acid residues at the outer segment of the TMD helices M1 and M4 of both the GluN1 and the GluN2B subunits. Single alanine substitution mutations included thefollowing: GluN1 (Q559A – V572A; F810A – V825A) and GluN2B (S555A – I568A; D814A – A830T). HEK cells expressing mutated receptors responded to glutamate application, with the exception of GluN1 (W563A)/GluN2B and GluN1/GluN2B (D816A) receptors, which were spontaneously active; and GluN1/GluN2B (W559A) receptors, which responded to glutamate with minimal currents (<5 pA). The effect of PES (100 μm) was tested during steroid coapplication with glutamate (1 μm) (Fig. 5A). WT receptor responses were potentiated by PES (104 ± 4%; n = 158). The effect of PES varied considerably in mutated receptors, ranging from inhibition (–33 ± 2%; n = 7) for GluN1/GluN2B (M824A) receptors to potentiation (292 ± 57%; n = 7) for GluN1 (G815A)/GluN2B receptors (Fig. 5A-C). The effect of PES at GluN1 (Q559A; W563A; L564A; N812A; F817A)/GluN2B and GluN1/GluN2B (M562A; V564A; L567A; M818A; G820A; F822A; L825A; M829A; A830T) receptors was significantly reduced compared with WT and PES inhibited responses induced in GluN1/GluN2B (D816A; Y823A; M824A) receptors (Fig. 5B,C). In contrast, PES potentiated GluN1 (S569A; G815A; M818A; L819A; G822A)/GluN2B receptor responses significantly more compared with WT (Fig. 5B). Transition from the steroid-induced potentiating effect to the inhibitory effect is not surprising given that PES has a dual action at the NMDAR: potentiating and inhibitory (Horak et al., 2004; Horak et al., 2006). It is expected that, if the potentiating effect is abolished, then the inhibitory effect will be unmasked.

Mutations in the M1 or M4 membrane domains of the GluN1 and GluN2B subunits affect PES potentiation. A, Representative recordings from WT, GluN1 (G815A)/GluN2B, and GluN1/GluN2B (M824A) receptors showing responses to glutamate (1 μm) before and in the presence of PES (100 μm). Open and filled bars represent duration of PES and glutamate application, respectively. B, C, Bar graph of the mean effect of PES (100 μm) ± SEM (+, potentiation; –, inhibition) on responses to 1 μm glutamate induced in GluN1/GluN2B receptors mutated in the M1 or M4 domains of GluN1 (B) and in the M1 or M4 domains of GluN2B (C) (n = 4-158 cells). Dashed line indicates the mean WT response. a, Spontaneously active receptors; b, nonresponding receptors. ‡Receptors had altered sensitivity to PES (one-way ANOVA, p < 0.001, followed by an unpaired t test for single comparisons vs WT, p < 0.050, and a significantly altered EC50 for glutamate; Table 1). *Receptors with altered sensitivity to PES (one-way ANOVA, p < 0.001, followed by an unpaired t test for single comparisons vs WT, p < 0.050) that had a similar EC50 for glutamate compared with WT (Table 1). D, E, Evaluation of the PES effect at mutated GluN1/GluN2B receptors with an altered EC50 for glutamate (B,C, ‡). Bar graph of the mean effect of PES (100 μm) ± SEM (+, potentiation; –, inhibition) on responses induced in mutated receptors by the glutamate concentration (0.08-3.20 μm) that induces ∼40% maximal response (n = 4-10 cells). *Altered PES effect at mutated receptors compared with WT (one-way ANOVA, p < 0.001, followed by an unpaired t test for single comparisons vs WT; p < 0.050). B-E, Red and green labeled bars represent mutations that changed the PES effect by >50% compared with WT. B, Inset, Side view of the TMD of the receptor. Gray represents GluN1; orange represents GluN2B (Cerny et al., 2019). Green represents amino-acid residues whose mutations increased the potentiating effect of PES by >50%. Red represents those that decreased the potentiating effect of PES by >50%, including those associated with a net inhibitory effect of PES.
Summary of the EC50 values for glutamate for WT and mutated GluN1/GluN2B receptors with an altered PES effect
The degree of the positive allosteric effect of PES on NMDARs is influenced by the glutamate dose (it is disuse-dependent) and involves a mechanism that increases the NMDAR ion channel open probability (Po) (Horak et al., 2004). Figure 5D, E shows results of experiments aimed to exclude indirect changes in the PES effect mediated by mutation-induced altered receptor affinity for glutamate. For mutations where the PES effect was significantly decreased or increased compared with WT receptors, glutamate EC50 was determined (Table 1). We found that receptors with mutated GluN1 (W563A; S569A; N812A; G815A; M818A; L819A) or GluN2B (M562A; L567A; D816A; M824A; L825A) subunits had a significantly altered EC50 for glutamate (Table 1). Therefore, the effect of PES (100 μm) on these receptors was reanalyzed at glutamate concentrations corresponding to the concentration used for testing WT receptors (∼40% maximal activation). Under these conditions, receptors with mutated GluN1 (G815A) and GluN2B (M562A; D816A; M824A) subunits were still significantly more potentiated or inhibited by PES compared with WT (Fig. 5D,E); however, the effect of PES at receptors with mutated GluN1 (W563A; S569A; N812A; M818A; L819A) and GluN2B (L567A; L825A) subunits was similar to that estimated for WT (Fig. 5D,E).
Next, we used GluN1/GluN2B (M824A) receptors to investigate whether the impact of mutations on the potentiating effect of PES will cover also steroids with a different structure. We selected androst-5-en-3α-yl hemisuccinate (Krausova et al., 2018) and 5α-androstan-3β-yl hemiglutarate (see Materials and Methods), steroids that potentiate NMDARs, however, which differ from PES in the structure and/or substituents at the A, B, and D rings of the steroid molecule. Androst-5-en-3α-yl hemisuccinate (30 μm) strongly potentiated responses of GluN1/GluN2B receptors to glutamate (1 μm) by 452 ± 46% (n = 5), whereas the responses of GluN1/GluN2B (M824A) receptors were potentiated only by 51 ± 8% (n = 7). Similarly, 5α-androstan-3β-yl hemiglutarate (30 μm) strongly potentiated responses of GluN1/GluN2B receptors to glutamate (1 μm) by 210 ± 75% (n = 9), whereas the responses of GluN1/GluN2B (M824A) receptors were inhibited by −12 ± 3% (n = 8) (Fig. 6). These results suggest the same or similar site of action for PES and its structural analogs.

Effect of PES analogs on the receptor containing GluN2B (M824A) subunit. Representative recordings from WT (GluN1/GluN2B) and GluN1/GluN2B (M824A) receptors show responses to glutamate (1 μm) before and in the presence of 30 μm androst-5-en-3α-yl hemisuccinate (AND-hSuc) (A) or 30 μm 5α-androstan-3β-yl hemiglutarate (AND-hGlut) (B). Open and filled bars represent duration of steroid and glutamate application, respectively. Bar graphs of the mean effect of steroid ± SEM (+, potentiation; –, inhibition) on responses to 1 μm glutamate induced in WT and GluN1/GluN2B (M824A) receptors (n = 6-9 cells). The difference in the effect of steroid at WT and mutated receptors was significant for both steroids (unpaired t test for single comparisons vs WT, p < 0.001).
Figure 7 shows results of experiments aimed to exclude indirect changes in the PES effect mediated by mutation-induced altered receptor Po. MK801, a use-dependent and virtually irreversible inhibitor of NMDAR ion channels, was used as a tool to compare the Po of WT and mutated receptors (Huettner and Bean, 1988; Jahr, 1992). In WT receptors, the responses to a saturating concentration of glutamate (1 mm) were blocked by MK801 (1 μm) with an onset best fitted by a double-exponential function with a weighted time constant (τw) 1443 ± 110 ms (n = 65), corresponding to the Po ∼10% (Chen et al., 1999; Vyklicky et al., 2018). Mutations in the GluN1 or the GluN2B subunit decelerated or accelerated the rate of the onset of MK801 inhibition, which we interpret as a decreased or increased Po, respectively (Fig. 7). No correlation was found between the effect of PES at mutated NMDARs and the relative rate of the onset of MK801 inhibition, even when only the data points for mutated receptors showing a significantly decreased potentiation were included (Fig. 7). This indicates that mutations in the TMD of the GluN1/GluN2B receptor can impact the sensitivity of the receptor to PES and/or the ion channel Po; however, changes in these two properties are independent.

Mutations in the M1 and M4 membrane domains of the GluN1 or GluN2B subunit affect NMDAR ion channel Po. A, Representative recordings from WT, GluN1 (L562A)/GluN2B, and GluN1 (V570A)/GluN2B receptors show the response to glutamate (1 mm) and the time course of the onset of MK801 (1 μm) inhibition. Right, The onsets are overlaid to show the difference in their kinetics. B, Graph represents the relative effect of PES (100 μm; ratio of the mean effect of the steroid on mutated receptors and the effect on WT receptors, 104% potentiation; +, potentiation; –, inhibition) plotted versus the relative onset of MK801 inhibition (ratio of the mean τw determined in mutated receptors to the mean τw of the WT receptors, 1.44 s). Data were fit by a linear regression. Black line indicates all data points (Pearson's correlation coefficient r2 = 0.0045; p = 0.608). Dotted line indicates the data points where the PES effect was significantly decreased compared with WT (open symbols; Pearson's correlation coefficient r2 = 0.0266; p = 0.596).
Although GluN2B (D816A) as well asthe double mutation GluN2A(D814A and D816A) (Kostakis et al., 2011) abolished the potentiating effect of PES, it is unlikely that this residue is a component of the neurosteroid binding site. First, this residue is part of the M4-S2 linker (Fig. 5B,C) that is not in direct contact with the cell membrane, on the route for PES access to the site(s) of action located at the TMD of the NMDAR (Fig. 1). Second, steroid orientation in the membrane is such that the sulfate moiety faces the aqueous solution (Fig. 4). Under these conditions, repulsion forces between the negatively charged sulfate group of PES and the negatively charged aspartate residue GluN2B (D816) will make any contact unlikely. Third, single amino-acid substitutions in the linker region have profound effects on NMDAR function (Ladislav et al., 2018). In agreement with this, GluN1/GluN2B (D816A) receptors have a 3.8-fold higher Po than WT receptors (Fig. 7) and are spontaneously active with an altered EC50 value for glutamate (Fig. 5; Table 1). Finally, MD simulation indicates high flexibility of linkers with a high content of extended conformations (Ladislav et al., 2018). Such structure is unlikely to be part of a binding site for rigid molecules, such as steroids. Together, our results indicate that, for the PES-potentiating action at NMDARs, amino-acid residues at the interface between GluN2B M1 and M4 helices play a critical role.
Location of the neurosteroid binding site
Sequence alignment of the M1 and M4 motifs from GluN1/2 subunits shows that GluN2B (M562; V564; M818; Y823; M824; and M829) residues, whose mutations decreased the steroid potentiating effect (Fig. 5), are conserved in homologous regions of the other members of the GluN2 family (Fig. 8A); consistent with this, PES potentiates GluN1/GluN2A-D receptor responses (Horak et al., 2006). In accord with this are results of experiments in which the effect of PES was assessed at GluN1/GluN2A(M823A) receptors, responses induced in GluN1/GluN2A receptors by 3 μm glutamate (concentration corresponding to 40 ± 2%, n = 6, of maximal activation) were potentiated by 100 μm PES by 51 ± 6% (n = 6), whereas responses induced in GluN1/GluN2A(M823A) receptors by 3 μm glutamate (concentration corresponding to 46 ± 3%, n = 6, of maximal activation) were inhibited by 100 μm PES by −58 ± 4%, n = 7). In contrast, GluN2B residues that are involved in the PES effect share little homology with the GluN3A family (Fig. 8A). To test the steroid effect on GluN1/GluN3A receptors, we transfected HEK cells with genes encoding for GluN3A andthe mutated GluN1-4a(F484A) subunit to reduce receptor desensitization (Smothers and Woodward, 2009). Responses of GluN1-4a(F484A)/GluN3A receptors to 100 μm glycine were inhibited (−68.0 ± 2.6%; n = 7) in the presence of PES (50 μm) (Fig. 8B). In addition, the effect of PES was assessed in WT GluN1-4a/GluN3A receptors. Although glycine (100 μm) responses induced in GluN1-4a/GluN3A receptors desensitized profoundly, PES (50 μm) had a clear inhibitory effect (−65.9 ± 8.5%; n = 4). In control experiments, responses of GluN1-4a(F484A)/GluN2B receptors to 1 μm glutamate and 100 μm glycine were potentiated (56.4 ± 5.7%; n = 6) in the presence of PES (50 μm). These results indicate that the positive allosteric effect of PES is confined to receptors containing a GluN2 subunit and is not observed in receptors containing the GluN3A subunit. Similar to GluN3A subunits, GluA1-4 and GluK subunits share little homology with sites at the GluN2B subunit important for the PES effect (Fig. 8A). Consistent with this, results of earlier electrophysiological experiments have shown that AMPA (GluA1 and 3) and kainate (GluK2) receptors are inhibited by PES (Yaghoubi et al., 1998; Malayev et al., 2002; Horak et al., 2006).

Mutations in the M3 helix of the GluN1 and M1 helix of the GluN2B subunit affect the positive allosteric effect of PES. A, Amino-acid sequence alignment showing M1, M3, and M4 regions across the glutamate receptor subunit family. Single amino-acid residues replaced with alanines are outlined in bold and underlined. Mutations that reduced or abolished PES potentiation (red), mutations that augmented PES potentiation (green), and homologous regions (yellow) are highlighted. B, Representative recordings from GluN1-4a(F484A)/GluN3A receptors show responses to glycine (100 μm) before and in the presence of PES (50 μm). C, Ribbon structure of a homology model of the GluN1/GluN2B receptor (Karakas and Furukawa, 2014; Lee et al., 2014) viewed from the extracellular side. Gray represents GluN1. Orange represents GluN2B. Residues GluN1 (S646; I642; G638) and GluN2B (W559) (blue) are located from GluN2B (M824) residue (red) at a distance 12.0, 6.0, 9.2, and 4.6 Å, respectively. M1, M3, and M4 transmembrane helices are indicated. For comparison, a stick model of PES with transparent surface is shown at the same scale as the M1-M4 transmembrane helices. D, E, Representative recordings from the GluN1/GluN2B (W559) (C) and GluN1 (G638A)/GluN2B (E) receptors, showing responses to glutamate (1 μm) before and during the application of PES (100 μm). F, Graph of the mean effect of PES (100 μm) ± SEM (+, potentiation; –, inhibition) on GluN1/GluN2B receptors mutated in the GluN1 (M3) and the GluN2B (M1) helix (n = 5-158 cells). *Significantly diminished positive allosteric effect compared with WT (one-way ANOVA, p < 0.001; unpaired t test for single comparisons vs WT, p < 0.050). G, Amino-acid residues whose mutations reduced the PES effect by > 50% compared with WT are depicted in red in a homology model of GluN1/GluN2B (Cerny et al., 2019). Side view of the TMD of the receptor. Gray represents GluN1. Orange represents GluN2B.
To explore the neurosteroid binding mechanism further, we used the homology model of the GluN1/GluN2B receptor (Ladislav et al., 2018) based on crystal structures (4pe5, 4tll, 4tlm) (Karakas and Furukawa, 2014; Lee et al., 2014) and searched for residues that are located within ∼15 Å (the steroid length) from GluN2B (Y823 and M824), the residues, whose mutations reduced the PES potentiation most profoundly. Two sites were suggested: (1) GluN2B (W559) at the M1 helix; and (2) GluN1 (G638; I642; and/or S646) at the M3 helix (Fig. 8C). Exploring the role of GluN2B (W559) in the positive allosteric effect of PES was complicated, since GluN1/GluN2B (W559A) receptors were nonfunctional. Therefore, we used the GluN2B (W559L) mutation that has been shown earlier to form functional receptors when expressed with GluN1 (Kashiwagi et al., 1997). Responses induced in GluN1/GluN2B (W559L) receptors by 1 μm glutamate were inhibited (−15 ± 2%; n = 7) in the presence of 100 μm PES (Fig. 8D,F). This mutation exhibited a glutamate EC50 similar to that determined for WT receptors (Table 1). Next, we analyzed the effect of PES on responses induced in receptors mutated in the M3 helix of the GluN1 subunit. The potentiating effect of PES was significantly reduced in GluN1 (G638A)/GluN2B and GluN1 (I642A)/GluN2B receptors, but not significantly altered in GluN1 (S646A)/GluN2B receptors (Fig. 8E,F). Receptors with mutated GluN1 (G638A; I642A) subunits exhibited glutamate EC50 values similar to WT (Table 1). These results are compatible with an interhelical binding site for PES formed by two residues at the M4 helix of GluN2B (Y823; M824) and those in close vicinity at the M1 helix of GluN2B (W559; M562), and the M3 helix of GluN1 (G638; I642) (Fig. 8G).
PES dose–response analysis in receptors containing GluN1 (G815A; G822A) subunits shows that the augmented potentiation is a consequence of increased steroid efficacy (Emax) rather than the apparent affinity (EC50) (Table 2). This is consistent with a modulatory role of residues in the M1 and M4 helix of the GluN1 subunit that affects the degree of the steroid potentiation rather than steroid binding to the receptor. This is further supported by experiments with GluN1 (W563A)/GluN2B receptors. Since receptors with alanine substituted for tryptophan 563 were spontaneously active (Kashiwagi et al., 2002; Vyklicky et al., 2015), we used a glutamate concentration of 80 nm that induced ∼40% of the maximum response (to 1 mm glutamate), comparable to WT receptors activated by 1 μm versus 1 mm glutamate. Responses induced by 80 nm glutamate in GluN1 (W563A)/GluN2B receptors were potentiated by 100 μm PES (77 ± 12%; n = 10; not significantly different compared with WT, unpaired t test, p = 0.109) (Fig. 5D). This contrasts with a role of homologous tryptophan at the M1 helix of the GluN2B subunit (Fig. 8A) rendering GluN1/GluN2B (W559L) receptors insensitive to the PES potentiating effect (Fig. 8D,F).
Summary of PES efficacy and potency from the dose–response analysis of WT receptors and receptors mutated in the GluN1 subunit
In silico analyses of PES binding
Our previous electrophysiological data have shown that the apparent affinity of the NMDAR for PES is affected by receptor activation (Horak et al., 2004). While the unliganded or single-agonist (glutamate or glycine)-bound states allow PES binding to the NMDAR with a high affinity, receptor activation by glutamate and glycine with the ion channel either closed or open reduces the apparent affinity for PES 5- to 50-fold (Horak et al., 2004). We took advantage of a model of the GluN1/GluN2B receptor that was recently built in our laboratory. The all-atom model was built using the available crystal structures (4pe5, 4tll, 4tlm) (Karakas and Furukawa, 2014; Lee et al., 2014); subsequently, GluN1/GluN2B receptor models in unliganded (with the ion channel closed) and liganded (with the ion channel closed and/or open) states were obtained from an MD simulations (see Cerny et al., 2019) (Fig. 9). Structural superpositions of helices forming the TMD in the unliganded receptor with the ion channel closed and the liganded receptor with the ion channel open show relative changes in the orientation of the M1-M4 helices and indicate that the most prominent changes occur in the outer segment of the M4 helix (Fig. 9A,B).

Structure of the TMD of the NMDAR and PES binding. A, B, Side (A) and top (B) view showing the helical organization (M1-M4) of the TMDs and a structural superposition of the unliganded (closed state, filled symbols) model of the GluN1 (gray)/GluN2B (orange) receptor and the liganded (open state, open symbols) model of the receptor from MD simulations (Cerny et al., 2019). C, Interaction of PES (green) with the TMD of GluN1/GluN2B receptor in the closed state obtained by steroid docking followed by a 100-ns-long all-atom MD simulation. Red sticks represent the experimentally identified residues involved in receptor potentiation. Sphere represents the position of the sulfate moiety of PES (green). D, Interaction of PES with the TMD of the GluN1/GluN2B receptor in the open state. E, The model of the TMD of the NMDAR with the ion channel open simulated in the absence of steroid. The GluN2B (M824) residue is in contact with the GluN2B (W559) residue.
In silico analyses of PES binding proceeded in two steps. First, PES interaction with the TMD of the unliganded NMDAR was analyzed by docking. The results indicated four preferential steroid interaction sites: two homologous sites at the GluN1 M1/M4 interface and two homologous sites at the GluN2B M1/M4 interface. Since docking neglects the lipid environment, in the subsequent modeling step, we used MD simulations to predict details of the PES interaction with the GluN1 M1/M4 and the GluN2B M1/M4 interface in the model membrane environment. The MD simulation of the unliganded receptor (the closed state) with four steroid molecules at docking-predicted sites confirmed that the steroid preferentially binds at the surface of the GluN1 M1/M4 and the GluN2B M1/M4 interfaces. Based on the 100 ns MD trajectory, we calculated the steroid binding affinity using MMPBSA. Two separate calculations were performed to take into account the structural relaxation of the steroid/TMD complex using data from the initial 30 ns part of the trajectory and from the converged final 30 ns part. The MMPBSA analysis of the GluN1 (M1/M4) PES binding site suggested the PES interaction energy of ∼−15 kcal mol−1 for both the initial and final parts of the MD simulation. The GluN1 (W563; L819) residues showed the largest contribution to this interaction energy, using van der Waals contacts with the steroid molecule. The MMPBSA analysis of the GluN2B (M1/M4) site showed PES binding energy of −17.5 kcal mol−1 in the initial 30 ns of the simulation increasing to −19.5 kcal mol−1 in the relaxed final 30 ns of the trajectory. The strongest contributing residues GluN2B (W559; M562; Y823) were involved in van der Waals contacts with the steroid molecule (Fig. 9C).
The MD simulation of the steroid and the liganded receptor with the ion channel in the open configuration predicted that the steroid binding to the GluN1 M1/M4 interface was not substantially changed during the simulation, in contrast to the GluN2B M1/M4 interface. Upon receptor activation, the M1 and M4 helices of the GluN2B subunit move apart, and a cavity surrounded by the GluN2B M1 and M4 helices and the GluN1 M3 helix at the bottom is formed (Fig. 9A,B). MD-based prediction indicates that PES binds into this cavity (PES binding energy −17.5 kcal mol−1 based on the final 30 ns of simulation using the open configuration) with the steroid making van der Waals contacts with GluN2B (W559; M562; Y823; M824) residues, and the C17 acetyl moiety at PES D-ring anchoring the steroid to the GluN1 M3 helix, interacting with G638 and I642 residues (Fig. 9D). Although in the final 30 ns of the simulation the steroid occupies a deeper cavity involving different residues, the PES binding energy calculated from the initial 30 ns of simulation using the open configuration is nearly identical for this interaction.
Comparison of the computational models of the liganded NMDAR with the ion channel in the open configuration with and without PES docked (Fig. 9D) indicates that the receptor interaction with the steroid leads to a more efficient rotation of the GluN2B M4 helix and a rearrangement of TMD residues promoting tighter contact between residues GluN2B (M824) and GluN1 (I642) that is part of the ion channel-lining M3 helix. In the absence of the steroid, GluN2B (M824) residue is in contact with GluN2B (W559) residue occluding the entrance to the cavity (Fig. 9E). The MMPBSA predicted higher binding affinity of the PES for the receptor in the closed state (−19.5 kcal mol−1) compared with the open state (−17.5 kcal mol−1), which agrees well with the PES disuse-dependent action described previously (Horak et al., 2004).
Discussion
In this study, we show that an endogenous neurosteroid PES acts at a discrete group of residues located at the TMD of the NMDAR. Structurally different classes of compounds with a positive allosteric effect on the NMDAR have been characterized and shown to act by enhancing the efficacy of the receptor without altering the agonist EC50, by shifting the agonist EC50 to lower values, or by a combination of both mechanisms (Hackos and Hanson, 2017). Steroids structurally similar to PES potentiate NMDAR responses with a potency differing 84-fold and a maximal efficacy differing by >4.5-fold (Weaver et al., 2000; Krausova et al., 2018). These include steroids with charged hemiesters of varying length attached to the C3 carbon, 5α-pregnanes, and compounds modified at the C17 and C20 to 20-oxo-pregn-5-ene, pregn-5-ene, androst-5-ene, 17-oxo-androst-5-ene. Providing that the site of action for charged steroids is the same or overlapping, it is likely that it can accommodate bulky substituents without obvious structural preferences for residues attached at the C3 and the C17.
Despite structural similarity among endogenous sulfated neurosteroids and sterols, these compounds have diverse effects at NMDARs. PES has a dual potentiating and inhibitory action; at receptors containing GluN2A-B subunits, the disuse-dependent positive allosteric effect is prevailing (Wu et al., 1991; Bowlby, 1993; Malayev et al., 2002; Horak et al., 2004; Gibbs et al., 2006; Horak et al., 2006). In contrast, PAS has a use-dependent inhibitory effect at a site located in the extracellular vestibule of the NMDAR ion channel (Park-Chung et al., 1994; Petrovic et al., 2005; Vyklicky et al., 2015). The site of action for sterols at the NMDAR has not been identified; but since the effect of cholesterol depletion on NMDARs could be reverted by 24(S)-HC but not by PES, site(s) of action of PES and sterols are likely different (Fig. 3). Previous work, using chimeric subunits that combined segments from NMDA and kainate receptors, suggested that the NMDA receptor TMD alone is necessary and sufficient to determine receptor modulation by 24(S)-HC; however, both the ligand-binding domain and the TMD are required for allosteric potentiation of NMDAR responses by PES (Wilding et al., 2016).
Mutations of residues GluN2B (W559; M562; Y823; M824) and GluN1 (G638; I642) markedly reduced the potentiating effect of PES on NMDARs, suggesting that these residues may constitute the steroid binding site and/or they may be important for the efficacy with which the signal from steroid binding is transferred to the increase in the NMDAR ion channel Po. The following results argue in favor of a PES binding site at the GluN2B M1/M4 interface: First, residues whose mutation to alanine abolished or strongly reduced the potentiation by PES are located close together and were also identified by the in silico analysis to form van der Waals contacts with the steroid molecule (Figs. 5, 8, and 9). Second, the comparison of the unliganded and liganded state of the receptor with the ion channel closed and open (Fig. 9A) indicates that, during ion channel opening, this site undergoes conformational changes that result in decreased steroid binding, a phenomenon that underlies the disuse-dependent mechanism of the potentiating effect of PES at the NMDAR (Horak et al., 2004). Finally, residues within the GluN2B (M1/M4) cavity whose mutations abolish or strongly reduce the positive allosteric effect of PES are highly conserved within the GluN2A-D subunit family (Fig. 8A) in agreement with experimental data showing that GluN2 subunit-containing NMDARs are potentiated by PES (Jang et al., 2004; Horak et al., 2006; Kostakis et al., 2011). In contrast, receptors containing GluA1-4, GluK1-2, and GluN3A subunits, which share little homology at this site, are not potentiated by PES (Yaghoubi et al., 1998; Malayev et al., 2002; Horak et al., 2006) (Fig. 8A).
The proposed steroid interaction site delimited by the GluN2B M1 and M4 helices and the GluN1 M3 helix undergoes structural rearrangement during receptor activation, and PES binding to this site affects its conformation (Fig. 9). In accordance with the experimental observation that mutations of GluN2B (W559; M562; Y823; M824) and GluN1 (G638; I642) markedly reduced the steroid potentiating effect (Figs. 5, 8), the MD simulation identified the same residues to interact with the steroid molecule. Based on the MD simulation, the PES interaction with the NMDAR leads to a more efficient rotation of the GluN2B M4 helix and rearrangement of TMD residues promoting a tighter contact between residues GluN2B (M824) and GluN1 (I642). Since GluN1 (I642) is part of the ion channel-lining M3 helix, it seems plausible to speculate that this interaction is the main structural background for the PES-induced increase of the NMDAR ion channel Po (Horak et al., 2004; Cerny et al., 2019). Once the steroid dissociates from its site of action, the M4 helix turns back to its normal position and GluN2B (M824) is in contact with GluN2B (W559) (Fig. 9E). This contact prevents PES reentering the site; at the macroscopic level, this underlies the disuse-dependent nature of the steroid effect at the NMDAR (Horak et al., 2004).
There is a potential for compounds with a positive allosteric effect at NMDARs to compensate for clinical symptoms that stem from the dysfunction of glutamatergic synaptic transmission with indirect link to NMDARs (e.g., idiopathic schizophrenia and autism) and directly linked to malfunction of the NMDAR brought about by mutations in genes encoding receptor subunits. Variants of human GRIN1 and GRIN2 genes (encoding the hGluN1 and hGluN2 subunits) were identified in individuals with neurodevelopmental disorders, including autism and schizophrenia (Burnashev and Szepetowski, 2015; Hu et al., 2016). The consequences of various de novo NMDAR subunit mutations include altered surface expression and a gain or a loss of receptor function (Burnashev and Szepetowski, 2015; Yuan et al., 2015; Hu et al., 2016; Swanger et al., 2016; Platzer et al., 2017; Fedele et al., 2018; Vyklicky et al., 2018). Since the potentiating effect of steroids at the NMDARs is similar regardless of the receptor GluN2 subunit composition (Krausova et al., 2018), these compounds may be useful in rectifying NMDAR hypofunction caused by mutations in the ubiquitous GluN1 subunit. These mutations are clinically characterized by developmental delay, intellectual disability, cortical visual impairment, as well as movement disorders (Lemke et al., 2016). Modulators with properties similar to PES could be beneficial particularly in therapeutic strategies where a deceleration of the deactivation time course and an increase in the current amplitude of NMDAR EPSC are desired. Surprisingly, recent results indicate that human NMDARs with rare variants in the agonist-binding domain (hGluN2A(V685G; D731N)) and in the TMD (hGluN2B (L825V)) are potentiated by PES and its synthetic analog considerably more than WT receptors (Swanger et al., 2016; Vyklicky et al., 2018). Knowledge of the PES site of action at the NMDAR may advance drug development and personalized therapies for individuals with genetic NMDAR variants characterized by a loss of receptor function.
Footnotes
The authors declare no competing financial interests.
This work was supported by Czech Science Foundation 17-02300S, 20-17945S, 16-03913Y; Technology Agency of the Czech Republic TN01000013; Charles University Grant Agency 1676119; ERDF/ESF project PharmaBrain CZ.02.1.01/0.0/0.0/16_025/0007444; Research Project of the AS CR RVO 67985823 and RVO 61388963; and Biotechnology and Biomedicine Center of Academy of Sciences and Charles University (Vestec), project supported from European Regional Development Fund. We thank Magda Kuntosova for excellent technical assistance; and Eva Zbornikova for light scattering analysis.
- Correspondence should be addressed to Ladislav Vyklicky at ladislav.vyklicky{at}fgu.cas.cz





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}