

Genes Regulated by Presynaptic Homeostatic Plasticity
Evan R. Harrell, Diogo Pimentel, and Gero Miesenböck
(see pages 3054–3067)
Neurons undergo homeostatic plasticity to maintain stable activity levels despite injury, disease, growth, or other perturbations. A neuron can influence its own activity by altering the expression of ion channels that regulate its excitability, changing the number of neurotransmitter receptors at postsynaptic sites, or releasing retrograde signals that alter presynaptic neurotransmitter release. Homeostatic changes in neurotransmitter release have been investigated extensively at neuromuscular junctions in Drosophila. Such studies have identified genes that contribute to presynaptic homeostatic plasticity by influencing presynaptic calcium influx, the size of the readily releasable vesicle pool, or the coupling between calcium influx and vesicle release. In most of these studies, homeostatic plasticity was induced by disrupting glutamate receptor function in the postsynaptic muscle. The extent to which the effects depended specifically on the disruption of glutamate receptors and on the postsynaptic cell type has been unclear.
Harrell et al. took a different approach to identify contributors to presynaptic homeostatic plasticity at a central olfactory synapse in Drosophila. Instead of altering glutamate receptor function, they reduced activity in antennal lobe projection neurons via conditional expression of an inwardly rectifying potassium channel, Kir2.1, which counters depolarization. They isolated antennal segments containing presynaptic olfactory receptor neurons at different times after the onset of Kir2.1 expression and compared gene expression profiles in these tissues to those expressing a nonconducting form of Kir2.1.
At 12 h after gene induction, 25 genes were differentially expressed in neurons expressing functional versus nonfunctional Kir2.1. These included several genes involved in cell-fate determination, morphogenesis, and synaptic structure, many of which have been linked to presynaptic homeostatic plasticity at the neuromuscular junction. By 48 h after Kir2.1 induction, genes involved in protein synthesis were upregulated, and by 96 h, gene expression profiles suggested that cell-preservation mechanisms were engaged. Importantly, however, most changes in gene transcription were relatively modest.
These results suggest that presynaptic homeostatic plasticity between Drosophila olfactory neurons involves mechanisms similar to those engaged at synapses between motor neurons and muscle. They also indicate that these changes do not require direct modification of postsynaptic glutamate receptors and that gene expression programs change over time with prolonged inactivity. Future work should delineate the pathways through which postsynaptic inactivity alters presynaptic gene expression.
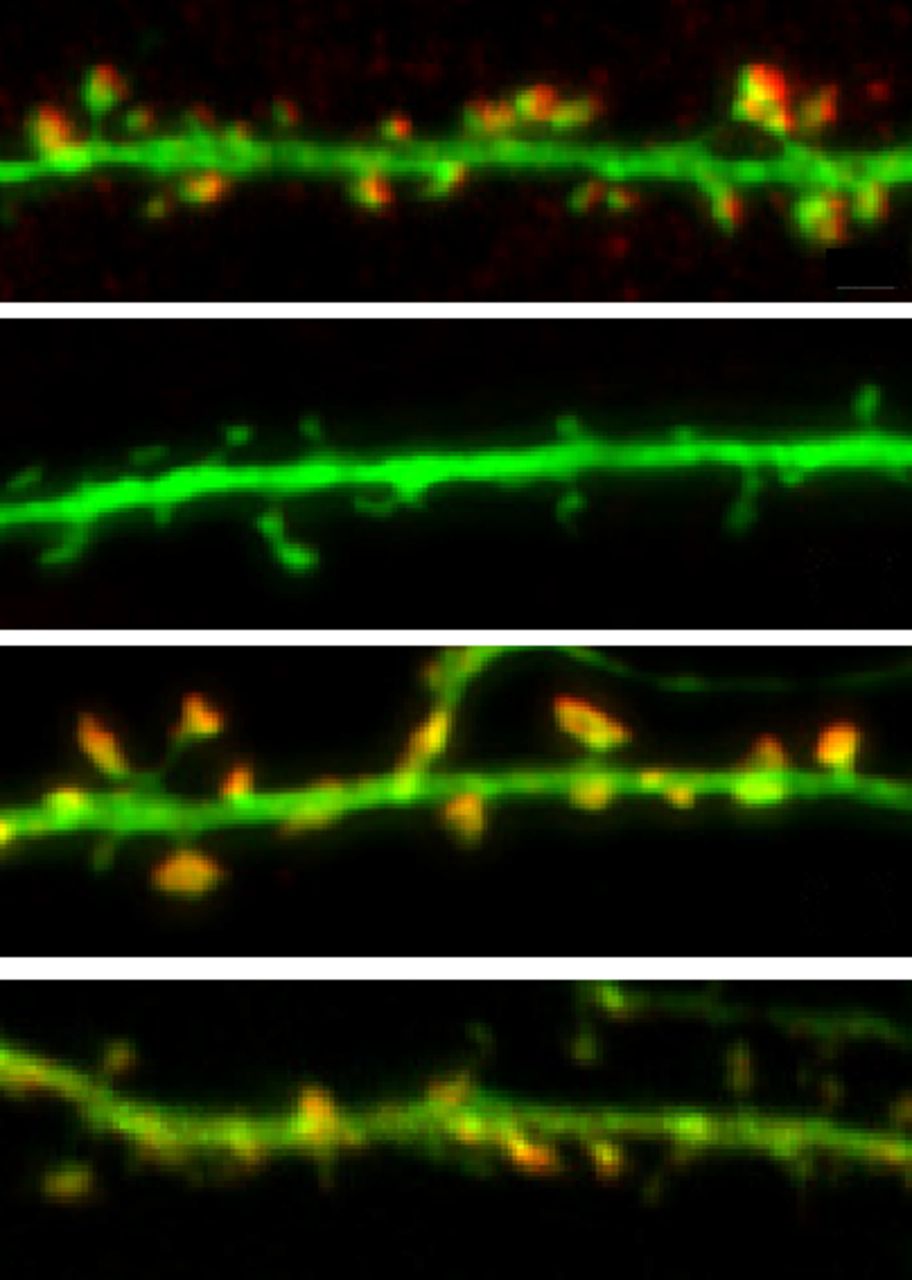
Compared with wild-type neurons (top), cortactin-deficient neurons have reduced spine density (2nd panel). Spine density is rescued by expressing an shRNA-resistant form of cortactin (3rd panel) but not by expressing cortactin with mutations in the SH3 domain (4th panel). See Shaw et al. for details.
Abl2, Cortactin, and the Stable Actin Pool in Spines
Juliana E. Shaw, Michaela B. C. Kilander, Yu-Chih Lin, and Anthony J. Koleske
(see pages 3068–3081)
Excitatory input to neurons typically occurs on dendritic spines. Spine shape and the molecular makeup of their postsynaptic densities are continually modified by synaptic activity and the resulting reorganization of the actin cytoskeleton. The actin network in spines is highly dynamic, with only ∼15% of filaments, mostly localized at the base of the spine, remaining in place for more than a few minutes. Shaw et al. suggest that interactions between the actin-binding nonreceptor tyrosine kinase Abl2 and another actin-binding protein, cortactin, help maintain this stable actin pool and that this is necessary for dendritic spine maintenance.
Consistent with previous work, Abl2 and cortactin were enriched in dendritic spines in cultured hippocampal neurons, and knocking down either protein reduced spine density. Knocking down cortactin also reduced Abl2 enrichment in spines by ∼45% and eliminated the stable pool of actin. Abl2 knockdown reduced cortactin enrichment by only ∼17%, and although it eliminated stable actin in small-headed spines, it increased the proportion of large-headed spines, and these spines retained cortactin and stable actin filaments.
Rescue experiments with mutated proteins showed that the enrichment of Abl2 in spines and its ability to promote cortactin enrichment and actin stabilization depended on its kinase activity and its SH2 protein-interaction domain, which associates with tyrosine-phosphorylated cortactin and other substrates. Similarly, the SH3 domain of cortactin, through which it binds Abl2 when tyrosine phosphorylated, was required for cortactin enrichment and maintenance of spines. Notably, tyrosine phosphorylation sites of cortactin were not required to enrich cortactin or stabilize actin in large-headed spines, although they were required in small spines. Finally, Abl2 was required for population-wide reductions in neuronal activity to increase spine density; and increasing neuronal activity rescued cortactin enrichment, spine density, and spine size in Abl2-deficient neurons.
These results suggest that Abl2 helps to stabilize small spines, particularly when neuronal activity is low, partly by increasing enrichment of cortactin and thus stabilizing actin filaments in spines. Although cortactin is required for Abl2 enrichment in spines, it can, in the absence of Abl2, support actin stability in large-headed spines, where its levels are highest. This Abl2-independent role of cortactin might allow it to compensate for loss of Abl2 when synaptic activity is high.
Footnotes
This Week in The Journal was written by Teresa Esch, Ph.D.





{kind=link}