

Abstract
Neuropathic pain is a chronic condition that occurs frequently after nerve injury and induces hypersensitivity or allodynia characterized by aberrant neuronal excitability in the spinal cord dorsal horn. Fibronectin leucine-rich transmembrane protein 3 (FLRT3) is a modulator of neurite outgrowth, axon pathfinding, and cell adhesion, which is upregulated in the dorsal horn following peripheral nerve injury. However, the function of FLRT3 in adults remains unknown. Therefore, we aimed to investigate the involvement of spinal FLRT3 in neuropathic pain using rodent models. In the dorsal horns of male rats, FLRT3 protein levels increased at day 4 after peripheral nerve injury. In the DRG, FLRT3 was expressed in activating transcription factor 3-positive, injured sensory neurons. Peripheral nerve injury stimulated Flrt3 transcription in the DRG but not in the spinal cord. Intrathecal administration of FLRT3 protein to naive rats induced mechanical allodynia and GluN2B phosphorylation in the spinal cord. DRG-specific FLRT3 overexpression using adeno-associated virus also produced mechanical allodynia. Conversely, a function-blocking FLRT3 antibody attenuated mechanical allodynia after partial sciatic nerve ligation. Therefore, FLRT3 derived from injured DRG neurons increases dorsal horn excitability and induces mechanical allodynia.
SIGNIFICANCE STATEMENT Neuropathic pain occurs frequently after nerve injury and is associated with abnormal neuronal excitability in the spinal cord. Fibronectin leucine-rich transmembrane protein 3 (FLRT3) regulates neurite outgrowth and cell adhesion. Here, nerve injury increased FLRT3 protein levels in the spinal cord dorsal root, despite the fact that Flrt3 transcripts were only induced in the DRG. FLRT3 protein injection into the rat spinal cord induced mechanical hypersensitivity, as did virus-mediated FLRT3 overexpression in DRG. Conversely, FLRT3 inhibition with antibodies attenuated mechanically induced pain after nerve damage. These findings suggest that FLRT3 is produced by injured DRG neurons and increases neuronal excitability in the dorsal horn, leading to pain sensitization. Neuropathic pain induction is a novel function of FLRT3.
Introduction
Neuropathic pain is a chronic condition that occurs after nerve damage or in various diseases (Basbaum et al., 2009). Nerve damage can lead to hypersensitivity, such as allodynia and hyperalgesia, which are often resistant to analgesics (Woolf and Salter, 2000; Kohno et al., 2005; Baron et al., 2010). Study investigators have proposed divergent mechanisms leading to chronic pain through alterations in nociceptive pathways in the peripheral nervous system and CNS (von Hehn et al., 2012). The spinal cord dorsal horn is a key region implicated in neuropathic pain development (Todd, 2010). The dorsal horn is not only a major transit point, where primary afferents engage projection neurons anatomically, but is also an important region for normal sensory processing and pathological conditions, such as neuropathic pain (Todd, 2010). Sensory information from the peripheral tissue is delivered to the dorsal horn via primary afferent neurons, after which it is processed and transmitted to secondary neurons connecting to the brain (Julius and Basbaum, 2001). It is generally thought that maladaptive changes in neural circuits occur in the dorsal horn after peripheral nerve injury (Kuner and Flor, 2016). However, the molecular and cellular mechanisms underlying neuropathic pain development in the dorsal horn neural circuits are complex and remain unclear, hindering the development of novel analgesic treatments.
Fibronectin leucine-rich transmembrane protein 3 (FLRT3) contains a fibronectin Type III domain and 10 leucine-rich repeats (Lacy et al., 1999). FLRT3 is expressed in the embryonic and adult nervous systems (Robinson et al., 2004). In the developing brain, FLRT3 functions as an axon-guidance factor by binding to the Unc5B receptor (Yamagishi et al., 2011; Seiradake et al., 2014). FLRT3 also regulates excitatory synapse formation by interacting with latrophilin 3 (LPHN3) (O'Sullivan et al., 2012). In the adult, FLRT3 expression was increased in the dorsal horn and injured nerve tissue after peripheral nerve transection (Robinson et al., 2004; Tsuji et al., 2004). Although ex vivo studies showed that FLRT3 promotes neurite outgrowth (Robinson et al., 2004; Tsuji et al., 2004), the function of FLRT3 in the adult CNS has not been characterized, and no reports have addressed the pathological function of FLRT3 in the dorsal horn after nerve injury.
In this study, our aim was to investigate the role of FLRT3 in spinal sensitization and chronic pain in the adult spinal cord. We examined FLRT3 expression in the dorsal horns of adult rats after peripheral nerve injury, as well as the effects of spinal FLRT3 on mechanical sensitivity using anti-FLRT3 antibody administration in a chronic pain model. Our results indicate that FLRT3 is involved in neuronal plasticity in the dorsal horn circuitry, leading to neuropathic pain.
Materials and Methods
Animals.
All animal experiments were performed in accordance with the Osaka University Medical School guide for animal care. All experimental protocols involving animals were approved by the institutional committee of Osaka University. We purchased 6- to 10-week-old male Wistar/ST rats from Japan SLC. The animals were fed food and water ad libitum.
Models for neuropathic or inflammatory pain.
To induce neuropathic pain, we performed partial sciatic nerve ligation (PSL) surgery for peripheral-nerve injury and antibody-administration experiments, or spinal nerve ligation (SNL) surgery for the Flrt3-knockdown experiment, as previously described (Seltzer et al., 1990). In brief, for the PSL model, we used 4–0 sutures (Alfresa Pharma) to tightly ligate the left sciatic nerve. For the SNL model, we carefully exposed the left L4 dorsal spinal nerve and tightly ligated it with 4–0 silk sutures around the spinal root (2 points, 1 mm apart). All surgical procedures were performed <2% isoflurane (Pfizer) anesthesia. Drugs to reduce postoperative pain were not administered.
Behavioral testing.
In the neuropathic pain model, we measured the mechanical paw-withdrawal threshold using the von Frey filament test, as previously described (Chaplan et al., 1994). Rats were individually placed in a plastic cage on wire mesh and habituated for 5–10 min. A calibrated von Frey filament (Semmes-Weinstein von Frey esthesiometer; Muromachi Kikai) was applied to the plantar surface of the hindpaw, and the 50% paw-withdrawal threshold was determined using the up-down method. Results are presented either as raw data or the percentage of the baseline threshold on day 0 in each rat. All behavioral analyses were performed under single-blind conditions. The rats that showed motor deficits or that were nonresponders to 15 g of von Frey filaments after the injury were excluded from the quantitative analysis. The wide range of baseline von Frey thresholds might be dependent on the difference in pain models, including PSL, SNL, catheter infusion, and virus infection.
Catheter implantation.
For recombinant protein or antibody administration, an intrathecal cannula was implanted as previously described (Maeda et al., 2009). Briefly, a polyethylene catheter (PE-10, Clay-Adams, Becton Dickinson) attached to the osmotic minipump (0.25 μl/h; model 1002, Alzet) was inserted between spinal cord vertebrae L5 and L6 to contact the lumbar enlargement. To verify proper placement of the catheter, lidocaine (AstraZeneca) was injected, followed by saline. Hindpaw paralysis indicated successful catheterization. The outer part of the catheter was plugged and fixed under the skin. All surgical procedures were performed <2% isoflurane anesthesia. Animals showing neurological deficits after catheter implantation were excluded from statistical analyses.
Recombinant protein and antibody administration.
Human recombinant FLRT3 protein (2795-FL, R&D Systems) was diluted to 200 ng/μl with saline. Under anesthesia, 10 μl of FLRT3 was slowly injected into the catheter, and the catheter was fitted to a saline-filled micro-osmotic pump (0.25 μl/h; model 1002, Alzet). The pump was subcutaneously fixed to the back of the rat. Goat anti-FLRT3 (AF2795, R&D Systems) and control goat IgG (144-09531, Wako) antibodies were diluted to 1 μg/μl with saline and intrathecally injected into the catheter (30 μg) on day 7 after the PSL operation using the same method.
Immunohistochemistry.
Immunohistochemistry was performed as described previously (Hayano et al., 2016). Rats were deeply anesthetized and transcardially perfused with 100 ml of PBS, followed by 150 ml of ice-cold 4% PFA in 0.1 m phosphate buffer. After perfusion, the L4–L6 spinal cord segments and bilateral L4–L6 DRG neurons were harvested and postfixed for 6 h at 4°C. The tissues were transferred to 15% sucrose in PBS for 12 h at 4°C, followed by incubation in 30% sucrose in PBS for 48 h at 4°C. Coronal spinal cord (20-μm-thick) and DRG (5-μm-thick) sections were cut on a cryostat. The sections were washed and incubated in blocking buffer (5% fetal BSA and 0.3% Triton X-100 in PBS) for 1 h at room temperature. For double-labeling immunofluorescence histochemistry, sections were incubated for 24 or 48 h at 4°C with antibodies against FLRT3 (1:250; AF2795, R&D Systems), neuron-specific Class III β-tubulin (Tuj1, 1:1000; MMS-435P, BioLegend), neuronal nuclei (NeuN, 1:1000; MAB377, Millipore), ionized calcium binding adaptor molecule 1 (Iba1; 1:500; 019-19741, Wako), GFAP (1:400; G-A-5, Sigma-Aldrich), c-Fos (1:1000; ABE457, Millipore), ATF-3 (1:500; sc-188, Santa Cruz Biotechnology), c-Myc (1:250; sc-40, Santa Cruz Biotechnology), isolectin B4 (IB4; 1:100; L2140, Sigma-Aldrich), calcitonin gene-related peptide (CGRP; 1:500; CA1134, Enzo Life Science), or vesicular glutamate transporter 1 (VGLUT1; 1:500; 135 304, Synaptic Systems). The sections were washed 3 times and incubated for 2 h at room temperature or overnight at 4°C with secondary antibodies (AlexaFluor-488/568; 1:500; Thermo Fisher Scientific). The sections were washed 3 times, slide-mounted, and coverslipped in mounting medium with the nuclear stain DAPI (Vector Laboratories). Anti-Tuj1 antibody has previously been used for the detection of DRG neurons (Rivat et al., 2018). Anti-Tuj1 antibody reacts with a structural protein that is a component of tubulin, whereas anti-NeuN antibody stains the nuclei of neurons. We therefore used anti-Tuj1 antibody for the assessment of the morphology of DRG neurons.
For the quantification of the immunostained cells, we prepared coronal sections of L4-L6 spinal cord that were 20 μm thick. Every 10th section (200 μm interval) was stained with the antibodies indicated, and 24 sections were used for analysis. For the quantification of the cellular area, we prepared L4-L6 DRG sections that were 20 μm thick. Every 10th section (200 μm interval) throughout the entire DRG was stained with anti-FLRT3 and anti-Tuj1 antibodies. Cells stained with DAPI in each section were submitted for cell area quantification. The cell area was measured using ImageJ software (National Institutes of Health). The Feret's diameters of c-Fos-positive cell nuclei were measured using ImageJ software.
The number of c-Fos-positive neurons in the superficial laminae I-II and deep laminae III-V was counted. As a stereological approach was not used in this study, quantification of the data may show a biased estimate of the actual number of cells.
Relative fluorescence intensities of FLRT3 and CGRP or IB4 were measured for the normalization of each intensity divided by each maximum intensity.
qPCR.
Total RNA was extracted from the spinal cord L4-L6 segments and bilateral L4-L6 DRGs with TRIzol reagent (Invitrogen) and purified with the RNeasy Micro Kit (QIAGEN). After cDNA synthesis (High Capacity cDNA RT Kit, Roche Diagnostics), gene expression was quantified with a TaqMan Gene Expression Assay (Applied Biosystems). The following primer pairs and universal probe were used: Flrt3, 5′-CTTCCTGGAGGTGCTCAGTCCGGCC-3′ (Rn01485365_m1); Unc5b, 5′-CCCATGTACTGGCGGAGA-3′ and 5′-GCGGAGGTTGGTGATCTTC-3′ (Universal Probe Library 76). Cycle threshold (Ct) values were calculated using the ΔΔCt method. All ΔCt values were normalized to rat GAPDH (4352338E, Applied Biosystems).
Western blot analysis.
Western blot analysis was performed as described previously (Hayano et al., 2016). Rats were killed under anesthesia, and the dorsal quadrant of the L4–L6 spinal cord or bilateral L4–L6 DRGs was dissected. The samples were homogenized in 50 mm Tris-HCl, pH 7.4, containing 150 mm NaCl, 1% NP-40, 0.1% SDS, protease inhibitor mixture (Complete; 11697498001, Roche Diagnostics), and a phosphatase inhibitor (PhosSTOP; 04906837001, Roche Diagnostics). The homogenate was centrifuged at 8000 × g for 10 min, and the supernatant was boiled in sample buffer at 95°C for 5 min. The lysates were separated on an SDS-polyacrylamide gel and transferred to PVDF membranes (Millipore). The membranes were blocked with 5% skimmed milk (Morinaga) in PBS-Tween 20 or TBS-Tween 20 buffer and incubated for 1 h at room temperature. The membranes were then incubated overnight at 4°C with antibodies against FLRT3 (1:500; AF2795, R&D Systems), NR2B (GluN2B) (1:500; TA309191, OriGene), phosphorylated (Tyr1472) NR2B (GluN2B; 1:500; TA309196, OriGene), β-actin (1:1000; 4970L, Cell Signaling Technology), or α-tubulin (1:1000; sc-5286, Santa Cruz Biotechnology). After washing in PBS-Tween or TBS-Tween, the membranes were incubated with an HRP-conjugated anti-rabbit IgG (1:1000; 7074s, Cell Signaling Technology), anti-mouse IgG (1:1000; 7076s, Cell Signaling Technology), or anti-goat IgG antibody (1:1000; sc-2020, Santa Cruz Biotechnology) for 1 h at room temperature. Protein bands were visualized using the enhanced chemiluminescence system (GE Healthcare), and densitometry analysis of the Western blot membranes was performed using ImageJ software (National Institutes of Health).
ELISA.
ELISA was performed using 96-well microplates (Thermo Fisher Scientific) that were coated with recombinant Unc5B /Human IgG Fc Chimera Protein or recombinant Human IgG Fc protein as a control (R&D Systems; 2 μg/ml, 100 μl/well) for 16 h at 4°C. After washing with PBS, the wells were blocked for 1 h with 5% BSA/PBS. Recombinant His-tagged FLRT3 protein (R&D Systems) was then added (0, 10, 20, 40, 80 ng/ml, 100 μl/well). Two hours after incubation, the plates were washed and anti-His antibody was added. HRP-conjugated secondary antibodies, the substrate reagent, and stop solution (R&D Systems) were used to detect protein binding. Absorbance was measured at 450 nm.
Cell surface-binding assays.
Cell surface-binding assays were performed as described previously (Seiradake et al., 2014). Human embryonic kidney 293T (HEK293T) cells were transfected with a pEGFP-N1 vector (TaKaRa) encoding full-length rat Unc5B cDNA (accession number U87306). At 48 h after transfection, the cells were washed with Dulbecco PBS and incubated for 1 h at 37°C with 2 μg/ml BSA (Sigma-Aldrich) and recombinant His-tagged human FLRT3 protein (FLRT3-His) (R&D Systems) in Dulbecco PBS containing 1% normal goat serum and 2 μg/ml heparin sodium. Anti-FLRT3 antibody or Control IgG was added to examine whether the anti-FLRT3 antibody can neutralize the binding of FLRT3-His to the cell surface-expressed Unc5b-GFP. The cells were fixed with 4% PFA in PBS for 15 min at room temperature, and permeabilized in PBS containing 5% BSA (Sigma-Aldrich) and 0.1% Triton X-100 (Wako). For the detection of the binding of recombinant His-tagged FLRT3 protein to cell surface-expressed Unc5B-GFP, the cells were immunostained for FLRT3 with His (1:1000; PM032, MBL) and GFP (1:1000; GF090R, Nacalai Tesque) antibodies, and an AlexaFluor-568-conjugated secondary antibody (1:500; Invitrogen). Images were quantified using ImageJ.
Adeno-associated virus (AAV) production.
AAV production was performed as described previously (Mochizuki et al., 2001). A full-length rat Flrt3 cDNA (accession number BC160843) was C-terminally tagged with a myc epitope by PCR and subcloned into a pCMV expression vector. The myc-tagged cDNA was cut out with PstI and HpaI and blunt-cloned into the Eco47III site of the pAAV-U6-ZsGreen vector after ZsGreen removal (TaKaRa).
For FLRT3-encoding AAV vector production, AAV293 cells were transfected with ZsGreen (control) or the FLRT3-myc-containing pAAV-U6 vector.
Candidate short-hairpin RNA (shRNA) constructs were designed using publicly available web tools (BLOCK-iT RNAi Designer, Thermo Fisher Scientific). The shRNA sequences were as follows: shFlrt3 #1, GCAAGAAGCAGAAGACATA; shFlrt3 #2, GCAGAAGACATAGCTATTT; shFlrt3 #3, GCAATTTACCTCAGGGTAT; and the previously described shFlrt3 #4, CTCGGCTGTTAGCATAGAA (O'Sullivan et al., 2012). To screen for target knockdown, HEK293T cells were cotransfected with shRNAs and the pCMV-FLRT3-AcGFP vector. The knockdown efficiency was determined using qPCR. The shRNAs with the first- and second-highest knockdown efficiencies (shFlrt3 #3 and #4, respectively) relative to an shLuc control were used in knockdown experiments. For shRNA-expressing AAV vector production, shRNAs were cut out with BamHI and EcoRI and cloned into a BamHI- and EcoRI-cut pAAV-CMV-tRFP vector. AAV293 cells were transfected with pAAV-shRNA, pHelper (TaKaRa), and pRC6 (TaKaRa) plasmids using transfection reagent. After a 5 d incubation, the cells were lysed, and the AAV was precipitated with ammonium sulfate solution by centrifugation at 8000 rpm for 70 min. The AAV-containing pellet was washed with PBS and concentrated with Amicon Ultra-15 10K centrifugal filter devices (Millipore).
Direct injection of AAVs into the DRG.
For DRG-targeted gene delivery, we performed direct injection into lumbar DRGs. Under anesthesia, the lateral aspect of the L4 vertebra was exposed and the laminar bone was partially removed. The L4 DRG was gently exposed and injected with 5 μl of virus vector solution using a Hamilton syringe (Sigma-Aldrich). In the Flrt3-knockdown experiment, SNL was performed at 2 weeks after AAV injection, and the paw-withdrawal threshold was evaluated on days 0, 7, 14, and 21 after SNL.
Experimental design and statistical analysis.
Statistical analysis was performed using the Student's t test for comparisons between two groups. For comparisons among multiple groups, the two-way ANOVA was used with the Tukey–Kramer post hoc test. Differences with p < 0.05 were considered statistically significant. Statistical analyses were performed using JMP (SAS Institute) and Prism software (GraphPad). All data are presented as the mean ± SEM.
Results
FLRT3 is expressed in lamina IIi of the spinal cord dorsal horn after peripheral nerve injury
Flrt3 mRNA was previously shown to be upregulated in peripheral neurons, and FLRT3 protein was distributed in the lumbar dorsal horn after sciatic nerve transection (Robinson et al., 2004). To determine whether FLRT3 is upregulated in the dorsal horn in neuropathic pain models, we examined FLRT3 expression in the spinal cord after PSL (Seltzer model) (Seltzer et al., 1990). After surgery, rats showed a significant decrease in the hindpaw-withdrawal threshold in the side ipsilateral to injury, compared with the threshold in the side contralateral to injury (Fig. 1A). We then examined FLRT3 protein expression and localization in this model by immunohistochemistry. Starting at 4 d after PSL injury, increased FLRT3 immunoreactivity was observed in the ipsilateral dorsal horn (Fig. 1B). We performed immunostaining for CGRP (Ju et al., 1987), IB4 (Silverman and Kruger, 1990), and VGLUT1 (Varoqui et al., 2002; Todd et al., 2003) to identify the central terminals of DRG neurons in the dorsal horn laminae (Lai et al., 2016). FLRT3 was distributed in laminae II-V, particularly in the region of IB4 loss at day 14 after injury (Fig. 1C, arrowheads). FLRT3 fluorescence was not significantly different between CGRP-positive and CGRP-negative areas, whereas it was significantly increased in IB4-negative areas compared with IB4-positive areas (Fig. 1D,E). We also quantitated FLRT3 protein levels in the dorsal horn by immunoblotting. The FLRT3 band density increased in tissue lysates from the side ipsilateral to the injury at days 7 and 14 after PSL injury (Fig. 1F). Further, the effects of PSL injury on Flrt3 mRNA expression were examined by qRT-PCR. Flrt3 mRNA levels increased markedly in DRG neurons on days 1–21 after PSL, whereas they remained constant in the spinal cord (Fig. 1G). Together, these results suggest that spinal FLRT3 protein expression might be derived from the DRG.

FLRT3 expression is increased in the dorsal horn after sciatic nerve injury in rats. A, The hindpaw-withdrawal threshold (g) was measured using the von Frey filament at day 0 (“pre”), 4, 7, and 14 after PSL. PSL decreased the paw-withdrawal threshold in rats (n = 6), as determined by two-way repeated-measures ANOVA followed by Tukey–Kramer test. *p < 0.05, **p < 0.01. B, FLRT3 expression (green) in the dorsal horn at days 0 (“pre”), 4, 7, and 14 after PSL. Dotted line indicates the border between the white and gray matter. Scale bar, 200 μm. C, Coimmunostaining of FLRT3 (green) with the DRG projection markers (red) CGRP (specific for peptidergic C/Aδ fibers in lamina I-outer II), IB4 (nonpeptidergic C fibers in inner dorsal lamina II), and VGLUT1 (Aβ fibers in inner lamina II-ventral V) in spinal cord sections prepared at 14 d after PSL. FLRT3 was distributed in CGRP/VGLUT1-positive laminae II-III, particularly in the IB4-negative areas (arrowheads). Scale bar, 200 μm. D, E, Representative graphs of relative fluoresce of FLRT3 and CGRP (D, left), or FLRT3 and IB4 (E, left). Relative fluorescence of FLRT3 in CGRP (E, right) or IB4 (E, right) positive and negative areas. FLRT3 fluorescence was increased in the IB4-negative area compared with the IB4-positive area (n = 3 rats). *p < 0.05 (Student's t test). F, FLRT3 immunoblotting of spinal cord lysates. At days 7 and 14 after PSL, the relative FLRT3 band density significantly increased in the ipsilateral side. The FLRT3 signal intensity was normalized to that of β-actin in each side, and then in the ipsilateral side relative to the contralateral side (n = 3 rats). **p < 0.01 (Student's t test). ***p < 0.001 (Student's t test). G, Relative Flrt3 mRNA expression in the DRG (top) and spinal cord (bottom). The Flrt3 mRNA levels increased in the ipsilateral side of the DRG but not the spinal cord compared with the contralateral side (n = 3 rats per time point). **p < 0.01 (Student's t test). ****p < 0.0001 (Student's t test). H, Coimmunostaining of FLRT3 (green) with the neuronal marker Tuj1 (red; top) or satellite cell marker GFAP (red; bottom) in the DRG at day 14 after PSL. FLRT3 was expressed in DRG neurons but not satellite cells. Graph represents the percentage of FLRT3-negative and FLRT3-positive neurons in Tuj1-positive neurons. Scale bar, 50 μm. I, DRG neuronal-size histogram of FLRT3-immunoreactive neurons (FLRT3+ neurons; black bars, n = 565) compared with the total neurons (white bars, n = 1103) in the L4/L5 DRGs at day 14 after PSL. ****p < 0.0001 (two-way ANOVA followed by Tukey–Kramer test). n.s., not significant.
Next, we examined FLRT3 expression in the DRG at day 14 after PSL. FLRT3 immunoreactivity was detected in the cell membranes of neuron-specific Class III β- Tuj1-positive DRG neurons, but not in those of GFAP-positive satellite cells (Fig. 1H). We found that ∼35% of neurons were FLRT3-positive. In addition, we classified the sizes of FLRT3+ DRG neurons (n = 565) and all DRG neurons (n = 1103). Cell sizes were classified as small (0–600 μm2), medium (600–1000/1200 μm2), or large (1000/1200–4000 μm2) (Harper and Lawson, 1985a,b). The FLRT3+ neuronal population comprised small-sized (<600 μm2; 73.6%) and medium-sized (600–1200 μm2; 22.7%) DRG neurons (Fig. 1I, black column). These results suggest that FLRT3 expression mainly increased in small- and medium-sized DRG neurons after PSL.
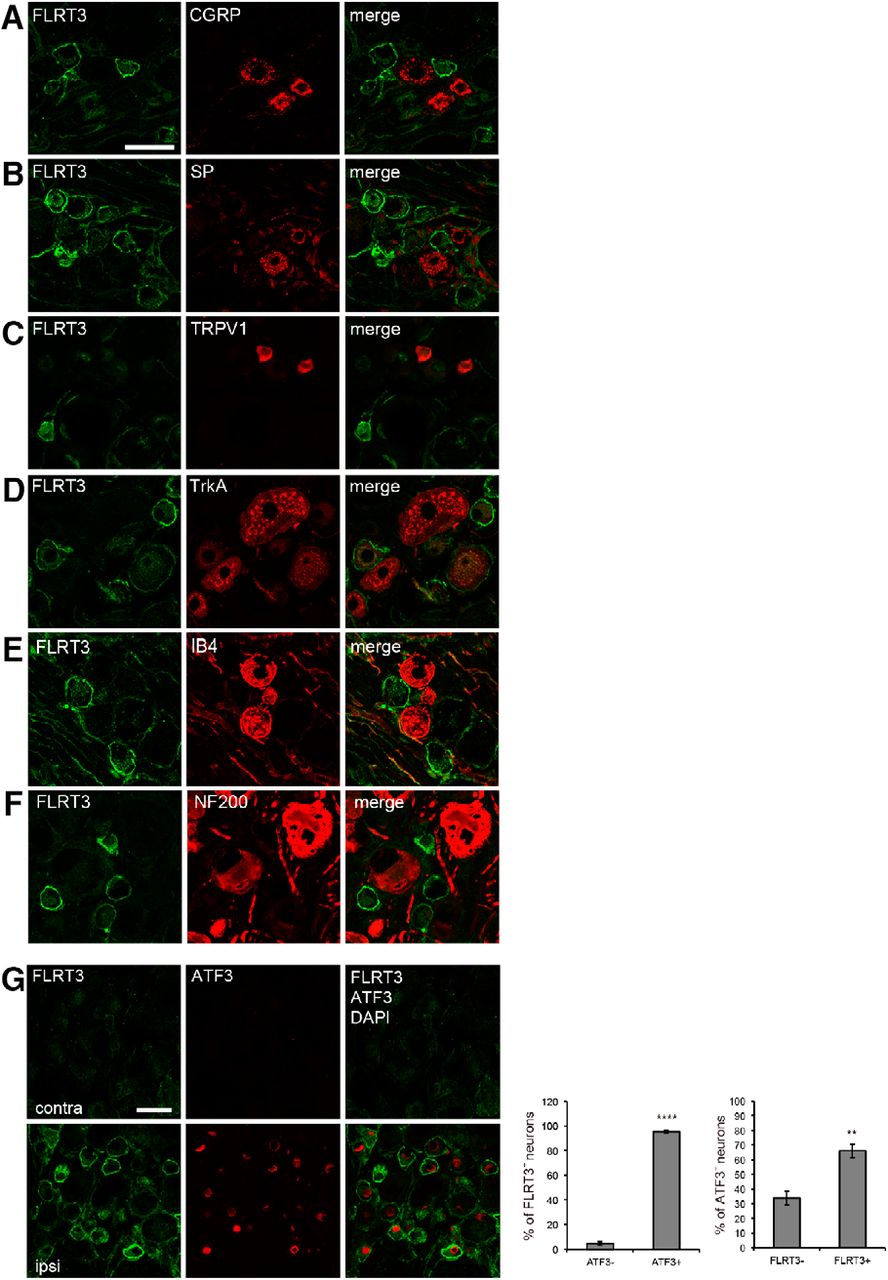
To examine the cellular subtypes of FLRT3+ neurons, we detected the expression of various neurochemical markers, including CGRP and substance P (SP) for peptidergic DRG neurons, transient receptor potential vanilloid 1, and tropomyosin receptor kinase A (TrkA) for nociceptive small DRG neurons (Averill et al., 1995), IB4 for nonpeptidergic DRG neurons, and neurofilament 200 (NF200) for myelinated large DRG neurons (Lawson and Waddell, 1991). Most FLRT3+ DRG neurons did not express the neurochemical markers CGRP, SP, NF200, or IB4 (Fig. 2A–F). In contrast, TrkA was expressed in FLRT3+ DRG neurons (Fig. 2D). These results indicate that FLRT3 was upregulated in nociceptive DRG neurons after peripheral nerve injury.

Nociceptive DRG neurons express FLRT3 after peripheral nerve injury. A–F, Coimmunostaining of FLRT3 (green) with neurochemical markers (red) of DRG neuronal subpopulations. Detection of CGRP (A) and SP (B) as peptidergic nonmyelinated neuronal markers, TRPV1 (C) and TrkA (D) as nociceptive markers, IB4 (E) as a nonpeptidergic nonmyelinated neuronal marker, and NF200 (F) as a large myelinated neuronal marker. FLRT3-positive DRG neurons expressed TrkA. FLRT3 immunoreactivity was not detected in CGRP-, SP-, IB4-, or NF200-labeled DRG neurons. Scale bar, 50 μm. G, Coimmunostaining of FLRT3 (green) with the injured-neuronal marker activating transcription factor 3 (ATF3; red) in DRG neurons at day 14 after PSL; FLRT3 was expressed in ATF3-positive DRG neurons. Nuclei were counterstained with DAPI (blue). Scale bar, 50 μm. **p < 0.01, ****p < 0.0001. contra, Noninjury side; ipsi, injury side.
Downregulation of neuropeptides and their receptors has been observed in nociceptive neurons after nerve injury (Hökfelt et al., 1994). In contrast, transcription factors and other peptides have been reported to be upregulated. Therefore, we hypothesized that FLRT3 might be upregulated in damaged nociceptive DRG neurons with reduced neuropeptides after nerve injury. To test this hypothesis, we performed immunostaining for FLRT3 and activating transcription factor-3 (ATF3), a marker of injured peripheral neurons (Tsujino et al., 2000). FLRT3 expression increased in ATF3+ neurons in the ipsilateral DRG on 14 d after injury, whereas FLRT3+ or ATF3+ DRG neurons were not detected in the contralateral DRG (Fig. 2G). Approximately 95.3% of FLRT3+ neurons were ATF3+ and 4.7% were ATF3−. Approximately 66.1% of ATF3+ neurons were FLRT3+ and 33.9% were FLRT3−. These results suggest that FLRT3 expression increased in damaged DRG neurons after peripheral nerve injury.
FLRT3 induces mechanical allodynia
To address the function of FLRT3 in mechanical sensitivity, we administered recombinant FLRT3 protein intrathecally via a catheter implanted in the lumbar subarachnoid space of intact rats (Fig. 3A). Mechanical sensitivity was evaluated at 12, 24, and 48 h after administering recombinant FLRT3 protein. FLRT3-injected rats showed a transient decrease in the paw-withdrawal threshold to mechanical stimuli compared with that in saline-injected rats (Fig. 3B). We previously reported that Netrin-4 injection induced an increase in the levels of phosphorylated GluN2B (pGluN2B), a subunit of the NMDAR, in the dorsal horn and enhanced mechanical sensitivity (Hayano et al., 2016). We therefore examined pGluN2B levels in the spinal cords of FLRT3-treated rats. Western blot analysis showed that FLRT3 administration tended to increase the relative pGluN2B band density at 12 h after injection, although the difference was not significant (Fig. 3C). Further, to examine neuronal activation, we compared the numbers of c-Fos-labeled cells in the dorsal horn before and after FLRT3 administration. More c-Fos-labeled cell profiles were detected in the FLRT3-injected group compared with the saline-injected group at 12 h after administration (Fig. 3D–F). FLRT3 treatment significantly increased the c-Fos-labeled cells in both the superficial lamina I-II and deep lamina III-V (Fig. 3G). The distributions of nuclear diameters are shown in the histogram and appear identical between control and treated groups, suggesting that larger cells were not sampled more frequently (Fig. 3H). These results indicate that spinal FLRT3 enhanced the sensitivity to mechanical stimuli and may have induced neuronal activation via GluN2B phosphorylation in neuropathic pain.

FLRT3 induces mechanical allodynia in vivo. A, Schematic and timeline of recombinant FLRT3 administration and behavioral testing. B, Relative hindpaw-withdrawal threshold after intrathecal administration of recombinant FLRT3 protein. The mean threshold in the FLRT3 group was significantly decreased compared with that in the saline (control) group, indicating that FLRT3 induced mechanical allodynia (n = 12–14 rats per group). *p < 0.05 (two-way repeated-measures ANOVA followed by Tukey–Kramer test). C, Representative immunoblots of NMDA-type glutamate receptor subunit GluN2B and pGluN2B in spinal cord lysates prepared at 12 h after FLRT3 administration. The pGluN2B signal intensity was normalized to that of total GluN2B (n = 3 rats per group), and signal intensity differences were analyzed by Student's t test. D, Double-immunofluorescence staining for the neuronal activation marker c-Fos (green) and neuronal marker NeuN (red) in the dorsal horn. Intrathecal FLRT3 administration increased c-Fos expression in dorsal horn neurons (arrows). Scale bar, 200 μm. E, Magnification images of Figure 4D. Scale bar, 10 μm. F, Total numbers of c-Fos-positive cells in dorsal horn laminae I-III at 12 h after administration in each group (n = 5 rats per group). *p < 0.05 (Student's t test). G, The c-fos-labeled cell profiles in lamina I-II and lamina III-V (n = 5 rats per group). *p < 0.05 (Student's t test). H, The frequency distribution of c-fos-positive cells is classified by nucleus diameter. n.s., not significant.
Increased FLRT3 expression in the DRG was sufficient to induce mechanical allodynia
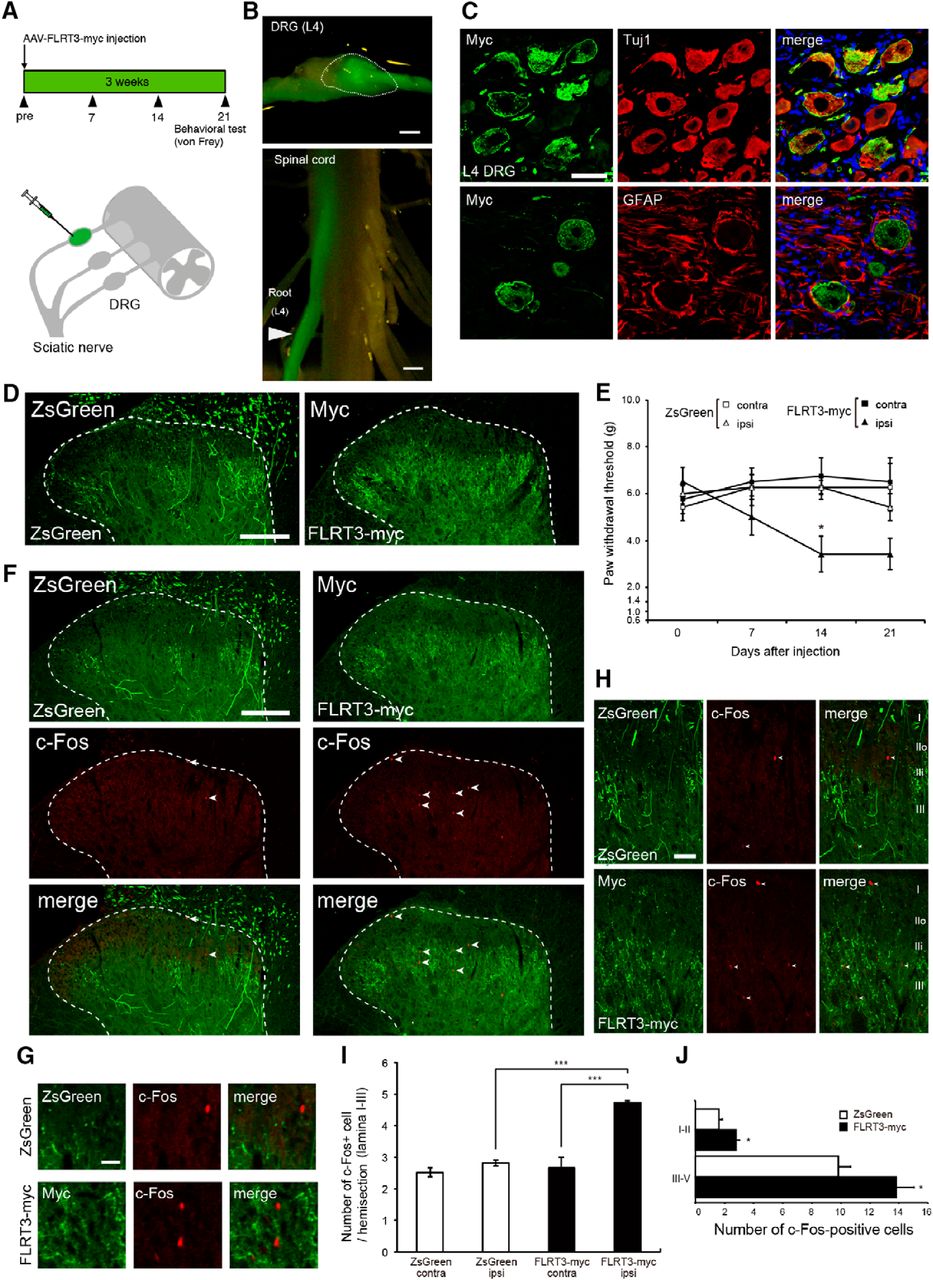
We found that FLRT3 expression increased in the DRG, but not in the spinal cord (Fig. 1C). To assess the effect of increased FLRT3 expression specifically in the DRG, an AAV encoding a C-terminally myc-tagged FLRT3 variant was injected into the L4 DRG (Fig. 4A). The AAV-mediated delivery of ZsGreen (used as a control) enabled confirmation of specific expression in the L4 DRG (Fig. 4B). ZsGreen overexpression was observed in Tuj1-positive neurons (Fig. 4C). ZsGreen and FLRT3-myc signals were also detected in laminae II-V of the dorsal horn (Fig. 4D). We then evaluated mechanical sensitivity at days 0, 7, 14, and 21 h after injecting ZsGreen or the FLRT3-myc-expressing AAV vector. FLRT3-AAV-injected rats showed a decrease in the paw-withdrawal threshold to mechanical stimuli compared with that of ZsGreen-AAV-injected rats at 14 d (Fig. 4E). Moreover, to determine whether DRG-derived FLRT3 triggered neuronal activation in the dorsal horn, we performed a c-Fos immunofluorescence analysis. The number of c-Fos-positive cells significantly increased in the FLRT3-myc group compared with that in the ZsGreen group (Fig. 4F–I). Furthermore, AAV-mediated FLRT3-myc injection significantly increased c-Fos-labeled cells in both superficial lamina I-II and deep lamina III-V compared with ZsGreen (Fig. 4J). Together, these results demonstrate that FLRT3 overexpression in the DRG induced mechanical allodynia.

AAV-mediated FLRT3 expression in the rat DRG neurons induces mechanical allodynia. A, Schematic and timeline of AAV injection in the DRG. FLRT3-myc-encoding AAV was injected into the left L4 DRG. ZsGreen-encoding AAV was used as a control. B, Representative images of a ZsGreen-AAV-injected L4 DRG (top, dotted line) and a dorsal root (bottom, arrowhead). Scale bar, 1 mm. C, Double immunofluorescence staining of Myc (green) and Tuj1 (red) or GFAP (red) in the DRG at day 14 after AAV injection. Nuclei were counterstained with DAPI (blue). Scale bar, 50 μm. D, ZsGreen (green) or Myc immunofluorescence (green) in the dorsal horn at day 14 after AAV injection. Dotted line indicates the border between the white and gray matter. ZsGreen- or Myc-expressing afferents were distributed to laminae II-V of the dorsal horn. Scale bar, 200 μm. E, Hindpaw-withdrawal threshold (g) after FLRT3-myc-encoding AAV injection. The mean threshold in the FLRT3-myc group (n = 8) was significantly decreased compared with that in the ZsGreen group (control; n = 7), indicating that FLRT3 upregulation induced mechanical allodynia; two-way repeated-measures ANOVA followed by Tukey–Kramer test. F, Representative image of coimmunostaining of ZsGreen (green) or myc (green) with c-Fos (red) in the dorsal horn at 14 d after AAV injection. c-Fos-positive neurons (arrowhead) increased in the dorsal horn of the FLRT3-myc group. Scale bar, 200 μm. Dotted line indicates the border between the white and gray matter. G, Magnification images of F. Scale bar, 20 μm. H, Distributions of AAV-ZsGreen- or AAV-FLRT3-myc-positive fibers and c-Fos-positive cells in dorsal horn laminae I-III. Scale bar, 50 μm. I, Graph represents numbers of c-Fos-positive cells in dorsal horn laminae I-III per section at day 14 after AAV injection in each group in F (n = 3 rats per group). *p < 0.05 (one-way ANOVA followed by Tukey–Kramer post hoc test). ***p < 0.001 (one-way ANOVA followed by Tukey–Kramer post hoc test). J, The c-fos-labeled cell profiles in lamina I-II and lamina III-V (n = 3 rats per group). *p < 0.05 (Student's t test).
FLRT3 antibody attenuates mechanical allodynia in a neuropathic pain model
An FLRT3 antibody was used to investigate whether functional inhibition of endogenous FLRT3 would reduce mechanical allodynia. Consistent with previous reports (Karaulanov et al., 2006; Söllner and Wright, 2009; Yamagishi et al., 2011; Seiradake et al., 2014), we found that recombinant FLRT3 bound to its receptor Unc5B in an ELISA experiment (Fig. 5A). Similarly, a cell surface-binding assay revealed that a His-tagged FLRT3 protein variant bound to Unc5B-GFP-expressing cells (Fig. 5B–D). We tested whether the FLRT3 antibody would block the interaction between FLRT3 and Unc5B in this assay. After coincubation with the FLRT3 antibody, the FLRT3-His signal significantly decreased compared with that in the control IgG group (Fig. 5B–D). These results suggest that the FLRT3 antibody inhibited the FLRT3-Unc5B interaction. To further examine whether blocking FLRT3 could attenuate neuropathic pain, we assessed the effect of this antibody after PSL surgery. Catheter insertion was performed 7 d before PSL induction, and a control antibody or FLRT3 antibody was administered 7 d after the PSL. Intrathecal injection of FLRT3 antibody suppressed the reduction in the paw-withdrawal threshold induced by PSL, suggesting that functional inhibition of FLRT3 attenuated allodynia in neuropathic pain (Fig. 5E,F).
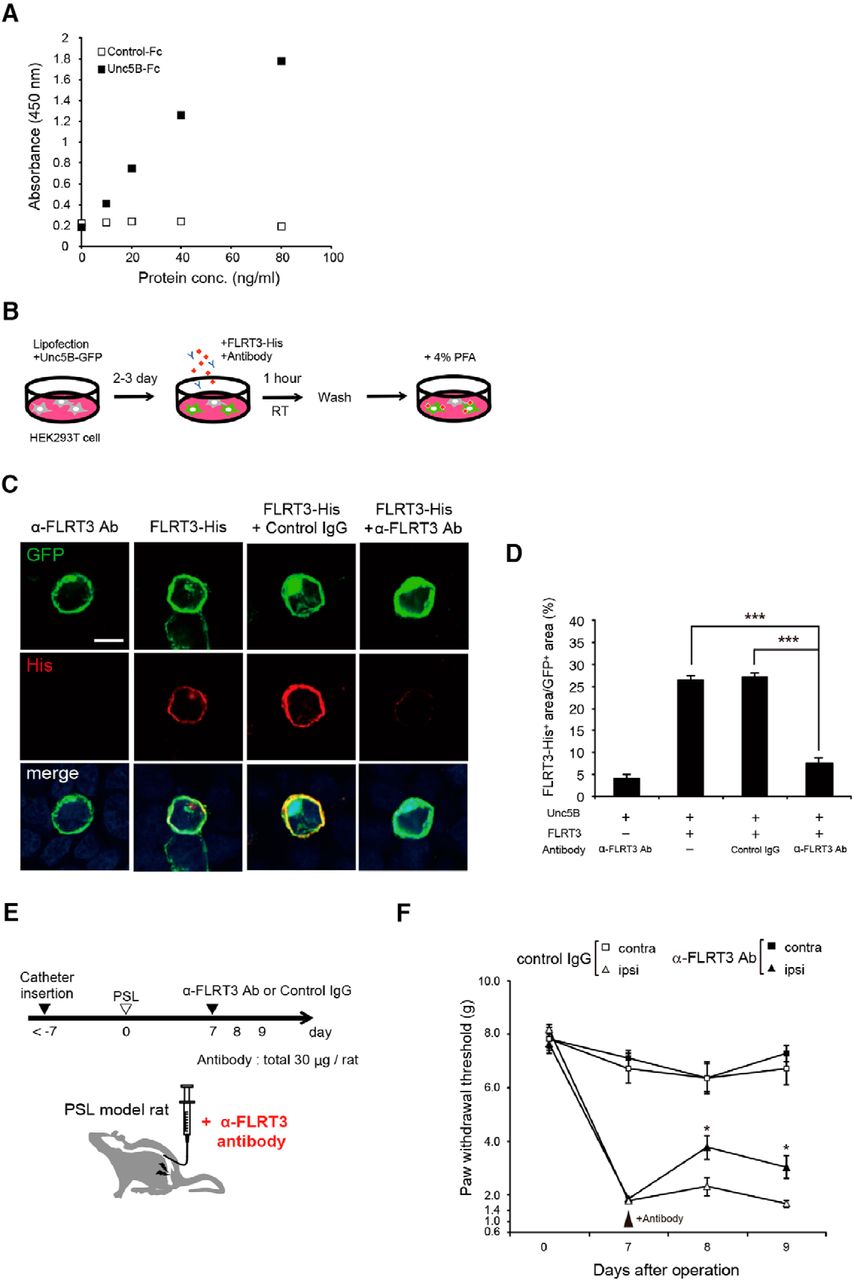
FLRT3 antibody produces an analgesic effect in rats. A, Recombinant FLRT3 interacted with Unc5b in an ELISA (n = 1). Control-Fc, recombinant Human IgG Fc protein; Unc5B-Fc, recombinant Unc5B /Human IgG Fc Chimera Protein. B, Schematic representation of the cell surface-binding assay. FLRT3-His, recombinant His-tagged human FLRT3 protein. C, Representative images of FLRT3-His immunoreactivity in HEK293T cells expressing Unc5B-GFP. Scale bar, 50 μm. D, Quantification of the FLRT3-His-positive area as a percentage of the GFP-positive area in each group (n = 10–12 fields per group); one-way ANOVA followed by Tukey–Kramer post hoc test. E, Schematic and timeline of FLRT3 antibody treatment in rats after PSL. F, Changes in the hindpaw-withdrawal threshold (g) after intrathecal administration of an anti-FLRT3 antibody through day 9 after PSL. FLRT3 antibody injection reversed the PSL-induced decrease in the paw-withdrawal threshold (n = 9–13 rats per group). Two-way repeated-measures ANOVA followed by Tukey–Kramer test: *p < 0.05; ***p < 0.001.
Knockdown of Flrt3 expression attenuates mechanical allodynia after nerve injury
We observed increased Flrt3 mRNA expression in the DRG but not spinal cord following pain induction (Fig. 1G). Since intrathecal injection delivers antibodies to the CSF, we cannot exclude the possibility that the FLRT3 antibody exerted its effect outside of the DRG because of the diffusion of the antibody toward the spinal cord. To investigate the effect of FLRT3 expression in the DRG, FLRT3 expression was suppressed specifically in the DRG using AAV-mediated delivery of FLRT3 shRNA. Although the location of sciatic nerve components ranges from L3 to L6 in rats (Rigaud et al., 2008), we found that the number of ATF3-positive neurons was highest in the L4 after PSL induction (Fig. 6A). It has also been reported that DRG neurons projecting to the sciatic nerve are mainly located in L4 and L5 in Wistar rats (Rigaud et al., 2008). We therefore focused on the L4 DRG to inhibit Flrt3 expression. To cause a lesion specifically in the L4 spinal nerve, we used the SNL model. FLRT3-shRNA-encoding AAV was injected into the L4 DRG 2 weeks before L4 SNL (Fig. 6B). Two weeks after injection, an AAV-encoded-RFP signal was specifically detected in the L4 DRG, suggesting successful AAV infection specifically in the L4 DRG (Fig. 6C). We designed and screened 4 FLRT3 shRNAs in vitro and in vivo (Fig. 6D,E). FLRT3 shRNA #3 reduced Flrt3 mRNA expression significantly more efficiently compared with an shRNA targeting luciferase, used as a negative control, at day 15 after SNL in vivo (Fig. 6E). We confirmed that FLRT3 shRNA #3 reduced FLRT3 protein expression in the DRG and dorsal horn (Fig. 6F,G) and found that this shRNA ameliorated the reduction in the withdrawal threshold induced by SNL (Fig. 6H). Thus, Flrt3 suppression in the DRG is sufficient to ameliorate neuropathic pain after injury.

Suppression of FLRT3 expression attenuates mechanical allodynia. A, Immunofluorescence staining of ATF3 in the L3-L6 DRG neurons at day 7 after PSL (left panels). Scale bar, 50 μm. The number of ATF3-positive cells per section in each DRG (n = 1; right graph). B, Schematic and timeline of AAV injection in the L4 DRG after SNL and AAV-FLRT3 shRNA plasmid construction. C, Immunofluorescence staining of RFP (red) with Tuj1 (green; top) or GFAP (green; bottom) in the DRG at day 14 after SNL injury (day 28 after AAV injection). Nuclei were counterstained with DAPI (blue). Scale bar, 50 μm. D, Knockdown efficacy of FLRT3 shRNA in vitro. Quantitative FLRT3 expression in the FLRT3-expressing HEK293T cells. HEK293T cells were transfected with an FLRT3-expression plasmid and either a control shRNA or four FLRT3 shRNAs for screening (n = 3). Tukey–Kramer test. E, Relative Flrt3 mRNA expression at day 15 after SNL injury. Injection of FLRT3-shRNA-encoding AAV (shFLRT3 #3) significantly reduced the Flrt3 mRNA levels compared with those in rats injected with control AAV (shLuc; n = 5 rats per group). *p < 0.05 versus control shRNA (one-way ANOVA with Bonferroni's multiple-comparison test). n.s., Not significant versus control shRNA. F, Double-immunofluorescence staining of FLRT3 (green) with RFP (red) in L4 DRGs injected with AAV-shLuc (top) or AAV-shFlrt3 #3 (bottom) at day 14 after SNL injury. AAV-shFlrt3 #3-infected cells (RFP-positive cells, bottom, arrowhead) show the reduced and diffused expression of FLRT3, whereas noninfected cells (RFP-negative cells, bottom, arrow) show cell surface expression of FLRT3. Scale bar, 50 μm. G, Double-immunofluorescence staining of FLRT3 (green) with RFP (red) in the dorsal horn injected with AAV-shLuc (left panels) or AAV-shFlrt3 #3 (right panels) at day 14 after SNL injury. Dotted line indicates the border between the white and gray matter. Bottom, left, Boxed areas are shown at higher magnification (right panels). FLRT3 immunoreactivity colocalized with RFP-positive afferent neurons and projection neurons in the dorsal horn of the control shRNA group (top, right). FLRT3 immunoreactivity decreased in the RFP-positive area of the dorsal horn of the FLRT3 shRNA group (bottom, right, arrowhead). Scale bars: left (low-magnification images), 200 μm; right (high-magnification images), 50 μm. H, Changes in the hindpaw-withdrawal threshold after SNL with intraganglionic injection (left L4 DRG) of AAV vectors encoding Flrt3 (shFlrt3 #3) or control (shLuc) shRNA 14 d before SNL injury. Flrt3 knockdown reversed the SNL-induced decrease in the paw-withdrawal threshold (n = 6–9 rats per group). *p < 0.05 (two-way repeated-measures ANOVA followed by Tukey–Kramer test).
Discussion
In this study, we showed that FLRT3 derived from injured sensory neurons induced mechanical sensitivity in the spinal cord dorsal horn. Four days after nerve ligation, Flrt3 mRNA expression increased in the DRG, where peripheral sensory neurons are located. Recombinant FLRT3 protein administered intrathecally increased the level of pGluN2B and led to mechanical allodynia. Furthermore, DRG-specific FLRT3 expression, achieved by transducing neurons with AAV encoding a rat FLRT3-myc fusion protein, induced mechanical sensitivity in naive rats. An antibody against FLRT3 suppressed mechanical allodynia after PSL, and knockdown of Flrt3 in the DRG partially suppressed mechanical allodynia after nerve injury. Our findings suggest a novel function of FLRT3 in inducing pain signals.
The PSL model started to show mechanical allodynia at day 4 after injury, and this condition lasted until 28 d after injury. FLRT3 protein expression in the dorsal horn was also first detected at day 4 after injury (coinciding with the development of mechanical sensitivity) and persisted for up to 21 d after injury. The Flrt3 gene was rapidly upregulated in injured sensory neurons, and FLRT3 protein expression was observed in ATF3-immunoreactive DRG neurons with varying diameters. These findings are consistent with those of a previous study, which used a nerve transection model (Lawson and Waddell, 1991; Robinson et al., 2004). Although Flrt3 mRNA expression increased in the DRG after nerve injury, the Flrt3 mRNA levels remained unchanged in a model of inflammatory pain induced by intraplantar complete Freund's adjuvant administration. Robinson et al. (2004) used an ultrastructural analysis to show that FLRT3 was localized to presynaptic axon terminals. Therefore, it is intriguing to hypothesize that FLRT3 protein produced in the DRG in response to injury is delivered to the dorsal horn.
Immunohistochemical staining performed with FLRT3 and dorsal horn lamina marker (CGRP, IB4, VGLUT1) antibodies showed FLRT3 staining in varicosity-like structures within the ipsilateral dorsal horn laminae I–V, particularly in the IB4-negative area (lamina IIID), where nonpeptidergic C fibers project (Silverman and Kruger, 1990; Lai et al., 2016). After nerve injury, C fibers showed synapse degeneration and a transient loss of IB4 binding (Bailey and Ribeiro-da-Silva, 2006). Given that the levels of adhesion molecules, such as L1, NCAM, and N-cadherin, are upregulated to promote neurite outgrowth after injury (Raivich and Makwana, 2007), it is possible that FLRT3 is transported to the terminals of axotomized DRG neurons. Indeed, FLRT3 has been reported to promote neurite outgrowth in DRG neurons in vitro (Robinson et al., 2004). Therefore, FLRT3 may function physiologically in regenerating injured DRG neurons.
It has been widely reported that glial activation contributes to the development and maintenance of neuropathic pain (Tsuda et al., 2003; Ji et al., 2013). In particular, nerve injury is known to cause the activation of microglia, leading to cytokine or chemokine production that increases the excitability of dorsal horn projection neurons (Tsuda et al., 2003; Pezet and McMahon, 2006; Calvo et al., 2012). Therefore, the involvement of FLRT3 in microglial activation could be a possible mechanism underlying the increased mechanical sensitivity. We demonstrate that anti-FLRT3 antibody, which suppresses the interaction between FLRT3 and Unc5b, ameliorates mechanical allodynia, suggesting that Unc5B mediates the FLRT3-induced pain signal (Fig. 5). We previously found that Unc5B expression was not observed in Iba1-positive microglia under pathological conditions (Hayano et al., 2016). In addition, the binding of Netrin-4 to Unc5b induced allodynia and neuronal excitability without the activation of microglia, and Netrin-4 induced allodynia was not inhibited by microglial inhibitor minocycline. These findings suggest that the activation of microglia might not be involved in FLRT3-induced allodynia via Unc5b.
Recombinant FLRT3 administration resulted in increased levels of phosphorylated GluN2B in the rat spinal cord. Phosphorylation of GluN2B, a subunit of the NMDAR, is involved in the regulation of excitatory synaptic transmission, and the NMDAR is a major player in synaptic alterations during central sensitization of the dorsal horn (Latremoliere and Woolf, 2009). GluN2B in the spinal cord is thought to be important for neuropathic pain (Guo et al., 2002; Tan et al., 2005). The phosphorylation of GluN2B contributes to neuronal activation (Iwata et al., 2007) and the development and maintenance of hyperalgesia (Guo et al., 2002). In addition, an increase in NMDAR-mediated synaptic current is also associated with neuropathic pain (Isaev et al., 2000; Yan et al., 2013). We previously found that Netrin-4 treatment increased GluN2B/NR2B phosphorylation via Unc5B signaling, and NMDAR-mediated currents were increased after Netrin-4 treatment (Hayano et al., 2016). Therefore, enhanced synaptic transmission by modulating NMDAR upon the binding of FLRT3 to Unc5b might partially mediate FLRT3-induced allodynia.
We show that recombinant FLRT3 is sufficient to induce hypersensitivity from 12 to 48 h, and GluN2B phosphorylation was induced 12 h after the FLRT3 injection (Fig. 3). The modulation of functional neural networks via axonal growth after neuronal injury is thought to take longer than 48 h. It is known that the expression of GAP-43, which is an axon growth-associated protein, is detectable 3 d after sciatic nerve injury in adult rats (Woolf et al., 1990). The alteration of neuronal activity seems to occur at an earlier phase following insult. The phosphorylation of NR2B increased 10 min after the induction of hyperalgesia (Guo et al., 2002). Therefore, recombinant FLRT3-induced mechanical hypersensitivity might be associated with synaptic excitability modulated by NMDAR, rather than the alteration of neural networks involving axonal growth.
Mechanical sensitivity was induced by intrathecal administration of recombinant FLRT3 protein (Fig. 3B). Consistent with this result, AAV-mediated FLRT3 expression in the DRG also induced mechanical allodynia (Fig. 4E). In contrast, knockdown of Flrt3 expression using shRNA partially attenuated mechanical allodynia, suggesting that Flrt3 expression in the dorsal horn may play a role in the development and persistence of neuropathic pain. These results suggest that FLRT3 may be a molecular target for pain relief therapeutics.
In the dorsal horn, FLRT3 may act as a synaptic adhesion molecule to modulate connectivity in the dorsal horn neuronal circuit (O'Sullivan et al., 2012) (e.g., to strengthen excitatory synaptic connections), leading to neuropathic pain. Although FLRT3 has been reported to bind to several molecules (e.g., FLRT3, Unc5B, and LPHNs) (Karaulanov et al., 2006; O'Sullivan et al., 2012; Seiradake et al., 2014), we could not identify the binding partner of FLRT3 in the dorsal horn after nerve injury in this study. FLRT3 has been reported to play a role in regulating glutamatergic synapse formation with LPHN3, its binding partner, in the hippocampus and in vitro (O'Sullivan et al., 2012). Recently, presynaptic FLRT3 binding to LPHN has been shown to mediate excitatory synapse formation (Sando et al., 2019). It is intriguing to hypothesize that the enhanced synaptic transmission induced by FLRT3-LPHN3 signaling is involved in neuropathic pain. Furthermore, we previously reported that Netrin-4, secreted from dorsal horn interneurons, induced mechanical allodynia via Unc5B signaling in dorsal horn neurons in the PSL model (Hayano et al., 2016). Unc5B is an axon-guidance molecule that interacts with other guidance molecules, including FLRT3, during embryonic development. Interestingly, Sema4-Plexin signaling is critically involved in axon guidance and was recently reported to modulate mechanical and thermal sensitivity development in peripheral sensory neurons in an inflammatory pain model (Paldy et al., 2017). In addition, the expression of other adhesion molecules, such as L1-CAM, ephrinB-EphBR, and CHL1, increased in the dorsal horn and was involved in neuropathic pain (Yamanaka et al., 2007, 2011; Song et al., 2008), suggesting that adhesion molecules may play an important role in the alteration of neural circuitry in the dorsal horn, resulting in neuropathic pain.
Intrathecal administration of an antibody against the extracellular domain of FLRT3 transiently attenuated mechanical sensitivity after the development of neuropathic pain following nerve injury. The antibody inhibited the binding between FLRT3 and Unc5B, suggesting that it may prevent the interaction of FLRT3 with other binding partners as well (Leyva-Díaz et al., 2014; Hayano et al., 2016). However, the mechanism underlying the analgesic effect of this antibody remains unclear.
This study has several limitations. First, because we were unable to identify the binding partner of FLRT3 mediating its function in neuropathic pain development, we cannot rule out the possibility of a nonspecific effect of the FLRT3 antibody used in our experiments. Second, AAV-mediated overexpression and recombinant protein administration may result in higher FLRT3 levels than those observed under physiological conditions. Third, we did not investigate potential differences among rat strains. Further investigation of the function of FLRT3 in neuropathic pain may contribute to the development of novel therapeutic strategies for chronic pain conditions.
Footnotes
This work was supported by Japan Society for the Promotion of Science KAKENHI Grant JP17H06178 to T.Y. and Japan Agency for Medical Research and Development-Core Research for Evolutional Science and Technology 18gm1210005h0001 to T.Y. We thank Drs. Hidenori Suzuki and Atsushi Sakai (Department of Anesthesiology, Graduate School of Medicine, Nippon Medical School) for helpful technical suggestions about AAV injection.
The authors declare no competing financial interests.
- Correspondence should be addressed to Toshihide Yamashita at yamashita{at}molneu.med.osaka-u.ac.jp.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}