
Abstract
Adenosine is a ubiquitous neuromodulator that increases sleep, inhibits seizures, and promotes neuroprotection. Many of these effects are mediated by A1 receptors, but A1 receptors are expressed in most brain regions, and distinguishing the precise site of action of adenosine is challenging. To test the role of adenosine in different hippocampal regions, we have used the Cre-loxP system and an adeno-associated viral (AAV) vector to focally delete endogenous adenosine A1 receptors in the hippocampus. Microinjection of an AAV vector containing the gene for Cre recombinase induced intense, focal, neuron-specific recombination in reporter mice. In a separate line of mice with loxP sites flanking the major coding exon for the adenosine A1 receptor, this AAV-Cre markedly reduced A1 receptor mRNA and focally abolished the postsynaptic response to adenosine without any change in basic electrophysiologic properties. Adenosine inhibits signaling between CA3 and CA1 neurons, but it is unclear from pharmacologic studies whether this response is caused by presynaptic or postsynaptic effects. Deletion of A1 receptors from CA3 neurons abolished this response to adenosine, but deletion of A1 receptors from CA1 neurons had no effect, demonstrating a presynaptic site of action. This transduction knock-out technique holds enormous potential for dissecting the functions of different CNS pathways.
- adenosine
- A1 receptor
- Cre recombinase
- adeno-associated virus
- AAV
- electrophysiology
- patch clamp
Introduction
Adenosine is a ubiquitous neuromodulator that increases sleep, inhibits seizures, and promotes neuroprotection (Dunwiddie and Masino, 2001). Many of these effects are mediated by A1 receptors (A1Rs), but A1Rs are expressed in most brain regions, and distinguishing the precise site of action of adenosine is challenging.
Focal manipulation of the A1R gene is necessary for detailed studies of the neurobiology of adenosine and many other signaling systems. The Cre-loxP system is well suited for this problem because it can be used to produce conditional gene deletions in particular cell types at specific times (Gu et al., 1994; Tsien et al., 1996a). Cre recombinase catalyzes recombination between short loxP sequences, thus deleting any intervening DNA (Sauer and Henderson, 1988). Mice with loxP sites flanking a gene can be crossed with mice expressing Cre only in certain brain regions to produce offspring with a regional lack of that gene.
Although this genetic approach is very useful, it has several critical limitations. First, it requires the use of a regionally specific promoter to drive the expression of Cre. Although some promoters have been identified that are selectively active in certain brain regions or in neurochemically distinct cells, such specific control is unavailable for many regions. In addition, even promoters with well defined activity may be expressed in unpredictable patterns (Tsien et al., 1996a), presumably because of differences in the sites at which the transgene is inserted. Finally, the timing of this conditional gene deletion is determined by the timing of the promoter activity that controls Cre expression.
Recombinant adeno-associated virus (AAV) vectors provide a useful tool to produce expression of Cre in specific brain regions. Recombinant AAV vectors cannot replicate, and thus transgene expression is limited to the area in which the virus was injected, lasting >6 months in nondividing cells such as neurons (Kirik et al., 2000). AAV vectors can transduce a wide range of host cells, and previous work has demonstrated that some AAV serotypes preferentially transduce neurons in vivo with almost no inflammatory response (Chamberlin et al., 1998; Davidson et al., 2000; Kaspar et al., 2002).
Here we describe a technique that allows anatomically specific deletion of the adenosine A1R gene using an AAV vector and the Cre-loxP system. We first tested whether microinjection of AAV-Cre could produce recombination in hippocampal neurons of reporter mice (Tsien et al., 1996a). To test the role of A1 receptors in different hippocampal regions, we then examined whether the injection of AAV-Cre into the hippocampus of mice with loxP sites flanking the major coding exon for the A1R could produce focal deletion of A1 receptors as demonstrated using anatomic and electrophysiologic techniques. This approach helps define critical sites for the effects of adenosine and should be useful in a wide variety of other studies requiring focal changes in gene expression.
Materials and Methods
Production of AAV-Cre and AAV-green fluorescent protein
AAV-Cre is based on AAV serotype 2 and was made using pSub201 (provided by R. J. Samulski, University of North Carolina, Chapel Hill). It contains an expression cassette consisting of the human cytomegalovirus immediate early promoter followed by an intron derived from the human β-globin second intron, and the polyadenylation signals from the human β-globin third exon (Fig. 1 A). The cassette is flanked by the AAV inverted terminal repeats and was modified from pMD.M (Ory et al., 1996). The Cre gene sequence, derived from pMC/CreN (provided by Andre Choulika, Institut Pasteur, Paris) (Choulika et al., 1996) and containing an N-terminal SV40 large T-antigen nuclear localization signal, was inserted in the cassette downstream of the intron (Fig. 1 A). AAV vector containing green fluorescent protein was made using a cassette with the UF5 version of green fluorescent protein driven by the CMV promoter and followed by the SV40 early region polyadenylation sequence (Chamberlin et al., 1998).

A, Construct for production of AAV-Cre. Expression of Cre is driven by the human CMV immediate-early promoter followed by a human β-globin intron. The β-globin polyadenylation (pA) sequence follows the Cre gene sequence, and the construct is flanked by inverted terminal repeats. SD, Splice donor site; SA, splice acceptor site. B, Schematic representation of the construct for production of the inducible A1 receptor knock-out mouse. A 12.8 kb construct was created with loxP sites flanking the major coding exon for the mouse A1 receptor; this exon is homologous to human A1 receptor exon 6.
Viral vectors were generated by tripartite transfection of AAV-rep/cap expression plasmid, adenovirus mini-plasmid, and AAV vector plasmid into 293A cells (Xiao et al., 1998) and purified by fractionation through two cycles of CsCl gradient centrifugation. Positive fractions were identified by dot blot hybridization, pooled, dialyzed against PBS, and titered by dot blot hybridization. The final titer of AAV-Cre was 1.3 × 10 12/ml, based on dot blot hybridization.
Production of inducible A1R knock-out mice
The targeting vector for generating the inducible A1R knock-out allele was constructed using plasmid pB-Not-XhoMaxi (pMaxi), which contains the major coding exon of the A1R [homologous to human exon 6; a gift from B. Johansson (Johansson et al., 2001)]. A loxP site along with HindIII and BamHI sites were inserted into the EcoR1 site 5′ to the exon. The 4.5 kb fragment from plasmid pGB128 (R. Mortensen, Brigham and Women's Hospital, Boston, MA) containing the cytosine deaminase and neomycin resistance genes was inserted into the NcoI site 3′ to exon 6. This fragment was flanked by loxP sites to allow removal of the cassette. All three loxP sites in the final targeting vector were in the same 5′ to 3′ orientation. A 1.6 kb fragment from plasmid pGKneobpAlox2PGKDTA (Frank Gertler, Massachusetts Institute of Technology, Cambridge, MA) containing the diphtheria toxin gene was inserted into the XhoI site of pMaxi. The final targeting vector was linearized with NotI and transfected into J1 embryonic stem (ES) cells derived from 129SvJ mice (Li et al., 1992) (Fig. 1 B).
To identify clones of ES cells that had undergone homologous recombination, we used a 1 kb BamHI–XhoI fragment that resides 3′ to the flanking sequences in the targeting vector as a Southern probe. A BamHI digest of wild-type DNA probed with this fragment produced a 20 kb band, whereas the homologously recombined allele gave a 6.3 kb band. Clones identified to have homologously recombined the 3′ portion of the targeting vector were further analyzed to identify the subset of clones that obtained the 5′ loxP site. HindIII or EcoRV digests were probed with the 800 bp EcoR1–EcoRv (E-E) fragment from the 5′ end of exon 6. HindIII digests of wild-type alleles produced a 5.2 kb band, whereas the homologous recombined alleles that contain the 5′ loxP site gave a 1.2 kb band. EcoRV digests of wild-type and homologous recombined alleles probed with the E-E probe generated a 9.6 kb band, whereas DNA resulting from random integrations gave aberrant-sized bands. Appropriate mutant cell lines were selected to make chimeras from C57BL/6 mice. We then crossed the chimeras with C57BL/6 mice and cross-bred the offspring to generate mice homozygous for the A1R gene flanked by loxP sites.
Animals and microinjections
Mice were housed in a pathogen-free barrier facility maintained at 21.5–22.5°C with lights on at 7 A.M. and off at 7 P.M. Mice had food and water available ad libitum. The Institutional Animal Care and Use Committees and Committee of Microbiologic Safety of Harvard Medical School approved all procedures.
Adult, male mice weighing 25–35 gm were anesthetized with chloral hydrate (450 mg/kg, i.p.), and 1 μl AAV-Cre or pyrogen-free saline was stereotaxically microinjected into the dorsal hippocampus (2 mm behind bregma, 2 mm lateral, and 1.6 mm below the dural surface for CA1, and 1.9, 2.1, and 2.1 mm, respectively, for CA3). To minimize tissue injury, these injections were performed using glass pipettes with a 10- to 20-μm-diameter tip, and AAV-Cre or saline was slowly injected over 1 hr using a pressure-injection system (Scammell et al., 1998).
Immunohistochemistry
Two to 5 weeks after these microinjections, the mice were deeply anesthetized with chloral hydrate (600 mg/kg, i.p.) and perfused with saline followed by 4% paraformaldehyde. Brains were postfixed for 2 hr, equilibrated in 20% sucrose, and cut at 30 μm on a sliding microtome. Immunohistochemistry was performed on free-floating sections as described previously (Estabrooke et al., 2001). Primary antibodies included rabbit anti-β-galactosidase (β-gal; 1:10,000 dilution; 3′5′, Inc.), rabbit glial fibrillary acidic protein (GFAP; 1:10,000; Sigma, St. Louis, MO), mouse anti-Cre (1:500; Covance Research Products), mouse anti-neuron-specific nuclear protein (NeuN) (1:1000; Chemicon), rat anti-CD45 (1:50; BD PharMingen), and rat anti-leukocyte common antigen (CD11b) (1:500; BD PharMingen). After incubation with primary antisera overnight, sections were rinsed and incubated for 2 hr in biotinylated anti-rabbit or anti-rat secondary antisera (Jackson ImmunoResearch) at 1:500 or 1:1000 dilutions. Biotinylated secondary antiserum from the MOM kit (Vector Laboratories) was used for Cre and NeuN immunostaining. Tissue was then reacted with avidin–biotin complex (Vectastain ABC Elite kit, Vector Laboratories) for 1 hr, and immunoreactive cells were visualized by reaction with 3,3′-diaminobenzidine/3% H2O2/0.01% NiSO4/0.01% CoCl2. Double-fluorescent immunostaining of NeuN and β-gal was performed using streptavidin-conjugated Alexa-488 (Molecular Probes) and Cy3-conjugated donkey anti-rabbit secondary antiserum (Jackson Laboratories) at 1:500 dilutions. Light and fluorescence microscopy were performed with a Zeiss Axioplan 2 microscope and a Bio-Rad MRC1024 confocal microscope, respectively.
The number of β-gal-immunoreactive (IR) cells was estimated using systematic random sampling in eight reporter mice (four each, 2 or 5 weeks after AAV-Cre injection) (Howard and Reed, 1998). To estimate the volume in which recombination had occurred, the length and breadth of the β-gal-expressing region were measured on 38 consecutive 30 μm sections of a 1:3 series using a microscope reticule. To estimate the density of β-gal-IR cells within this region, counting boxes (42 × 210 μm) were placed at 840 μm intervals across this region. The total number of cells was then calculated by multiplying the estimated volume by the cell density. The relative numbers of GFAP-IR and CD45-IR cells per section were counted in a 420 × 420 μm region centered on the injection site of mice treated with saline or AAV-Cre or the same region of uninjected mice (n = 2–3 in each group). In inducible A1R knock-out mice, the relative number of NeuN-IR neurons per section was counted on confocal images of a 200 × 200 μm area ipsilateral and contralateral to the AAV-Cre injection site (n = 5).
In situ hybridization
The A1R riboprobe was generated from a Bluescript II SK(+) plasmid containing bases 436–900 of the rat A1R cDNA sequence (a kind gift of D. Weaver, University of Massachusetts Medical School) (Reppert et al., 1991). Across this fragment, the rat and mouse cDNA sequences are 96% homologous [basic local alignment search tool (BLAST)]. This plasmid was linearized with EcoR1 and transcribed with T7 to produce antisense probe or with HindIII and T3 to produce sense probe. The hippocalcin riboprobe corresponded to bases 59–503 of the rat hippocalcin cDNA, a region in which the rat and mouse sequences are 97% homologous. In situ hybridization procedures were identical to previously described methods (Chou et al., 2001). No hybridization signal above background was evident with A1R sense probes used on A1R transgenic mice or with A1R antisense probes used on brain tissue from three constitutive A1R knock-out mice (Johansson et al., 2001).
The expression of A1R hybridization signal after injection of AAV-Cre into five inducible A1R knock-out mice was measured by comparing the optical densities of the ipsilateral CA1 region with the corresponding contralateral region. First, the center of the injection site was identified on sections immunostained for Cre. Then, the optical density of A1R signal was measured within this region on a film autoradiogram of an adjacent section. Measurements were performed within this central 50 × 200 μm area using NIH Image 1.61, and optical densities were normalized by subtracting the background optical density of the corpus callosum at the midline.
Electrophysiological recordings
Electrophysiological recordings were made in the CA1 region in hippocampal slices obtained from wild-type mice or from inducible A1R knock-out mice 2–6 weeks after AAV-Cre injection.
Hippocampal slice preparation. Coronal, 300 μm slices were prepared from isoflurane-anesthetized animals and maintained at 4°C in artificial CSF. This solution was saturated with 95% O2 and 5% CO2 and consisted of (in mm): 128 NaCl, 3 KCl, 0.5 NaH2PO4, 1 MgCl2, 1.5 CaCl2, 23.5 NaCO3, 30 glucose, pH 7.35, 315–320 mOsm. Slices were recorded submerged and perfused with this solution (1.8 ml/min) at 32°C.
Patch-clamp recordings. Whole-cell patch-clamp recordings were performed using an Axoclamp-2A amplifier (Axon Instruments, Foster City, CA) and infrared differential interference contrast microscopy (Stuart et al., 1993) (Axioscop 2 FS, Zeiss; Scion Image acquisition and display software). Signals were filtered at 0.5–1 kHz and digitized on-line at 2–5 kHz with Digidata 1200 hardware and pClamp 8.2 software (Axon Instruments). Patch electrodes (4–8 MΩ) were filled with (in mm): 120 K-gluconate, 10 KCl, 3 MgCl2, 10 HEPES, 2 MgATP, 0.2 NaGTP, pH 7.2, adjusted with KOH, 280 mOsm). Lucifer yellow CH ammonium salt (0.1%) was added to the pipette solution to label recorded cells. After recording, slices were fixed and cut at 30 μm. The location of labeled cells was confirmed by confocal microscopy for lucifer yellow and Cre immunolabeled with Alexa-350 and A1R in situ hybridization on adjacent sections.
Extracellular field recordings. Field EPSPs (fEPSPs) were recorded from the CA1 stratum radiatum using glass electrodes filled with 2 m NaCl (2–4 MΩ). Signals were filtered at 1–2 kHz and digitized at 40 kHz with Digidata 1200 hardware and pClamp 8.2 software (Axon Instruments). Using a bipolar electrode placed in the stratum radiatum adjacent to CA2, the Schaffer collateral fibers were stimulated every 10–15 sec with single, constant current pulses of 0.2 msec duration. Stimulation pulses were delivered with a constant-current source (Iso-flex; A.M.P.I. Jerusalem, Israel), triggered by Clampex software (Axon Instruments), and stimulus strength was adjusted to give ∼75% of maximum fEPSP amplitude ranging between 0.5 and 2.0 mV. Evoked field potentials were quantified as the slope of the fEPSP measured between 10 and 90% of maximum fEPSP amplitude.
Results
Recombination in transgenic reporter mice
To determine the transduction efficiency of AAV-Cre in vivo, we injected AAV-Cre into the hippocampus of Cre-excision reporter mice (a kind gift of D. Anderson, California Institute of Technology). These mice have a transgene in which the expression of β-gal is driven by a chicken β-actin promoter, but a stop sequence upstream of the lacZ gene prevents β-gal expression (Tsien et al., 1996a). This stop sequence is flanked by loxP sites, and Cre recombinase can remove the stop sequence, allowing expression of β-gal.
Two weeks after stereotaxic microinjection of 1 μl AAV-Cre into the hippocampus of reporter mice, the injection site contained numerous Cre-IR cells (Fig. 2). Cre immunoreactivity was located mainly in the cell-dense pyramidal and granular layers but also was common in cells scattered throughout the stratum oriens and stratum radiatum. Rare Cre-IR cells had morphology suggestive of astrocytes. No Cre immunoreactivity was evident in saline-injected mice.

Injection of AAV-Cre into the dorsal hippocampus of a reporter mouse results in extensive expression of Cre immunoreactivity in neurons throughout the CA1 region and dentate gyrus as well as in interneurons of the stratum oriens and stratum lucidum. Scale bar, 250 μm.
Production of Cre in these cells induced intense, focal expression of β-gal, indicative of Cre-mediated recombination (Fig. 3). This β-gal immunoreactivity was most evident within the nuclei of cells in the pyramidal cell layer of the CA fields and in the granule cell layer of the dentate gyrus, with lighter staining in the cytoplasm and proximal cell processes. Using stereologic methods, we found that injection of 1 μl of AAV-Cre (∼109 vector particles) resulted in β–gal expression in ∼105 CA field and dentate gyrus cells, typically extending laterally across more than half of the dorsal hippocampus and up to 2–3 mm in the rostral–caudal direction. β-gal-IR somata were much less common in the stratum oriens and stratum radiatum, even in mice with many Cre-IR cells in those regions. Although these presumed interneurons may have undergone recombination, they may produce β-gal less effectively than CA field neurons. β-gal-IR cells were very rare in the contralateral hippocampus, although a few β-gal-containing cells were usually present in the hilus of the dentate gyrus bilaterally. The stop sequence that normally prevents β-gal expression may be less effective in these cells because a few β-gal-IR cells were present even in the dentate gyrus of uninjected mice. Most brains contained a few β-gal-IR pyramidal neurons in the entorhinal cortex just below the rhinal fissure; these were probably retrogradely labeled perforant pathway neurons (Chamberlin et al., 1998; Kaspar et al., 2002). Injections of AAV-Cre into the striatum or thalamus also induced robust, local expression of β-gal immunoreactivity.

Injection of AAV-Cre into the dorsal hippocampi of reporter mice induces the expression ofβ-galactosidase immunoreactivity in thousands of neurons. A, Injection site centered on the dentate gyrus and CA3 field. B, A different case with the injection site centered on the CA1 field and dentate gyrus. Scale bar, 1 mm.
Recent work suggested that high concentrations of Cre can be cytotoxic (Loonstra et al., 2001; Pfeifer et al., 2001; Silver and Livingston, 2001), with a marked decrease in the number of β-gal-expressing cells 5 weeks after injection of a lentiviral vector containing Cre (Pfeifer et al., 2001). To determine whether AAV-Cre produced similar cell loss, we examined β-gal expression in the hippocampi of reporter mice 2 and 5 weeks after injection of AAV-Cre. Mice killed 2 weeks after injection had 104,000 ± 31,000 β-gal-IR cells, and mice killed 5 weeks after injection had 101,000 ± 38,000 cells (n = 4 in each group; Mann–Whitney, p = NS).
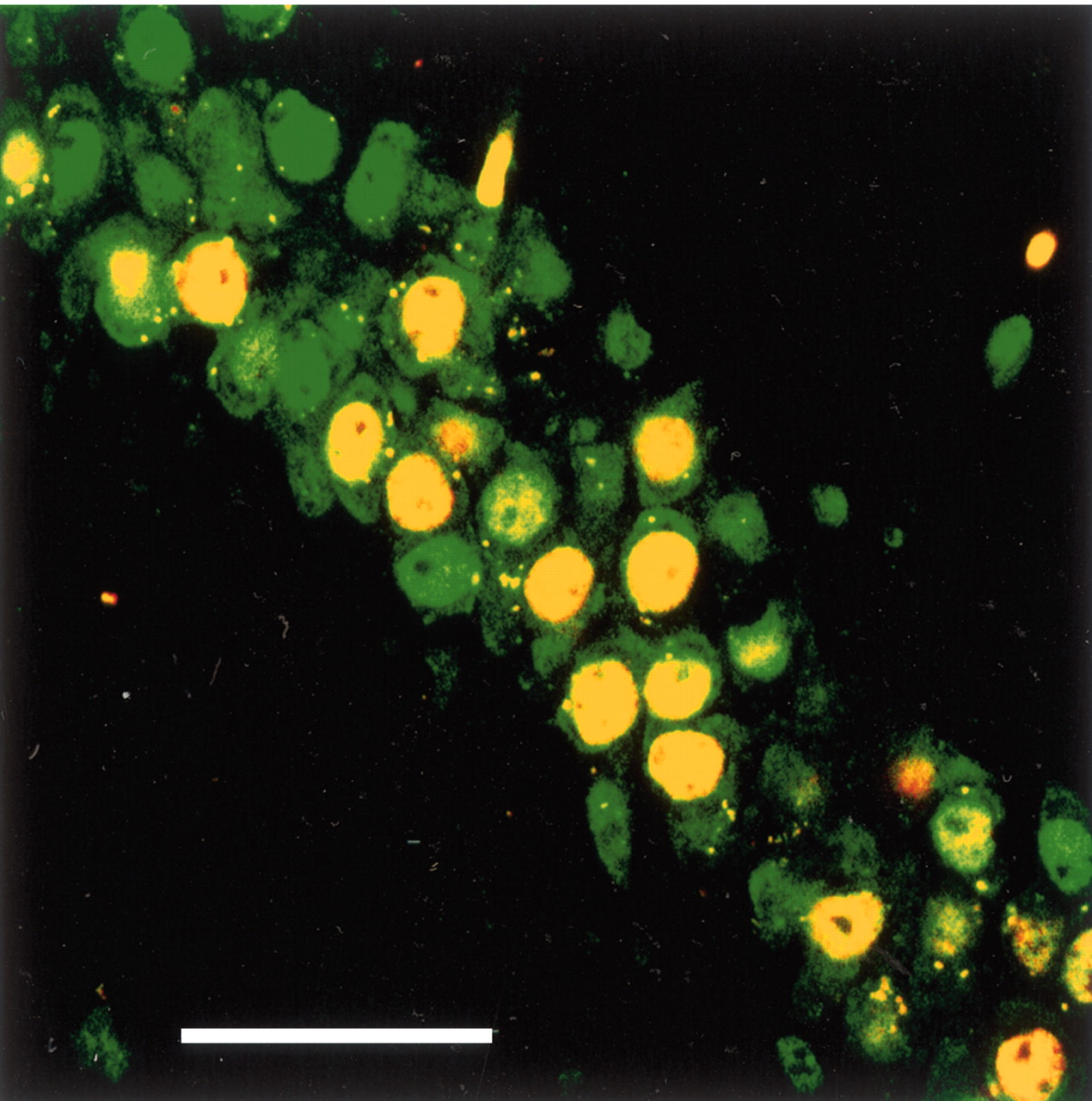
To determine whether β-gal was induced in neurons, we double immunostained hippocampal sections from four reporter mice for β-gal and the neuronal marker NeuN. Within a 400 × 400 μm region centered on the injection sites, at least 62% of neurons produced β-gal, and 99% of β-gal-IR cells stained for NeuN (Fig. 4). Double labeling for the astrocyte marker, GFAP, and β–gal demonstrated only very rare β–gal-IR astrocytes. Double immunostaining for β–gal and CD45 (leukocyte common antigen) produced no double-labeled cells. This neuronal selectivity most likely reflects the neurotropic nature of AAV serotype 2 (Davidson et al., 2000).

Confocal microscopic image of β-gal and NeuN immunoreactivity in the CA1 region of a reporter mouse injected with AAV-Cre. Green, NeuN immunoreactivity is apparent in the nucleus and to a lesser degree in the cytoplasm of neurons. Red, β-gal immunoreactivity is found only in neurons as indicated by yellow double labeling. Scale bar, 50 μm.
These injections were performed over 1 hr using a glass pipette with a 20 μm tip. This approach produced little tissue injury and resulted in much higher rates of transduction than seen with faster injections using a Hamilton syringe [see also Mastakov et al. (2001)]. Even so, most animals that received injections of AAV-Cre or saline had a small increase in the number of astrocytes and a clear increase in the number of microglial cells (Fig. 5). In a 420 × 420 μm region centered on the CA1 injection sites, the number of GFAP-IR somata per 30 μm section was 92 in uninjected mice, 134 in saline-injected mice, and 154 in AAV-Cre-injected mice (p = 0.07 by ANOVA; n = 3–4 in each group). The astrocytes of injected animals often had an activated appearance, with enlarged soma and thick, numerous processes. In uninjected animals, this region contained an average of 28 CD45-IR microglia, but saline- and AAV-injected animals had 78 and 122 cells, respectively (p = NS). Nearly all of these cells also stained for CD11b, confirming that they were microglia. This microgliosis was variable but usually did not extend beyond the injected hippocampus. CD45-IR lymphocytes were very rare in uninjected brains, but AAV-Cre or saline injection sites typically contained two to four cells per section. Thus, although this injection technique caused little overt tissue injury, it did produce a small amount of astrogliosis and microgliosis. However, compared with saline, injection of AAV-Cre caused little additional inflammation.

Stereotaxic microinjections produce a mild inflammatory response. In the hippocampus of an uninjected reporter mouse (A), GFAP-immunoreactive astrocytes are moderately common, but in the hippocampus of reporter mice injected with either saline (B) or AAV-Cre (C), the number of astrocytes is slightly increased. These sections have also been immunostained for β-gal that is evident in the CA1 region of the AAV-Cre-injected reporter mouse. The hippocampi of uninjected reporter mice contain rare CD45 immunoreactive microglia (D), whereas moderate numbers of microglia are evident in saline- or AAV-Cre-injected mice (E, F). Occasional CD45-immunoreactive lymphocytes are present after injection of saline or AAV-Cre. Sections immunostained for CD45 were not double labeled for β-gal. Scale bar, 200 μm.
Recombination in A1 receptor transgenic mice
The adenosine A1R is expressed at high levels in hippocampal CA field neurons. Through these receptors, adenosine tonically inhibits pyramidal cell activity (Greene and Haas, 1985), providing protection against neural injury from hypoxia, hypoglycemia, or seizures (Dunwiddie and Masino, 2001). To enable focal deletion of the A1R gene, we produced an inducible A1R knock-out mouse. The mice were phenotypically normal and appeared with the expected Mendelian frequency.
Injection of AAV-Cre into the hippocampus of these mice induced local expression of Cre and a marked decrease in A1R mRNA as indicated by in situ hybridization (Fig. 6). The mean optical density of A1R hybridization signal in the CA1 field injected with AAV-Cre was only 8% of that seen in the contralateral, uninjected hippocampus (n = 5; p = 0.002 by ANOVA). These AAV-Cre injections substantially decreased A1R expression over 30–50% of the dorsal hippocampus, extending 1–2mm in the rostral–caudal direction. Staining of adjacent sections revealed that this decrease in A1R mRNA was precisely coextensive with the expression of Cre.

AAV-Cre focally deletes A1 receptor mRNA. A, Injection of AAV-Cre into the brain of an inducible A1 receptor knock-out mouse induces the expression of Cre in the CA1 field and the dorsal leaf of the dentate gyrus. B, A1 receptor mRNA is markedly reduced in the same regions on an adjacent section. In a different inducible A1R mouse, AAV-Cre deletes A1R mRNA in the CA1 field and dentate gyrus (C), yet the expression of hippocalcin mRNA is normal on an adjacent section (D). Scale bars, 1 mm.
This substantial decrease in A1R expression was not caused by a reduction in the number of CA field neurons. Within a 200 μm by 200 μm region centered on the AAV-Cre injection site, the relative number of NeuN-IR neurons was 55 per section, compared with 57 in the matching contralateral region (n = 4; p = NS by ANOVA). This reduction of A1R mRNA also was not caused by a nonspecific decrease in mRNA production because in situ hybridization for hippocalcin was unaltered (Fig. 6D). Control injections of saline or AAV vector containing the gene for green fluorescent protein did not alter A1R expression.
Electrophysiology after deletion of A1 receptors
Adenosine inhibits CA1 and other CNS neurons via postsynaptic A1 receptors by inducing a G-protein-activated K+ current (Trussell and Jackson, 1987; Luscher et al., 1997). Two to 6 weeks after the unilateral injection of AAV-Cre into the CA1 region of inducible A1R knock-out mice, we examined the effect of adenosine (50 μm)on in vitro hippocampal slices. At a holding potential of –60 mV, all neurons recorded from the CA1 region contralateral to the AAV-Cre injection responded to adenosine with a robust, outward current (Fig. 7A) (n = 5) that was identical to that recorded from CA1 neurons in uninjected, wild-type mice (n = 3). Nine CA1 pyramidal neurons were recorded from within AAV-Cre injection sites. In seven of these neurons, adenosine had no effect (Fig. 7B,C), just as seen in constitutive A1R knockout mice (Johansson et al., 2001). In the remaining two cells, adenosine induced the typical outward current. This all-or-none response to adenosine is most consistent with A1R gene deletion.

Deletion of A1 receptors abolishes the postsynaptic effects of adenosine on CA1 hippocampal neurons. A, Under voltage-clamp control (Vh = –60 mV), adenosine (50 μm) induces a robust outward current in neurons from wild-type C57BL/6 mice (□, n = 3) or in neurons contralateral to an injection of AAV-Cre into the CA1 region of an inducible A1 receptor knock-out mouse (•, n = 5). B, In contrast, adenosine had no effect on seven CA1 neurons within the AAV-Cre injection site (•), although two cells had normal responses to adenosine (□, ○). Holding currents (Ihold) are presented as the change from the baseline during the first 3 min of each recording. Grouped cells are displayed as means with SDs. C, Current–voltage relationships of the adenosine-induced current of two representative CA1 neurons: one recorded within an AAV-Cre injection site (thick line) and one recorded contralateral to an AAV-Cre injection (thin line). Each line represents the difference between current in the presence of adenosine and artificial CSF alone. Signals were recorded during voltage ramp protocols from –110 to –60 mV (7 sec duration) and averaged from three recordings.
Although these neurons lacked any response to adenosine, their basic electrophysiologic properties were normal. Comparison of neurons responsive and unresponsive to adenosine revealed no differences in action potential threshold, duration or amplitude, resting membrane potential, or input resistance (Fig. 8, Table 1).

Deletion of A1 receptors does not affect basic electrophysiologic properties of CA1 neurons. A, Under current clamp, a representative neuron in the CA1 region contralateral to an injection of AAV-Cre responds to 1 sec current pulse injections with a series of accommodating, single action potentials. B, Ipsilateral to an injection of AAV-Cre, neurons lacking any response to adenosine had similar properties. C, Expanded view of the first (left) and third (right) action potentials shown in A and B show no differences in action potential duration, amplitude, or afterhyperpolarization. Action potentials recorded from the AAV-Cre injection site are represented with a bold line.
Electrophysiologic characteristics are unaltered in CA1 pyramidal neurons lacking A1 receptors (n = 5)
Stimulation of the CA3 Schaffer collateral input to the CA1 neurons elicits an fEPSP that is inhibited by adenosine. A1 receptors are present on CA1 and CA3 neurons, and it is unclear whether this response to adenosine occurs presynaptically or postsynaptically. To determine the site at which adenosine acts, we injected AAV-Cre into the CA1 or CA3 regions of inducible A1R knock-out mice. Electrophysiologic responses to adenosine were analyzed in all slices with histologically confirmed deletion of A1 receptors. Contralateral to the injection of AAV-Cre, adenosine (50 μm) reduced the fEPSP to ∼40% of its baseline amplitude, and a similar decrease occurred in slices lacking A1 receptors in the CA1 neurons (Fig. 9). In contrast, deletion of A1 receptors from CA3 neurons abolished the effect of adenosine, demonstrating a presynaptic site of action.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
fEPSPs in the CA1 region are inhibited by presynaptic effects of adenosine. Schaffer collaterals from CA3 neurons were stimulated, and fEPSPs were recorded in the stratum radiatum of the CA1 region. A, In a slice lacking A1 receptors in the CA1 region, adenosine decreased the fEPSP to ∼40% of its baseline amplitude, and a similar response was seen in the contralateral, uninjected hippocampus. B, In a slice lacking A1 receptors in the CA3 neurons, adenosine had almost no effect on the fEPSP. C, Mean fEPSP responses to adenosine with focal deletion of A1 receptors from the CA1 or CA3 regions (n = 3–5 slices). *p < 0.01 by ANOVA with post hoc Fisher's test.
Discussion
The transduction knock-out technique described here allows efficient and focal deletion of endogenous genes or transgenes. In our reporter mice, AAV-Cre induced robust recombination almost exclusively in neurons. In the inducible A1R knock-out mice, hippocampal injection of AAV-Cre markedly reduced A1R mRNA. Most importantly, focal deletion of A1 receptors from CA1 neurons blocked any postsynaptic response to adenosine, and deletion of A1 receptors from CA3 neurons blocked the effects of adenosine on the fEPSP.
Although this method is useful for focally manipulating gene expression, it has some limitations. Even large or multiple injections are unlikely to knock out a gene within all neurons of a region, but because stereotaxic injections differ slightly in the distribution of recombined cells, this variability can provide useful anatomic controls. In addition, injection-related inflammation, the transduction of neurons, or the expression of vector genes might alter the behavior of neurons. However, we and others (Ehrengruber et al., 2001) have found no changes in the electrophysiologic properties of neurons transduced with AAV. Finally, AAV-Cre predominantly transduces neurons, but some experiments may require altered gene expression in non-neuronal cells. Fortunately, capsids that direct binding and entry differ in other serotypes of AAV. For example, AAV4 efficiently transduces ependyma, and AAV5 transduces astrocytes and neurons (Davidson et al., 2000). Use of cell type-specific promoters could further enhance selectivity.
This approach provides several advantages when compared with other molecular methods currently in use. First, it has broader applicability than previous methods for regional gene deletion. Most genetic approaches require cell- or region-specific promoters to drive the expression of Cre (Lakso et al., 1992; Tsien et al., 1996a,b; Schwenk et al., 1998; Iwasato et al., 2000), and such promoters are currently available for only a limited number of brain regions. In contrast, AAV-Cre can simply be injected into any brain area, thus allowing one to easily test the function of a gene in many brain regions (Kaspar et al., 2002). For example, A1Rs are hypothesized to play a role in the production of sleep (Alam et al., 1999; Strecker et al., 2000), but it is unknown which brain regions mediate this response. Focal deletion of A1R from different brain regions could help identify these critical sites. These injections of AAV-Cre also enable one to delete a gene unilaterally, thus allowing the contralateral region to serve as a control.
Second, this technique allows creation of a regional knock-out at any age, enabling great flexibility in experimental design. As with other types of inducible gene expression systems, one can compare an animal's behavior before and after the knock-out. Deletion of a gene in adult animals avoids concerns about abnormal development or developmental compensations for the knock-out. Although the microinjections might be more challenging, AAV-Cre also could be used to test the role of a gene earlier in development.
These injections of AAV-Cre produce very little toxicity or inflammation. Methods producing high levels of Cre can be complicated by cell toxicity (Loonstra et al., 2001; Pfeifer et al., 2001; Silver and Livingston, 2001), but with AAV-Cre, we and others (Kaspar et al., 2002) found no decrease in the number of β-gal-expressing cells over time. In addition, the AAV-Cre injection site contained the same number of neurons as the corresponding contralateral region, most likely because our vector induces relatively low, nontoxic levels of Cre. As described previously (Blomer et al., 1997; Kaspar et al., 2002), injections of AAV-Cre produced no more gliosis than seen with saline injections. Most likely, this modest gliosis is related to the 1 μl volumes used in our study, because 50 nl AAV produces no gliosis at all (Chamberlin et al., 1998). Production of AAV with higher titers (Bennett et al., 1999) should allow injection of smaller volumes to minimize this inflammatory response. Our use of glass pipettes and slow infusion rates were clearly helpful; in preliminary experiments using Hamilton syringes and faster infusion rates, injury and inflammation were much more pronounced, and the rates of transduction were much lower [also see Mastakov et al. (2001)]. Recent studies using lentiviral vectors appear encouraging (Pfeifer et al., 2001), but compared with previous reports of Cre-containing vectors derived from herpes virus or adenovirus (Wang et al., 1996; Brooks et al., 1997), AAV-Cre appears to produce higher rates of recombination with much less inflammation (Kaspar et al., 2002).
This technique allowed us to focally delete the gene coding for the adenosine A1 receptor. In situ hybridization provided clear evidence of A1R gene deletion, and most neurons within the AAV-Cre injection site had no postsynaptic response to adenosine. Although adenosine antagonists can acutely depolarize hippocampal neurons (Greene et al., 1985), our adenosine-unresponsive neurons had normal electrophysiologic properties, suggesting that postsynaptic A1 receptors have little lasting effect on the tonic activity of CA1 neurons in vitro. Hippocampal neurons also contain A2a and A3 adenosine receptors (Sebastiao and Ribeiro, 1996; Dunwiddie et al., 1997), but the absence of any response to adenosine in neurons lacking A1 receptors indicates that signaling through these other receptors must depend on A1 receptors (Johansson et al., 2001; Lopes et al., 2002).
Focal deletion of A1 receptors allowed us to examine the presynaptic and postsynaptic effects of adenosine on hippocampal neurons. Deletion of A1 receptors from postsynaptic CA1 neurons did not alter the adenosine-mediated inhibition of the fEPSP. In contrast, deletion of A1 receptors from presynaptic CA3 neurons abolished this effect of adenosine. This result agrees with previous pharmacologic studies that suggest that adenosine inhibits the fEPSP through presynaptic A1 receptors on CA3 Schaffer collaterals (Lupica et al., 1992).
The AAV-Cre technique efficiently deletes A1 receptors without altering the basic electrophysiology of neurons. In the future, this technique could be used to focally knock out other endogenous genes or to produce knock-ins or focal rescue of a traditional knock-out animal. A transgenic animal with a loxP-flanked stop sequence upstream of a gene would have the phenotype of a knock-out, but injection of AAV-Cre could induce expression of the gene in a specific brain region, thus allowing one to define brain regions sufficient for rescuing the knockout phenotype. This type of experimental approach would have wide applicability in neuroscience.
Footnotes
- Received March 6, 2003.
- Revision received March 6, 2003.
- Accepted April 14, 2003.
This study was supported by United States Public Health Service Grants MH01507, HL60292, and MH62589, National Institutes of Health Grant P30 HD18655-20, the Department of Veterans Affairs, and a grant from the Nancy Marks Family Foundation. Jeng-Shin Lee and John Gray of the Harvard Gene Therapy Initiative graciously created the AAV-Cre vector. We appreciate the efforts of Matt Beaudet, Bin Du, and Navita Kaushal for their essential help with preliminary experiments. Maria Papadopoulou, Yiping Chou, Hong Ye, Quan Ha, Mihn Ha, Ivy Estabrooke, Marie McCarthy, and Courtney Sears provided excellent technical assistance.
Correspondence should be addressed to R. Greene, Department of Psychiatry, Veterans Affairs Medical Center, 4500 Lancaster Road, 116A, Dallas, TX 75216. E-mail: robertw.greene{at}utsouthwestern.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/235762-09$15.00/0