
Abstract
Calsenilin has been identified as a presenilin-binding protein, a transcription factor regulating dynorphin expression, and a β-subunit of Kv4 channels and could, thus, be a multifunctional protein. To study these functions of calsenilin in vivo and to determine the neuroanatomical expression pattern of calsenilin, we generated mice with a disruption of the calsenilin gene by the targeted insertion of the β-galactosidase gene. We found that calsenilin expression (as represented by β-galactosidase activity) is very restricted but overlaps better with that of presenilins and Kv4 channels than with dynorphin, suggesting that calsenilin may regulate presenilin and Kv4 channels in brain. Aβ peptide levels are reduced in calsenilin knock-out mice, demonstrating that calsenilin affects presenilin-dependent γ-cleavage in vivo. Furthermore, long-term potentiation (LTP) in dentate gyrus of hippocampus, in which calsenilin is strongly and selectively expressed, is enhanced in calsenilin knock-out mice. This enhancement of LTP coincides with a downregulation of the Kv4 channel-dependent A-type current and can be mimicked in wild-type animals by a Kv4 channel blocker. The data presented here show that lack of calsenilin affects both Aβ formation and the A-type current. We suggest that these effects are separate events, caused by a common mechanism possibly involving protein transport.
- KChIP
- DREAM
- presenilin
- APP
- Aβ
- apoptosis
- calcium
- neuronal calcium sensor
- Kv4 channel
- long-term potentiation
- A-type current
Introduction
Calsenilin was originally isolated as a presenilin-interacting protein and can coimmunoprecipitate with both presenilin (PS)-1 and PS-2 in transfected cells and at endogenous levels in brain (Buxbaum et al., 1998; Zaidi et al., 2002). In vitro calsenilin enhances apoptosis by increasing intracellular Ca2+ stores (Lilliehook et al., 2002) and increases presenilin-dependent γ-cleavage of the Alzheimer amyloid protein precursor (APP), elevating formation of the β-amyloid peptide (Aβ42) (Jo et al., 2001, 2003). Calsenilin, under the name DREAM, has also been identified as a regulator of transcription from the dynorphin gene that binds DNA in a calcium-dependent manner (Carrion et al., 1999; Osawa et al., 2001; Cheng et al., 2002), and DREAM was, thus, suggested to act as a calcium-regulated transcription factor. Finally, calsenilin has also been identified as a Kv4 channel-interacting protein (KChIP3). Calsenilin has three homologs, KChIP1, KChIP2, and calsenilin-like protein (CALP), the latter also known as KChIP4. KChIPs increase the transient A-type current and are thought to modulate both the kinetic properties and cell surface accumulation of Kv4 channels (An et al., 2000; Ohya et al., 2001; Beck et al., 2002; Morohashi et al., 2002; Takimoto et al., 2002). CALP/KChIP4 was isolated in a yeast two-hybrid screen using a domain of PS-2 as bait and is, thus, like calsenilin, thought to regulate both presenilins and Kv4 channels (Morohashi et al., 2002).
Calsenilin belongs to a family of small (∼20-25 kDa) neuronal calcium sensors (NCS), widely but differentially expressed in the nervous system and currently consisting of 13 genes in humans (for review, see Braunewell and Gundelfinger, 1999; Burgoyne and Weiss, 2001). NCS show 25-35% identity with calmodulin but are able to bind calcium with higher affinity (∼10-fold higher than calmodulin) and are, thus, likely able to respond quicker to minor Ca2+ changes in the resting Ca2+ concentration. Calsenilin and its closest homologs, KChIP1, KChIP2, and CALP, differ from other NCS in having extended and variable NH2-terminal domains, typically disrupting NH2-terminal myristoylation sites. KChIPs show ∼70% identity within the subgroup and ∼35-45% identity with other NCS (Burgoyne and Weiss, 2001). Frequenin, also known as NCS-1, regulates Kv4 channels in a similar manner as the KChIPs (Nakamura et al., 2001).
The independent reports on calsenilin and its putative functions have raised the question of whether calsenilin is a multifunctional protein or whether some, or all, of the functions are related. Not only are the functions quite diverse, but the postulated roles played by calsenilin may also place it in three different cellular compartments: as a presenilin interactor in the endoplasmic reticulum/Golgi, as a transcription factor in the nucleus, and as a Kv4 channel subunit at the plasma membrane. Thus, calsenilin would have to localize in all three of these compartments, or have the ability to translocate. To investigate whether in vivo calsenilin regulates presenilins, Kv4 channels, and dynorphin transcription and to resolve the neuroanatomical expression pattern of calsenilin, we generated a calsenilin knock-out mouse with the β-galactosidase (β-gal) gene inserted into the knock-out locus under control of the endogenous calsenilin promoter.
Materials and Methods
Generation of calsenilin knock-out mice. A targeting construct directed at the second exon (containing the translation start codon) was made from a genomic fragment of calsenilin isolated from the 129SVJ mouse genomic library (Wattler et al., 1999). The targeting construct included the β-gal marker gene downstream of an internal ribosome entry site (IRES) and the neomycin resistance gene under the control of the MC1 promoter. Homologous recombinants were identified with Southern blot analysis, and mutant clones were expanded and microinjected into J1 129SV blastocysts that were subsequently transferred to pseudopregnant females. Generation of chimeras and resultant heterozygous knock-out animals was performed by Lexicon Genetics (Woodlands, TX). Chimeric male mice were crossed with, and progeny outbred with, C57BL/6. Mice null for calsenilin were born with an expected Mendelian frequency, were fertile, and were indistinguishable from wild-type littermates.
Behavioral Studies. Calsenilin knock-out mice, 6-8 months of age, and heterozygous and wild-type littermates were subjected to a suite of motor, cognitive, and sensory tasks. Beam walking, rod hanging, paw grasping, righting, eye lid closing, and reaching were recorded following standard protocols. In the rotorod task, animals were tested at incrementing speed (starting at 21 rpm with 1 rpm increments) and at fixed speed (23 rpm). The Morris water maze place-learning paradigm was performed as described (Santucci et al., 1995). Movement activity was assessed in a chamber with infrared detectors mounted to the ceiling (Coulbourn Instruments, Allentown, PA). Animals were assessed twice for 30 min. Fear and ambulatory behavior was assessed in an open-field maze and in a round maze, and time spent in open versus closed areas was monitored. Exposing animals to an electric foot shock, maximum intensity of 0.08 mA, assessed shock sensitivity. Shock intensity was increased in 10% gradations and responses (paw withdrawal, running, jumping, vocalization, and tail wagging) were noted absent or present. The tail-flick assay was conducted using a tail-flick analgesia meter (Columbus Instruments, Columbus, OH).
Detection of β-gal activity in embryos and brains of adult calsenilin knock-out mice. Embryos were dissected from the uterus of pregnant females on ice in PBS and were then washed extensively in PBS/0.01% Triton X-100. Whole embryos were fixed for 2-10 min on ice (2% formaldehyde, 100 mm potassium phosphate buffer, pH 8, 5 mm EGTA, and 2 mm MgCl2), washed in solution C (100 mm potassium phosphate buffer, pH 8, 5 mm EGTA, 2 mm MgCl2, 0.01% Na deoxycholate, and 0.02% NP-40), and incubated overnight at 37°C in solution D (solution C, 10 mm K3[Fe(CN)6], 10 mm K4[Fe(CN)6], and 1 mg/ml X-gal). Subsequently, the embryos were washed three times in solution C and photographed using a Zeiss AxioCam. For histological analysis of β-gal staining, stained embryos were postfixed overnight in 4% paraformaldehyde, cleared through a series of graded ethanols and Americlear, and embedded in paraffin. Twenty-five micrometer sections were collected with a microtome and counterstained with 1% eosin or cresyl violet. For postnatal and adult mice, animals were anesthetized and perfused with potassium phosphate buffer, pH 8.0. Adult animals were then perfused with 2% paraformaldehyde, whereas brains from pups were dissected out and fixed on ice for 20 min. Whole brains or sections were stained in the β-gal reaction buffer, as described above, and postfixed overnight. For histological analysis, stained whole brains were embedded in paraffin and sectioned with a microtome. Because the β-gal reaction might not be homogeneous in all regions when performed on larger tissues such as whole brains from postnatal and adult mice, cryostat sections from unstained samples were also processed for β-gal activity. Sections were counterstained with 1% eosin.
Quantification of Aβ40 and Aβ42 peptide levels. Cortex, cerebellum, and hippocampus from wild-type and calsenilin knock-out littermates were dissected out in ice-cold PBS. Tissue was homogenized in DEA buffer (0.2% diethylamine and 50 mm NaCl; 1:10 w/v) in a Dounce homogenizer using pestle B. The homogenate was centrifuged for 1 hr at 100,000 × g at 4°C, and the supernatant was neutralized to pH 8.0 with 0.5 m Tris, pH 6.8 (1:10 vol) (Rozmahel et al., 2002). The DEA extracts were analyzed for endogenous Aβ levels with ELISA using Aβ40 and Aβ42 murine-specific antibodies JRF/rAβ1-15/2 (Janus et al., 2000; Mercken et al., 2002). For each sample, the levels of Aβ40, Aβ42, and total Aβ were standardized to brain tissue weight and expressed as pmol Aβ/gm (brain tissue, wet weight).
SDS-PAGE and immunoblotting. Cortex, cerebellum, and hippocampus from wild-type and calsenilin knock-out mice were homogenized in immunoprecipitation assay buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 2 mm EDTA, 1% Triton X-100, 1% NP-40, and 0.25% sodium deoxycholate) with protease inhibitors (Complete 1; Boehringer Mannheim, Indianapolis, IN). Lysates were solubilized for 1 hr with rocking at 4°C and centrifuged at 14,000 × g for 10 min to remove insoluble material. For each sample, 75 μg of protein was separated on a 14% Tris-glycine gradient gel (Invitrogen, Carlsbad, CA) and then transblotted onto polyvinylidene difluoride membrane. Membranes were blocked in 5% nonfat dry milk in TBS-T (25 mm Tris, pH 7.5, 135 mm NaCl, and 0.15% Tween 20) for 2 hr, followed by an overnight incubation with the appropriate antibody at 4°C. Blots were then washed three times for 15 min with TBS-T and incubated with secondary antibody conjugated with horseradish peroxidase (1:7500) at room temperature. After washing with TBS-T, blots were developed using the ECL detection system (Amersham Biosciences, Piscataway, NJ) according to the manufacturer's protocol.
Electrophysiological recordings. Calsenilin knock-out mice, 2-4 months of age, and wild-type littermates, anesthetized with isoflurane, were decapitated, and their brains were removed rapidly and placed in ice-cold Ringer's solution (125 mm NaCl, 2.5 mm KCl, 1.3 mm MgSO4, 1.0 NaH2PO4, 26.2 mm NaHCO3, 2.5 mm CaCl2, and 11.0 mm glucose, pH 7.2-7.3; osmolarity, 310 ± 2 mOsm). Hippocampus was dissected out and cut in 350 μm slices using a tissue chopper, and the slices were incubated in Ringer's solution bubbled with 95% O2/5% CO2 for at least 1 hr before recordings. During electrophysiological recordings, slices were submerged and perfused at 2 ml/min. Electrophysiological experiments were performed using conventional microelectrode techniques (Bozdagi et al., 2000). Glass micropipettes (3-20 MΩ) were filled with 3 m KCl or 3 m NaCl for intracellular or extracellular recordings, respectively. The potentials were recorded and amplified using an Axoclamp 2A amplifier (Axon Instruments, Foster City, CA), digitized by an Axon analog-to-digital/digital-to-analog converter. Field EPSPs (fEPSPs) were recorded from the dentate gyrus molecular layer, evoked by a stimulation of the medial perforant pathway (MPP) to the dentate gyrus with a bipolar tungsten electrode. The position of the pipette was adjusted to obtain an intensity of responses just suprathreshold for MPP stimulation response, with pulses 300 μsec in duration. The EPSP initial slope (mV/msec) was determined from the average waveform of four consecutive responses. Long-term potentiation (LTP) was induced by applying 100 Hz, 1 sec tetanic stimulation as two trains, separated by a 20 sec interval. Membrane currents were recorded from dentate gyrus granule cells using a whole-cell configuration of the patch-clamp technique. Data were acquired using an Axopatch 200B amplifier and PClamp software (Axon Instruments). Recordings were filtered at 2 kHz and sampled at 200 μsec. Capacitance and series resistance compensation were performed using the built-in amplifier circuitry. Eighty percent of series resistance was compensated for in this manner. The patch pipettes had a resistance of 3-5 MΩ when filled with pipette solution. The pipette solution contained (in mm): 130 K-gluconate, 2 NaCl, 20 HEPES, 4 MgCl2, 2 EGTA, 0.4 NaGTP, and 4 Na2ATP, pH 7.3 (280 mOsm). A-type charge density (pC/pF) was calculated by dividing A-type charge transfer (pC) by whole-cell capacitance (pF) as a measure of cell size. Three to eight slices per condition were used for all electrophysiological recording experiments. The Kv4 channel-specific antagonist phrixotoxin-2 (1 μm; Alomone Labs, Jerusalem, Israel) was bath applied for the durations indicated in the figure legends. All data are presented as mean ± SEM.
Results
Generation of calsenilin knock-out mice
To study the role of calsenilin in vivo, mice with a disrupted calsenilin (Csen) gene were generated. A construct targeted against exon 2 of the Csen gene (Fig. 1A) that incorporates an IRES and the β-gal gene was electroporated into embryonic stem cells. Positive clones, identified by Southern blot analysis (Fig. 1B) using 5′ and 3′ probes, were used to establish chimeric mice carrying the Csen mutation. Mice null for calsenilin were born with an expected Mendelian frequency, were fertile, and were indistinguishable from wild-type littermates. Northern blot analysis showed that calsenilin mRNA levels were reduced in heterozygous mice and absent in knock-outs, and Western blot analysis showed that knock-out mice lacked calsenilin protein (Fig. 1C,D).

Molecular characterization of the calsenilin knock-out mouse. A, Gene targeting strategy. Top, Wild-type calsenilin locus showing exon 2 and the position of the 5′ and 3′ probes used for genotyping. Bottom, Mutant locus in which exon 2 has been replaced with an IRES, the marker gene β-gal, and the neomycin (neo) resistance gene, the latter under the control of the MC1 promoter. B, Southern blot analysis of genomic DNA from calsenilin +/+, +/-, and -/- mice. Left, HindIII digest; right, NcoI digest. C, Northern blot analysis of mRNA extracted from the brains of calsenilin +/+, +/-, and -/- mice. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is included as a loading control. D, Immunoprecipitation-immunoblot analysis of 1% SDS extracts from the brains of calsenilin +/+, +/-, and -/- mice. Immunoprecipitation of calsenilin was performed with a polyclonal antibody directed against human calsenilin (Choi et al., 2001; Zaidi et al., 2002). The precipitates were then subjected to SDS-PAGE and immunoblotted with a monoclonal antibody directed against human calsenilin (Zaidi et al., 2002). Mouse calsenilin migrates with a slightly higher apparent molecular weight than human calsenilin, extracted from H4 cells stably expressing human calsenilin (Lilliehook et al., 2002). The asterisk indicates a band migrating just above mouse calsenilin, and the position of IgG (from the immunoprecipitation) is also indicated.
Behavioral assessment of calsenilin knock-out mice
In our behavioral studies, calsenilin knock-out mice were indistinguishable from wild-type littermates with regard to motor coordination, balance, and fine motor control of the forelimbs. In the tail-flick assay, calsenilin knock-outs showed increased latency (Fig. 2A). In contrast, we observed increases in shock sensitivity in calsenilin knock-out mice (Fig. 2B). Calsenilin knock-out mice also demonstrated increased movement activity (Fig. 2C), and in the Morris Water Maze, calsenilin knock-out mice consistently exhibited shorter escape latencies than wild-type littermates (data not shown).

Behavioral assessment of calsenilin knock-out mice. A, Tail-flick test. Values represent latency to withdraw tail. B, Shock sensitivity. Values represent percentage of maximum shock level (0.08 mA) required to elicit two stereotypical behavioral responses, running and vocalizing. C, Movement activity. Values represent number of movements detected with infrared monitors during a 30 min test session. All data are presented as mean ± SEM, and p values were calculated using the Student's t test. Wild-type mice (WT), n = 8; heterozygous mice (HET), n = 5; knock-out mice (KO), n = 8. *p < 0.05.
β-Gal activity in calsenilin knock-out mice
With the identification of three calsenilin homologs and the resultant potential cross-reactivity of some of the antibodies to calsenilin, the precise expression of calsenilin needed to be reexamined. To resolve the regional expression of calsenilin during embryonic development and in the adult brain, we used β-gal activity as a marker for sites of calsenilin expression.
β-Gal activity during embryonic development
The β-gal reaction product could be detected as early as embryonic day (E) 12 (data not shown). In E14 embryos, β-gal activity was very prominent in the midbrain (Fig. 3A) and could be detected in two regions curving along the rostro-caudal axis. The broader region corresponds to the ventricular zone of the tectum, and the thinner region maps to the area of epithalamus (Fig. 3B,D). In E16 embryos, the midbrain expression becomes more defined and appears to be restricted to a subpopulation of cells in the tectal neuroepithelium, likely the lateral inferior colliculus and brachium (Fig. 3H). The midbrain expression remains high throughout embryonic development (Fig. 3E) but decreases in postnatal development (Fig. 4A). β-Gal activity in the forebrain is comparatively weaker but is present in the ventricular zones of both hippocampus and cortex (Fig. 3C,F,G). We were unable to detect any β-gal activity in non-neuronal tissues or in the spinal cord during development.

β-Gal as a marker for sites of calsenilin expression during embryonic development. A, Whole-mount-stained E14 embryo showing strong and specific β-gal activity in the midbrain and weaker β-gal activity in the forebrain. B, Sagittal section through E14 embryo. C, Dorsal view of head from whole-mount-stained E15 embryo. D, Coronal section through E14 embryo. β-Gal activity is prominent in a subpopulation of cells in the tectal neuroepithelium and in the epithalamus and can also be detected in the ventricular zone of the developing cortex. E, Whole-mount-stained E18 embryo cut along the midline. β-Gal activity in the midbrain remains high throughout development and appears to be absent from the hindbrain; however, in the developing cerebellum, β-gal activity can be detected in the EGL. F-H, Coronal sections through E16 embryo. β-Gal activity can be detected in the ventricular zones of both hippocampus (F) and cortex (G) and in the lateral inferior colliculus (H).

β-Gal as a marker for sites of calsenilin expression during cerebellar development and in adult brain. A, Whole-mount staining of P4 brain with a dorsal view of midbrain, developing cerebellum, and brainstem. B, Sagittal section through P8 brain. C, Sagittal view of β-gal activity in P20 cerebellum. During postnatal development, β-gal activity decreases in the midbrain and increases in the hindbrain. In developing cerebellum, β-gal activity is strong in the external germinal layer and appears to accumulate in the granule cell layer. By P20, β-gal activity is expressed in an anteroposterior gradient in the granule cell layer. D, Whole-mount-stained section from adult brain. β-Gal activity is strong in layer VI of cortex, in piriform cortex, and in dentate gyrus. β-Gal activity can also be detected in layer IV of visual cortex. E, Coronal section through caudal cortex (bregma, -4.36). β-Gal activity is strong in layer II of entorhinal cortex and present in subiculum. F, Sagittal section through adult hippocampus. In hippocampus, β-gal activity appears to be restricted to the granule cell layer in dentate gyrus.
β-Gal activity in postnatal cerebellum
Cerebellar granule cells are born in a specialized germinal region called the rhombic lip. After birth, this external germinal layer (EGL) becomes highly proliferative, and immature granule cells migrate into the cerebellar cortex, in which they form the dense granule cell layer. β-Gal activity is strong in the rhombic lip and can be seen in the EGL of postnatal day (P) 4 and P8 pups, in which it appears to accumulate in the granule cell layer (Fig. 4A,B). By P20, the development of cerebellum is complete, and at this stage, β-gal activity is restricted to the granule cell layer. Calsenilin appeared not to be present in the Purkinje cell layer, in agreement with a previous report (Zaidi et al., 2002). Interestingly, the anterior part of cerebellum appears to show a more prominent β-gal activity than the posterior part, which could reflect a gradient of expression of calsenilin. The gradient-like expression pattern of calsenilin in cerebellum was reported earlier using in situ hybridization (Spreafico et al., 2001) and can also be observed for the Kv4 channels (Serodio and Rudy 1998). β-Gal activity in the cerebellar granule cell layer remains high in juvenile and adult animals but decreases in older and aging animals (data not shown).
β-Gal activity in the adult brain
In adult mouse brain, calsenilin has a restricted expression pattern. In coronal sections throughout the brain, β-gal activity is prominent in layer 6 of cortex, in piriform cortex, and in the dentate gyrus of hippocampus (Fig. 4D). β-Gal activity is also weakly present in layer 4 of visual cortex, in amygdala and in the CA3 region of hippocampus. Furthermore, calsenilin expression is strong in layer 2 of entorhinal cortex and present in subiculum (Fig. 4E). The expression in dentate gyrus follows an interesting temporal pattern: β-gal activity is absent during postnatal development but is strong in the adult and remains strong in old and aging mice (Fig. 4F). Using β-gal activity as a marker for calsenilin expression, we were unable to detect any β-gal activity in the spinal cord from calsenilin knock-out mice, and calsenilin was also absent from the olfactory bulb, basal ganglia, and thalamus (Table 1).
Comparison of calsenilin expression pattern with reported expression patterns of presenilins, dynorphin, and Kv4.2/Kv4.3
APP processing and Aβ peptide levels in calsenilin knock-out mice
Calsenilin expression in mouse is more restricted than presenilin expression, but coexpression in select cortical areas, hippocampus, and cerebellum provides a neuroanatomical basis for a functional interaction between calsenilin and presenilin (Table 1). Changes in the levels of Aβ40 and Aβ42 peptides can reflect an alteration in presenilin function. We, therefore, quantified these peptides in brain lysates from calsenilin mutant mice using ELISA with Aβ40- and Aβ42-specific antibodies (Mercken et al., 2000). As shown in Figure 5, A and B, levels of both Aβ40 and Aβ42 peptides were decreased in cortex and cerebellum from calsenilin knock-out mice compared with wild-type littermates. The lesser reduction of Aβ peptide levels in cortex compared with cerebellum likely reflects the lower levels of calsenilin expression in cortex. Aβ peptide levels also showed a trend toward reduction in hippocampus from calsenilin knock-out mice (data not shown), however, the smaller amount of hippocampal tissue made these measurements more difficult. Levels of mature and immature APP appeared similar between knock-out and wild-type animals, as did the levels of processed PS-1 and PS-2 (Fig. 5C).

Aβ40 and Aβ42 levels in calsenilin knock-out mice. Aβ40 (A) and Aβ42 (B) in brain lysates from calsenilin knock-out mice and wild-type littermates were quantified using ELISA. In cortex, in which calsenilin is mainly expressed in layer 6 and piriform cortex, levels of Aβ40 decreased in the calsenilin knock-out mouse (n = 9). In cerebellum, in which calsenilin is strongly expressed, levels of both Aβ40 and Aβ42 peptides are reduced by approximately one-half in calsenilin null brain samples (n = 6). Data are represented as relative units (wild type, 100%); the range for Aβ40 samples was 1.8-12.2 pmol Aβ/gm brain tissue and 0.7-5.8 pmol Aβ/gm brain tissue for Aβ42 samples (wet weight) *p < 0.05. C, Western blot analysis of SDS lysates (75 μg) from cerebellum (CRB), cortex (CTX), and hippocampus (HIP) from calsenilin (KO) mice and wild-type (WT) littermates. Protein levels and processing of PS1, PS2, and APP appear similar in knock-out and wild-type animals.
LTP in dentate gyrus of calsenilin knock-out mice
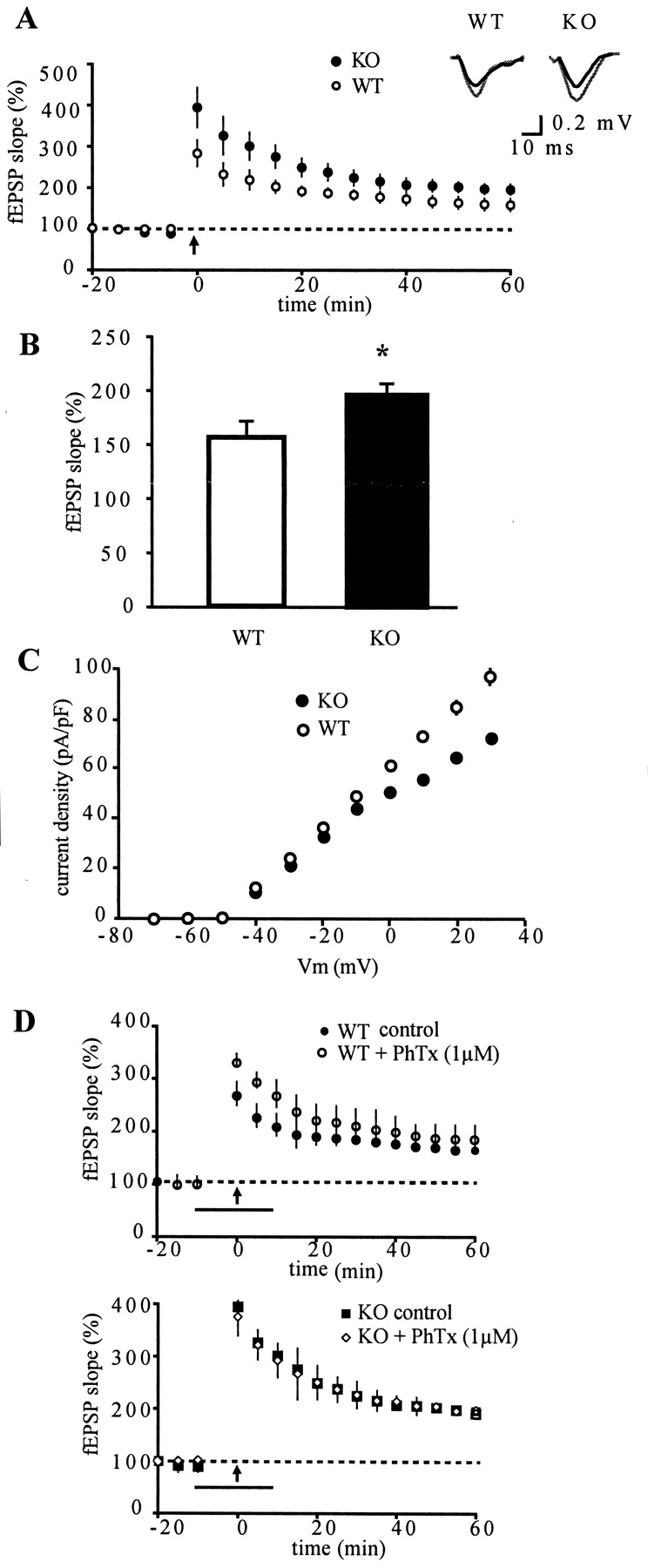
Calsenilin expression is high in the granule cell layer of dentate gyrus as well as in entorhinal cortex; these two synaptically coupled regions form the first link in the hippocampal trisynaptic circuit. Because there is yet no evidence that calsenilin can influence synaptic plasticity, we investigated plasticity of synaptic transmission at perforant path-dentate gyrus molecular layer synapses in acute hippocampal slices taken from calsenilin knock-out and wild-type mice. We induced LTP by two trains of 100 Hz tetanic stimulation applied to the dentate gyrus molecular layer, which resulted in a potentiation of fEPSPs. Figure 6A shows the time course of changes in the fEPSP initial slope and reveals that LTP was significantly greater in knock-out animals than in wild-type controls. The average potentiation at 60 min after tetanic stimulation is shown in Figure 6B (knock-out, 194 ± 12%; wild type, 157 ± 15%). These effects on LTP were specific to dentate gyrus, because LTP in CA1 pyramidal cells, induced by tetanic stimulation of Schaffer collaterals, remained unaltered in the calsenilin knock-out animal in comparison with wild-type animals (data not shown). The enhanced LTP could not be attributed to changes in basal membrane properties, synaptic transmission, or short-term plasticity. No differences between genotypes were observed in resting membrane potentials (-64 ± 2 mV and -62 ± 3 mV in wild-type and knock-out mice, respectively) or input resistance (325 ± 54 MΩ for wild-type mice and 332 ± 51 MΩ for knock-out mice). Furthermore, the input-output relation of fEPSPs revealed no difference in the mean amplitudes at stimulus intensities of 2-20 V in knock-out mice compared with wild type. Finally, to investigate effects on short-term plasticity, we examined paired pulse facilitation, a form of plasticity that is thought to reflect presynaptic mechanisms. Paired pulse stimulation (50 msec interpulse interval), however, did not produce any detectable differences between wild-type and knock-out mouse slices (P2/P1 = 0.79 ± 0.03 in wild-type animals and 0.78 ± 0.04 in knock-out animals, respectively). Taken together, these data indicate that basic synaptic transmission and one measure of short-term plasticity were normal in calsenilin knock-out mice.

LTP and the transient A-type current in calsenilin knock-out mice. LTP of synaptic transmission at perforant path-dentate gyrus molecular layer synapses was induced in acute hippocampal slices taken from calsenilin knock-out (KO) and wild-type (WT) animals. A, Potentiation of fEPSP slope after tetanic stimulation (arrow) is larger in KO mice in comparison with WT mice. Inset, Superimposed representative fEPSPs taken before (black) and 60 min after (gray) tetanus for wild-type and calsenilin knock-out mice. B, Mean percentage increase of LTP 60 min after tetanus. Data are presented as mean ± SEM (n = 8; p = 0.009; paired t test). C, Graph showing relationship between mean current density and membrane potential for WT and calsenilin KO mice determined in whole-cell voltage-clamp experiments (n = 4). Membrane currents were recorded using voltage-clamp steps from a holding potential of -80 mV to depolarized potentials in 10 mV steps. Note that mean current density was significantly lower in dentate gyrus granule cells from KO animals in comparison with cells from WT animals at all membrane potentials more positive than -30 mV (p = 0.001 at +30 mV). D, Twenty-minute bath application of 1 μm phrixotoxin-2 (PhTx) increases the initial degree of potentiation in WT mice (top) compared with control slices but has no effect on potentiation in calsenilin KO mice (bottom). Bar, Duration of drug application; arrow, time point for tetanic stimulation.
Transient A-type current in calsenilin knock-out mice
In myocytes from KChIP2 knock-out mice, the Kv4 channel current was completely absent (Kuo et al., 2001). Whereas KChIP2 is the dominating KChIP in heart, calsenilin as well as other KChIPs are expressed in hippocampus; thus, a complete lack of the A-type current was not to be expected in the calsenilin knock-out animals. To test the hypothesis that a decrease in IA may contribute to the increase in LTP in calsenilin knock-out mice, K+ currents were recorded using the whole-cell voltage-clamp technique. Figure 6C shows that the fast inactivating outward K+ current amplitudes are lower in knock-out mice compared with wild-type controls. The peak current-voltage relationship between wild-type and knock-out mice shows a significant difference in the amount of the current for membrane potentials greater than -30 mV. We surmised that the enhanced LTP observed in dentate gyrus of calsenilin knock-out mice could be a result of the decrease in the A-type current. Possible IA contribution in increase in LTP in calsenilin knock-out mice was further characterized by using phrixotoxin-2, a spider venom that specifically blocks Kv4 channels. Application of 1 μm phrixotoxin-2 facilitated the induction of LTP in wild-type animals (Fig. 6D, top), in agreement with a recent study (Ramakers and Storm, 2002). The mean increase in the response amplitudes for the first post-tetanus minute were 267 ± 10% in wild-type control slices and 330 ± 12% in the presence of phrixotoxin-2. In contrast, in calsenilin knock-out animals, the induction phase of LTP was not altered by an inhibition of the transient A-type current (Fig. 6D, bottom). This is consistent with the hypothesis that the enhanced LTP observed in mice lacking calsenilin results from a decrease in the transient A-type current.
Discussion
To be able to regulate presenilins, dynorphin transcription, or Kv4 channel function in vivo, calsenilin must be expressed in the same brain regions as these suggested molecular partners. The expression pattern of calsenilin in mouse, as determined by β-gal activity, is both restricted and specific. Temporally, calsenilin expression appears to be turned on in three consecutive waves, the first in the midbrain during embryonic development, the second in the germinal layer of postnatal cerebellum, and the third in adulthood in the dentate gyrus of hippocampus and distinct cortical areas. Presenilins and Kv4 channels are expressed in most regions of the CNS, whereas dynorphin has its major sites of expression in the spinal cord and basal ganglia (Table 1). From our experiments, we conclude that calsenilin expression in adult mouse brain overlaps better with that of presenilins and Kv4 channels than dynorphin.
While our studies were in progress, it was reported that mice lacking calsenilin (termed “DREAM” in that report) showed a decreased sensitivity to pain in several behavioral paradigms. The authors proposed a model in which dynorphin levels in the spinal cord are upregulated in the absence of calsenilin, thus increasing the pain threshold for calsenilin knock-out mice (Cheng et al., 2002). In our study, we found no evidence for calsenilin expression in the spinal cord using β-gal activity; thus, calsenilin may be absent from or present in low amounts in the spinal cord, although β-gal activity may be a less sensitive measure of expression than other methods. However, we were unable to see any changes in dynorphin mRNA levels in adult brain using microarray analysis (data not shown).
In our behavioral analysis, we first observed increases in shock sensitivity, which was our measure of nociception. When it was then reported that Cheng et al. (2002) observed a reduction in pain sensitivity using the tail-flick assay, we performed this analysis as well. We also observed increased latency in the tail-flick assay in our studies, indicating that the absence of calsenilin results in a mixed behavioral phenotype in terms of models for nociception. Pain transmission in the CNS is undoubtedly complex, and variations in pain sensitivity could perhaps be explained by the alterations in electrophysiological properties of calsenilin knock-out mice described here. Where tested, mice heterozygous for the calsenilin deletion displayed alterations in behavioral assays, indicating that levels of calsenilin are limiting.
Presenilins can be detected by reverse transcription (RT)-PCR as early as E8.5 (Lee et al., 1996) and is prominently expressed in ventricular zones of developing cortex and hippocampus (Moreno-Flores et al., 1999). The presence of calsenilin in early embryonic development seems likely to be related to presenilin function, because the temporal expression in ventricular zones seems to overlap, although it is unknown whether Kv4 channels are expressed in embryonic development. The calsenilin homolog KChIP2 was barely detectable by in situ hybridization analysis of E15 embryos or Northern blotting of E13-E15 hearts but could easily be seen in E17 embryos (Kuo et al., 2001). The authors concluded that KChIP2, thus, likely participates in physiological, rather than developmental, processes. Because we detected β-gal activity as early as E12 in calsenilin knock-out mice and an earlier report detected calsenilin with RT-PCR at E10.5 (Spreafico et al., 2001), calsenilin may participate in developmental processes, possibly related to presenilins.
The expression of calsenilin in postnatal cerebellum, as resolved using β-gal activity, is quite striking and could potentially be functionally related to both Kv4 channels and presenilins. In adult rat cerebellum, Kv4.2 is enriched in posterior lobules and Kv4.3 in anterior lobules (Serodio and Rudy, 1998). This differential expression pattern is also seen in heart, in which murine Kv4.3 channels are expressed in a gradient across the cardiac wall, resulting in larger transient outward K+ currents in the epicardium compared with the endocardium. In human heart, the Kv4.3 mRNA is evenly distributed, whereas KChIP2 is expressed in a gradient, thus maintaining the electrophysiological differences across the ventricular wall (Rosati et al., 2001). In a similar manner, the graded expression of calsenilin in adult cerebellum might contribute to variations of the A-type current along the anterior-posterior axis.
During cerebellar development, granule cell precursors proliferate in the EGL, migrate through the molecular or Purkinje cell layer, and complete synaptogenesis in the internal granule cell layer. Presenilin expression peaks at P5/P7 and is mainly distributed along the EGL but is also present in granule cells, either en route to or in the internal granule cell layer (Moreno-Flores et al., 1999). Interestingly, patients with the E280A missense mutation in the PS-1 gene display severe accumulation of amyloid plaques in the cerebellum (Lemere et al., 1996). In our β-gal activity studies, we observed an expression pattern for calsenilin overlapping with presenilins in cerebellum. Strikingly, we found that in this brain region, Aβ40 and Aβ42 peptide levels were reduced by ∼50% in the calsenilin knock-out, strongly implying that calsenilin affects γ-secretase cleavage in vivo. Changes in Aβ levels were not accompanied by detectable changes in APP processing, consistent with what is observed for familial mutations in presenilin, which modulate Aβ formation without a dramatic effect on APP processing. In cultured cells, we had previously observed alternate processing of presenilins in the presence of calsenilin (Buxbaum et al., 1998). In the calsenilin knock-out, however, levels and processing of the presenilins seemed similar to the wild-type, possibly because of the presence of other presenilin-interacting factors in vivo.
The increase of LTP in calsenilin mutant animals is best explained with calsenilin-modulating Kv4 channel density, and, hence, the A-type current. In the KChIP2 knock-out, Ito (the A-type current equivalent in heart) was completely absent in myocytes isolated from KChIP2 null mice, and these animals were also more susceptible to ventricular tachycardia induced by electrical stimulation. The authors concluded from these studies that in the absence of KChIP2, Kv4 channels failed to reach the plasma membrane, and, as a result, the myocardium became more excitable (Kuo et al., 2001). KChIPs increases the plasma membrane localization of Kv4 channels in transfected cells (Bahring et al., 2001; Ohya et al., 2001; Takimoto et al., 2002); hence, this is a plausible model. According to this model, calsenilin knock-out animals would be expected to have less functional Kv4 channels targeted to the somato-dendritic membrane than wild-type animals. It is unlikely that the A-type current would be completely absent in mutant animals; the calsenilin homologs KChIP1, KChIP2, and CALP are also expressed in hippocampus and would be expected to be, more or less, functionally equivalent. In our β-gal activity studies, we found strong and specific expression of calsenilin in the granule cell layer of dentate gyrus. This suggests that calsenilin could be the dominant KChIP in this brain area, and its absence may, therefore, be reflected as a decrease in the A-type current, consistent with what we observed. Johnston et al. (2000) have proposed a model for how inactivation of the A-type current contributes to the induction of LTP in CA1 pyramidal cells (Hoffman et al., 1997; Magee and Johnston, 1997; Watanabe et al., 2002), and the same reasoning may apply for the granule cells of dentate gyrus. In this model, activated Kv4 channels act as “shock absorbers” (i.e., dendritic excitability is controlled so that incoming EPSPs will only generate action potentials in the axon hillock region). When inactivated, the Kv4 channel block on the dendritic membrane is absent. Incoming EPSPs may, then, generate action potentials in the dendrites, which could result in larger influxes of calcium, unblocking of NMDA receptors, and induction of LTP. Granule cells with less functional Kv4 channel reducing the excitability of the somato-dendritic membrane would, thus, be expected to exhibit a larger increase in long-term potentiation, in agreement with our findings.
Our data suggest that calsenilin regulates both Aβ formation and the transient A-type current. The molecular evidence for interactions between calsenilin and presenilin as well as between calsenilin and Kv4 channels are equally strong. Previous reports show that calsenilin coimmunoprecipitates with both PS-1 and PS-2 and Kv4 channels and that calsenilin translocates from the cytosol to membranous compartments when coexpressed with presenilins or Kv4 channels (Buxbaum et al., 1998; An et al., 2000; Choi et al., 2001; Takimoto et al., 2002). In addition, there is now data showing coprecipitation of endogenous calsenilin and presenilin proteins from brain (Zaidi et al., 2002), as well as coprecipitation of the calsenilin homolog CALP with presenilins (Morohashi et al., 2002). Many proteins interact with presenilins, however, γ-secretase cleavage of APP and Notch requires four components: presenilin, nicastrin, APH-1, and PEN-2 (Francis et al., 2002). Coexpression of these components reconstitutes γ-secretase activity in yeast (Edbauer et al., 2003); thus, calsenilin is not necessary for catalytic activity. We suggest that the regulation of Aβ formation and Kv4 channels by calsenilin are independent events occurring by a common mechanism, as shown in Figure 7. In this scheme, we propose that calsenilin regulates transport, assembly, or maturation of Kv4 channels and presenilin-γ-secretase complexes through the secretory pathway.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Model for how calsenilin may regulate both Aβ formation and Kv4 channels. Functional γ-secretase complexes and Kv4 channel complexes are assembled in the endoplasmic reticulum. Calsenilin, in a cytoplasmic pool, associates with membrane-bound presenilins and Kv4 channels and regulates vesicular transport through the secretory pathway to the cell surface. In the absence of calsenilin, dendrites of granule cells contain less functional Kv4 channels, resulting in increases in dendritic excitability and LTP. Similarly, in the absence of calsenilin, fewer functional γ-secretase complexes reach the cell surface and less APP can be proteolytically processed into Aβ peptides. Gray cylinders, γ-Secretase complex; black bar, APP; single gray cylinder, Kv4 channel complexes; ▵, calsenilin.
Footnotes
- Received April 7, 2003.
- Revision received August 14, 2003.
- Accepted August 14, 2003.
We are grateful to the National Institutes of Health, National Institute on Aging (Grants AG10491 to S.G. and J.D.B.; AG15801 and AG05138 to J.D.B.), and the Alzheimer's Association (Grant 11RG-99-1508 to J.D.B.) for funding the study. We also thank Dr. Marc Mercken of Johnson & Johnson Pharmaceutical Research and Development/Janssen Pharmaceutica for the Aβ monoclonal antibodies and Stephen D. Schmidt at the Nathan Kline Institute for advice and protocols for the Aβ ELISAs. We are deeply indebted to Drs. Tomas Lufkin, Ruth Simon, Victor Friedrich, and Sheila Harroch for sharing expertise in the β-galactosidase activity studies.
Correspondence should be addressed to Dr. Joseph D. Buxbaum, Department of Psychiatry, Box 1668, Mount Sinai Medical Center, One Gustave L. Levy Place, New York, New York 10029. E-mail: Joseph.Buxbaum{at}mssm.edu.
Copyright © 2003 Society for Neuroscience 0270-6474/03/239097-10$15.00/0