
Abstract
The wake-promoting neuropeptides orexins (hypocretins) play a crucial role in controlling neuronal excitability and synaptic transmission in the CNS. In this study, using whole-cell patch-clamp recordings in an acute dorsal raphe nucleus (DRN) slice preparation, we report that orexin B (Orx-B) depresses the evoked glutamate-mediated synaptic currents in DRN 5-HT neurons. The Orx-B-induced depression is accompanied by an increase in the paired-pulse ratio and the coefficient of variance, suggesting a presynaptic site of action. Orx-B also reduces the frequency but not the amplitude of miniature EPSCs, indicating that depression of glutamatergic transmission is mediated by a decrease in glutamate release. Surprisingly, the Orx-B-induced inhibition of glutamatergic transmission is abolished by postsynaptic inhibition of G-protein signaling with GDPβS, suggesting that this effect is signaled by postsynaptic orexin receptors and expressed presynaptically, presumably through a retrograde messenger. Interestingly, the Orx-B-induced depression of glutamate release is mimicked and occluded by the cannabinoid receptor agonist WIN 55,212-2, and is abolished by the CB1 cannabinoid receptor antagonist AM 251. These results imply that the Orx-B-induced depression of glutamatergic transmission to DRN 5-HT neurons is mediated by retrograde endocannabinoid release. Examination of downstream signaling pathways involved in this response indicates that the effect of Orx-B requires the activation of phospholipase C and DAG lipase enzymatic pathways but not a rise in postsynaptic intracellular calcium. Therefore, our findings reveal a previously unsuspected mechanism by which postsynaptic orexin receptors can modulate glutamatergic synaptic transmission to DRN 5-HT neurons.
- raphe
- serotonin
- orexin
- cannabinoid
- glutamate
- synaptic transmission
Introduction
The orexins (Orxs)-hypocretins (orexin A and B or hypocretins 1 and 2) are two neuropeptides synthesized by a group of neurons located in the dorsolateral hypothalamus (de Lecea et al., 1998; Sakurai et al., 1998). These peptides are the endogenous agonists of two orexin receptors (OxRs), Ox1R and Ox2R, which belong to the G-protein-coupled receptors family (Sakurai et al., 1998). Several lines of evidence indicate that the orexin system is involved in the regulation of normal sleep-wake cycles and that the dysfunction of this system is linked to the pathology of narcolepsy, a disorder characterized by a dysregulation of rapid eye movement (REM) sleep and an inability to maintain wakefulness (Scammell, 2003). For instance, a mutation in the Ox2R gene appears to be the primary cause of canine narcolepsy (Lin et al., 1999). Mice lacking Orxs or Ox2R exhibit narcoleptic symptoms (Chemelli et al., 1999; Hara et al., 2001; Willie et al., 2003). Moreover, human narcoleptic patients have low levels of Orxs in their CSF and reduced numbers of Orx-containing neurons (Peyron et al., 2000; Thannickal et al., 2000).
Hypothalamic fibers containing Orxs provide dense projections to several brain regions known to be involved in the regulation of the sleep-wake cycle, including the dorsal raphe nucleus (DRN) (Peyron et al., 1998). DRN 5-HT neurons express high levels of both Ox1R and Ox2R (Marcus et al., 2001) and provide a major ascending serotoninergic pathway to the forebrain. Evidence for the involvement of these neurons in the regulation of arousal comes from early animal studies showing that their activity is closely linked to REM sleep and arousal. Thus, they discharge at a high frequency during waking, discharge at a lower rate during non-REM sleep, and become electrically silent during REM sleep (McGinty and Harper, 1976; Trulson and Jacobs, 1979). Furthermore, direct inhibition of the activity of DRN 5-HT neurons increases REM sleep, whereas stimulation of these neurons increases wakefulness (Portas et al., 1996). Previous studies examining the influence of Orxs on the excitability of DRN 5-HT neurons have reported that both Orx-A and Orx-B depolarize and increase the activity of DRN 5-HT neurons (Brown et al., 2001, 2002; Liu et al., 2002b). This excitatory effect is thought to contribute at lease in part to the wake-promoting effect of Orxs (Saper et al., 2001).
The glutamatergic input to DRN has also been implicated in both the regulation of the activity of 5-HT neurons and the sleep-wake cycle. For instance, iontophoretic application of ionotropic glutamate receptor agonist increases the activity of DRN 5-HT (Levine and Jacobs, 1992) but reduces REM sleep and increases wakefulness (Sakai and Crochet, 2001). However, the influence of Orxs on glutamatergic transmission to DRN 5-HT neurons remains unknown. In the current study, we show that Orx-B depresses glutamatergic EPSCs by inhibiting glutamate release. This effect is mediated by the stimulation of postsynaptic OxRs and involves a retrograde endocannabinoid signal. As such, these results outline a novel mechanism by which Orxs modulate the function of glutamate synapses in the DRN 5-HT neurons.
Materials and Methods
Brain slice preparation. Coronal brainstem slices containing the DRN were prepared from 4- to 6-week-old male Sprague Dawley rats as described previously (Haj-Dahmane, 2001). This procedure was in accordance with National Institutes of Health and American Association for Accreditation of Laboratory Animal Care guidelines and was approved by the University at Buffalo Institutional Animal Care and Use Committee. Briefly, animals were anesthetized with halothane and killed by decapitation. The brain was quickly removed and cooled in ice-cold standard artificial CSF (aCSF) solution of the following composition (in mm): 119 NaCl, 2.5 KCl, 2.5 CaCl2, 1.3 MgSO4, 1 NaH2PO4, 26.2 NaHCO3, and 11 glucose (saturated with 95% O2/5% CO2). A block of tissue containing the DRN was dissected, and coronal slices (300-400 μm) were cut with a vibratome (Lancer series 1000; Ted Pella, Irvine, CA). Slices were stored for at least 1 h in a holding chamber filled with the aCSF solution and continuously bubbled with 95% O2/5% CO2. Slices containing the DRN were then transferred one at a time to a recording chamber, in which the slice was held submerged between two nylon nets and continuously perfused with the standard aCSF solution (1-2 ml/min) saturated with 95%O2/5% CO2 at 30 ± 1°C.
Whole-cell recordings. Whole-cell recordings were performed from DRN neurons using the blind patch technique (Blanton et al., 1989). Low-resistance path electrodes (3-5 MΩ) were made from borosilicate glass tubing (Warner Instruments, Hamden, CT) using a P97 horizontal puller (Sutter Instruments, Novato, CA). Recording pipettes were filled with an internal solution containing the following (in mm): 120 potassium gluconate, 10 KCl, 10 Na2-phosphocreatine, 10 HEPES, 1 MgCl2,1 EGTA, 2 Na2-ATP, and 0.25 Na-GTP, pH 7.3 (adjusted with KOH; osmolarity, 280 mOsm/L). In some experiments, the calcium-buffering capacity of the internal solution was increased by replacing 25 mm potassium gluconate with 25 mm 1,2-bis(2-aminophenoxy)ethane-N, N,N′,N′-tetra-acetic acid (BAPTA). Miniature EPSCs (mEPSCs) were recorded with an internal solution in which potassium gluconate was substituted for cesium gluconate. In experiments examining the role of postsynaptic G-proteins in the Orx-B modulation of evoked glutamatergic EPSCs (eEPSCs), GTP was replaced with either GDPβS (200 μm) or GTPγS (200 μm). The membrane potentials were corrected for the liquid junction potentials of +4 and +12 mV recorded with potassium- and cesium gluconate-based internal solutions, respectively.
Stimulation and recordings. Stainless-steel bipolar stimulating electrodes (Frederick Haer Company, Bowdoinham, ME) were placed 100-200 μm dorsolateral to the recording site. Single-pulse electrical stimuli (10-30 V in amplitude and 100-200 ms in duration) were used to evoke postsynaptic currents. eEPSCs were recorded in the continuous presence of the GABAA and glycine receptor antagonists bicuculline (20 μm) and strychnine (10 μm), respectively. Membrane currents were recorded with an Axoclamp 2B amplifier (Axon Instruments, Foster City, CA), filtered at 10 kHz, digitized at 20 kHz with the Digidata 1200 interface board (Axon Instruments), and stored on a hard disk for additional analysis using the Origin 7.0 software (Microcal Software, Northampton, MA). Access resistance (10-20 MΩ) was monitored on-line during the course of the experiments with 5 mV hyperpolarizing steps (200-500 ms duration). Experiments were discarded when series resistance changed by >10%.
Data analysis. The amplitude of eEPSCs was calculated by measuring the average current duringa2ms time window taken at the peak of each EPSC and subtracted from the current obtained during a 5 ms window taken immediately before the stimulus artifact. All eEPSC amplitudes were normalized to the mean baseline amplitude recorded for at least 10 min before Orx-B application. Paired stimuli were given at 30 ms intervals, and the ratios of the second and the first eEPSC were averaged for at least 60 trials in the absence and presence of Orx-B. For the calculation of coefficient of variance (CV), the background noise variance, obtained from interleaved traces collected without a stimulus, was subtracted from the eEPSC amplitude variance (60 consecutive trials in the absence and presence of Orx-B for each cell). The CV is then given by the following ratio: (VareEPSC - Varnoise)/(eEPSC mean amplitude).
Glutamate-mediated mEPSCs were recorded in the presence of tetrodotoxin (TTX) (1 μm), bicuculline (20 μm), and strychnine (20 μm) (Liu et al., 2002a) and analyzed off-line using the Mini Analysis software (Synaptosoft, Decatur, GA). The events were selected using amplitude, rise time, and area criteria. The amplitude detection and area threshold were set at 5 pA and 50 fC, respectively. All detected events were further checked individually by visual inspection to prevent noise from compromising the analysis. Results in the text and figures are presented as mean ± SEM. Statistical analyses were performed using the Student's paired t test for within-cell comparison, the Student's unpaired t test for comparison between group, and the nonparametric Kolmogorov-Smirnov (K-S) test for comparison of mEPSC distributions. p < 0.05 was considered statistically significant.
In most experiments, drugs were administered in the bath dissolved at known concentrations by gravity perfusion. U 73122 and U 73343 were first reconstituted in chloroform into single-use aliquots. The aliquots were evaporated to dryness, dissolved in DMSO, and then added to the aCSF solution at the desired concentration. In experiments aimed at examining the effect of Orx-B on the sensitivity of postsynaptic glutamate receptors, a patch pipette containing 100 μm glutamate was placed above the recorded neuron, and glutamate was administered by pressure application (10 psi and 100 ms duration). Most chemicals and drugs were purchased from Sigma-Aldrich (St. Louis, MO). 6,7-Dinitroquinoxaline-2,3-dione (DNQX), AM 251, WIN 55,212-2, CGP 55845, and anandamide were purchased from Tocris Cookson (Ellisville, MO). TTX was purchased from Alomone Labs (Jerusalem, Israel), and Orx-B was from American Peptide (Sunnyvale, CA). RHC-80267, U 73122 and U 73343 were obtained from Biomol (Plymouth Meeting, PA).
Results
To examine the effect of Orx-B on glutamatergic synaptic transmission in the DRN, we performed whole-cell recording from DRN neurons in acutely prepared brainstem slices. Only neurons (n = 125) that exhibited membrane properties typical of putative 5-HT neurons were included in this study. These properties include a resting membrane potential ranging between -60 and -70 mV, slow spiking activity in response to suprathreshold depolarizing pulses, a large afterhyperpolarizing potential (AHP), and a membrane hyperpolarization induced by the stimulation of somatodendritic 5-HT1A receptors (Aghajanian and Vandermaelen, 1982; Haj-Dahmane, 2001). Glutamatergic EPSCs were evoked by local electrical stimulation in the DRN in the presence of GABAA and glycine receptor antagonists bicuculline (20 μm) and strychnine (20 μm), respectively. As illustrated in Figure 1A, local stimulation in the DRN evoked fast EPSCs in DRN 5-HT neurons voltage clamped at -70 mV. These eEPSCs were blocked by the non-NMDA glutamate receptor antagonist DNQX (20 μm; n = 7) (Fig. 1A), indicating that they were mainly mediated by AMPA/kainate receptors. As expected for AMPA/kainate-mediated synaptic currents, holding neurons at more depolarized potentials resulted in a reduction of the eEPSC amplitude and revealed an apparent reversal potential of ∼2.5 ± 4.5 mV (n = 10) (Fig. 1B).

Orx-B causes a reversible depression of glutamate-mediated eEPSCs in DRN 5-HT neurons. A, eEPSC amplitude elicited by local stimulation of the DRN and recorded from 5-HT neurons voltage clamped at -70 mV in the absence and presence of DNQX (20 μm). Each data point represents the peak amplitude of a single eEPSC. Inset depicts averaged eEPSC traces (30 trials) collected before and during DNQX application. Calibration: 50 pA, 10 ms. B, eEPSC traces recorded at different membrane potentials (left) and the corresponding current-voltage (I-V) relationship (right). Note that the I-V curve is linear and shows an apparent reversal potential at ∼2 mV. Calibration: 50 pA, 10 ms. C, Bath application of 300 nm Orx-B (bar) decreased the amplitude of eEPSCs. Each dot represents the peak amplitude of a single eEPSC. Top illustrates averaged traces (30 trials) collected before, during, and after Orx-B application. Calibration: 100 pA, 10 ms. D, Summary graph of the time course and magnitude of Orx-B-induced depression of eEPSC amplitude (n = 20). Each data point represents the mean ± SEM of eEPSC amplitude normalized to the average amplitude before Orx-B application. In this and the remaining figures, the stimulus artifacts were digitally blanked.
Orx-B presynaptically inhibits glutamatergic EPSCs in DRN 5-HT neurons
We found that bath application of Orx-B (300 nm) reduced the amplitude of eEPSCs in all putative DRN 5-HT neurons tested. A representative experiment showing the effect of Orx-B on eEPSC amplitude is illustrated in Figure 1C. On average, the amplitude of eEPSCs was depressed to 53 ± 5% of baseline (n = 20; p < 0.01; paired t test) (Fig. 1D). In our recording conditions, the depression of eEPSCs reached its peak within 5 min of Orx-B application and recovered to 92 ± 4.5% of baseline after 30-40 min washout.
In addition to the depression of eEPSCs, Orx-B (300 nm) induced an inward current ranging from 10 to 80 pA in amplitude (data not shown) and was associated with little change in the membrane input resistance (control, 383 ± 15.5 MΩ; Orx-B, 372 ± 13.9 MΩ; n = 15; p > 0.05; paired t test). Because Orx-B did not significantly affect the cell input resistance, it is unlikely that the depression of eEPSC was secondary to an alteration of the membrane conductance. Consistent with this view, we found no correlation between the ability of Orx-B to induce an inward current and the magnitude of Orx-B-induced inhibition of eEPSC. In addition, the time constant (τ) of the rising phase of eEPSCs recorded in control condition was essentially identical to that obtained in the presence of Orx-B (τ control, 1.26 ± 0.56 ms; τ Orx-B, 1.31 ± 0.44 ms; n = 20; p > 0.05; paired t test).
In principle, Orx-B could reduce the amplitude of eEPSCs by reducing the sensitivity of postsynaptic glutamate receptors, inhibiting glutamate release, or a combination of these effects. To determine the mechanism underlying the Orx-B-induced depression of eEPSC amplitude, we first sought to examine the effect of Orx-B on the amplitude of the inward current induced by exogenous application of glutamate. Pressure application of glutamate (100 μm; 100 ms duration) to DRN 5-HT neurons elicited an inward current that was blocked by DNQX (20 μm; n = 4) (Fig. 2A1). Administration of Orx-B (300 nm) had no significant effect on the amplitude of glutamate-induced current (98.1 ± 3.5% of control; n = 4; p > 0.05; paired t test) (Fig. 2A2). These results suggest that the depression of eEPSC amplitude was not attributable to an alteration of postsynaptic AMPA/kainate receptor function.
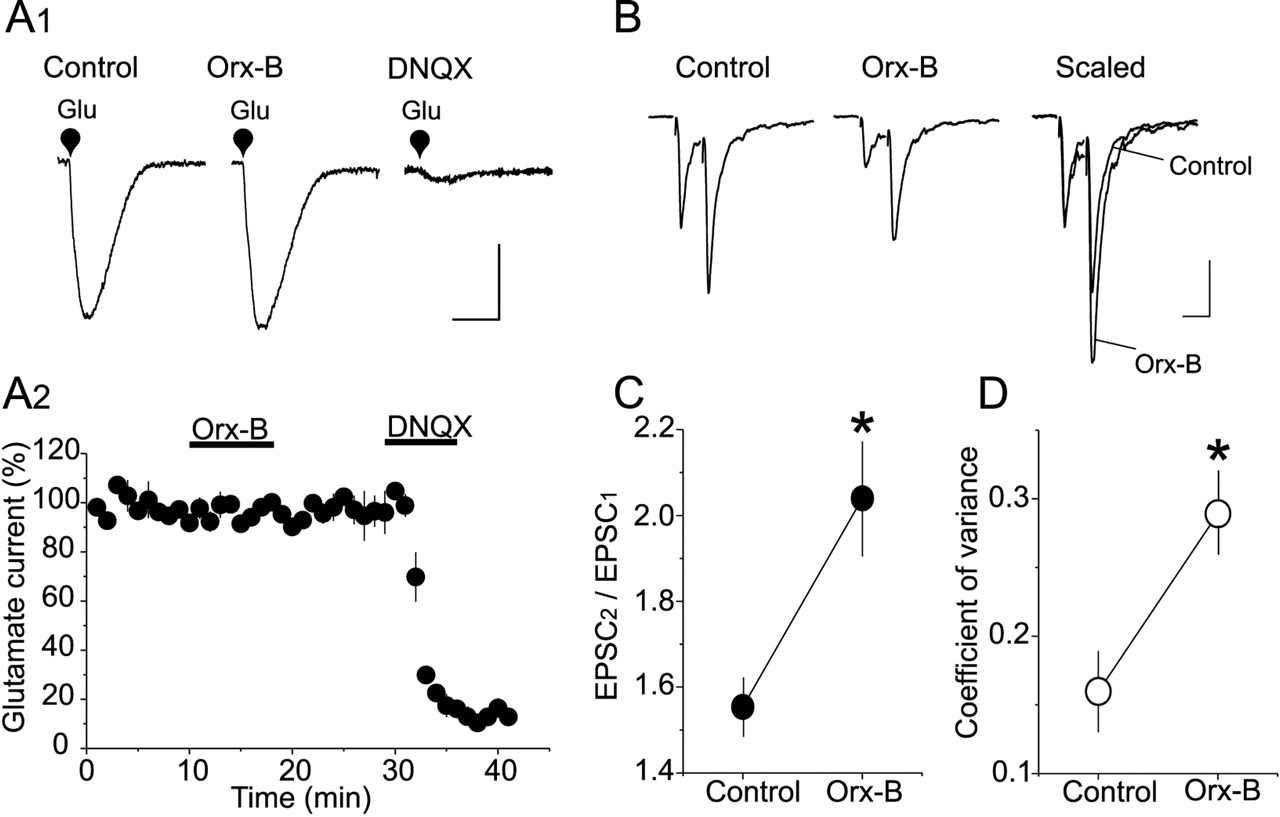
Orx-B depresses eEPSC amplitude by a presynaptic mechanism. A1, Orx-B had no significant effect on the amplitude of l-glutamate-induced current. Averaged inward currents (10 consecutive trials) elicited by pressure application of l-glutamate (Glu) (100 μm, 250 ms) in control condition (left trace), in the presence of 300 nm Orx-B (middle trace), and in the presence of 20 μm DNQX (right trace). Calibration: 50 pA, 200ms. A2, Summary graph depicting the effect of Orx-B on the mean amplitude (n = 4) of l-glutamate-induced current normalized to average amplitude before Orx-B application. B, Orx-B-induced depression of eEPSC amplitude is associated with an increase in the PPR. Representative eEPSC traces (average of 30 consecutive trials) evoked by paired stimuli (30 ms interval) in control condition (left trace) and in the presence of 300 nm Orx-B (middle trace). Right traces depict superimposed and scaled EPSC traces obtained in the absence (control) and presence of Orx-B. The first eEPSC during Orx-B application is scaled to the amplitude of the first eEPSC collected in control condition. Calibration: 100 pA, 30 ms. C, Summary plot of the mean PPR obtained in the absence and presence of 300 nm Orx-B (*p < 0.05; n = 5). D, The decrease in EPSC amplitude is also associated with an increase in CV. Each data point depicts the average CV of EPSC amplitude determined in control condition and in the presence of 300 nm Orx-B (*p < 0.05; n = 5).
We next examined the effect of Orx-B on the paired-pulse ratio (PPR) of eEPSCs, a measure that reflects changes in neurotransmitter release at central synapses (Perkel and Nicoll, 1993; Dobrunz and Stevens, 1997). Two consecutive stimuli with a 30 ms interstimulus interval elicited a pair of eEPSCs with the second eEPSC amplitude significantly larger than the first (Fig. 2B). This paired-pulse facilitation (PPF) of eEPSCs was observed in all 5-HT neurons tested (n = 7) (Fig. 2C). Administration of Orx-B (300 nm) reduced the amplitude of both eEPSCs. However, the depression of the first eEPSC was more profound than the second eEPSC, resulting in a significant increase in PPF (PPF control, 1.55 ± 0.07; PPF Orx-B, 2.04 ± 0.13; n = 7; p < 0.05; paired t test) (Fig. 2C). The inhibition of eEPSC amplitude was also accompanied by a significant increase in CV (control, 0.16 ± 0.025; Orx-B, 0.29 ± 0.026; n = 20; p < 0.05; paired t test) (Fig. 2D). The increase in these presynaptic parameters suggests that the inhibition of eEPSCs induced by Orx-B in DRN 5-HT neurons is predominantly mediated by a presynaptic mechanism.
To determine the locus of Orx-B-mediated inhibition of glutamatergic synaptic transmission, we examined its effect on the frequency and amplitude of mEPSCs. Bath application of Orx-B (300 nm) reversibly decreased the frequency of mEPSCs (p < 0.01; K-S test) (Fig. 3A1-A3). The average mEPSC frequency was reduced from 2.7 ± 0.66 Hz in the control condition to 1.36 ± 0.42 Hz in the presence of Orx-B (n = 5; p < 0.05; paired t test) (Fig. 3A4). In contrast, no significant change was detected in the amplitude distribution of mEPSCs in the presence of Orx-B (300 nm) (p > 0.05; K-S test) (Fig. 3B1). The average amplitudes of all of the selected events recorded in the absence and presence of Orx-B were 21.67 ± 1.77 and 21.10 ± 1.31 pA, respectively (n = 5; p > 0.05; paired t test) (Fig. 3B2). These results clearly indicate that Orx-B causes reversible inhibition of glutamate release from glutamatergic terminal synapsing on DRN 5-HT neurons.
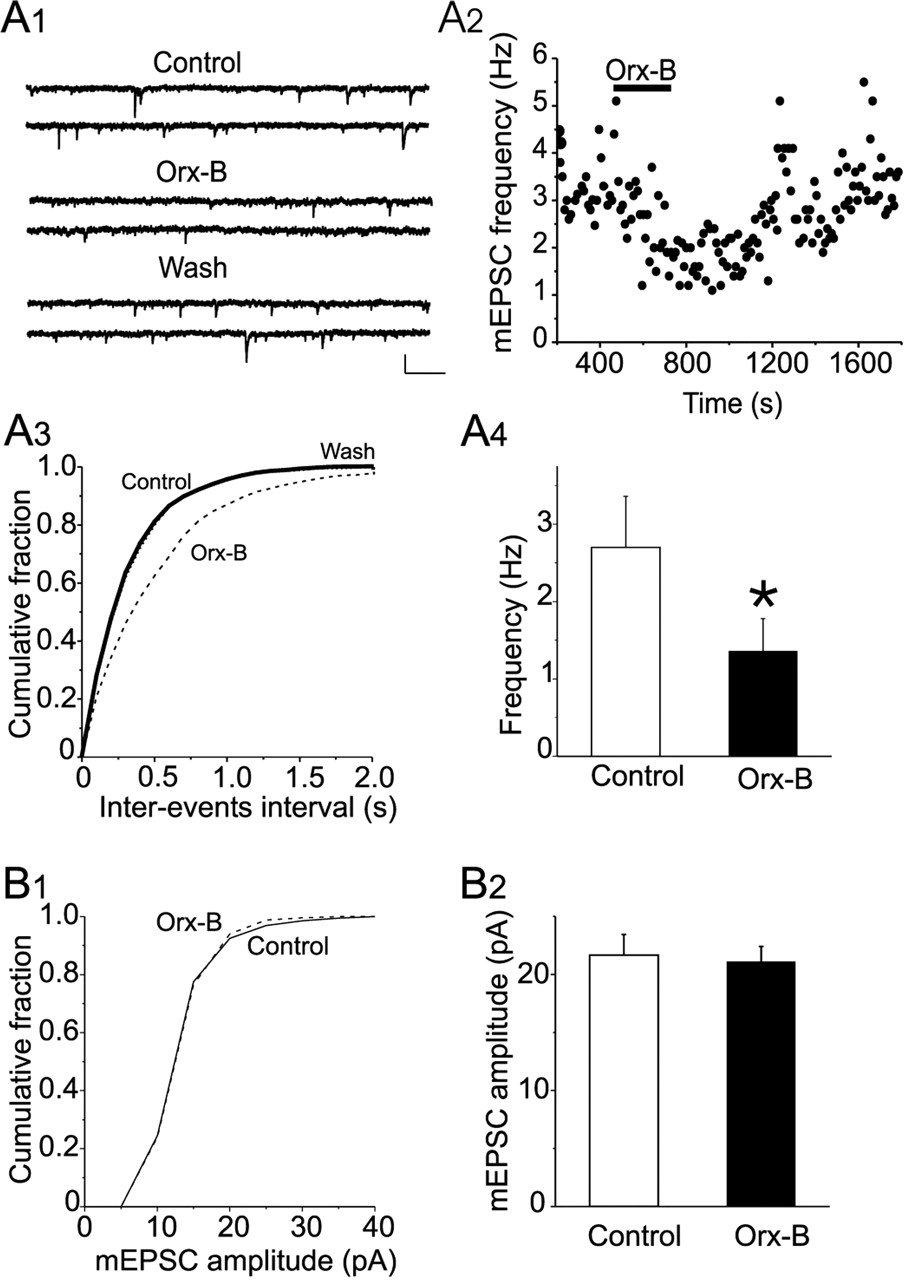
Orx-B inhibits the frequency but not the amplitude of mEPSCs. A1, Sample current traces showing mEPSCs recorded in control condition (top traces), in the presence of Orx-B (300 nm; middle traces), and during wash (bottom traces). Calibration: 20 pA, 250 ms. A2, Plot of the frequency of mEPSCs as a function of time obtained from the same neuron illustrated in A1. A3, Plot of the cumulative distribution of the interevent interval for the neuron illustrated in A1 showing that Orx-B significantly reduced the frequency of mEPSC (p < 0.05; K-S test). A4, Summary graph of the average mEPSC frequency in control condition and in the presence of 300 nm Orx-B (*p < 0.05; n = 5). B1, Plots of cumulative distribution of mEPSC amplitude obtained in the absence (solid line) and presence (dashed line) of Orx-B. Application of Orx-B (300 nm) did not significantly affect the distribution of mEPSC amplitude (p > 0.05; K-S test). B2, Summary graph of the mean amplitude of all events selected in control condition and in the presence of 300 nm Orx-B (p > 0.05; n = 5).
The depression of EPSCs is mediated by postsynaptic OxRs through a retrograde endocannabinoid release
A possible interpretation of the above results is that Orx-B would reduce glutamate release by stimulating OxRs located on glutamatergic terminals impinging on 5-HT neurons. If this interpretation were correct, blockade of postsynaptic OxR signaling should have no effect on the depression of eEPSCs induced by Orx-B. Unexpectedly, intracellular application of the membrane-impermeable G-protein inhibitor GDPβS (200 μm; for at least 10 min) via the patch pipette completely abolished the depression of eEPSCs induced by 300 nm Orx-B (102 ± 2.5% of control; n = 7; p = 0.15; paired t test) (Fig. 4A1,A2). The inhibition of G-protein with GDPβS also resulted in a complete blockade of Orx-B-induced inward current (n = 7; data not shown), a response known to be mediated by stimulation of postsynaptic OxRs (Brown et al., 2001; Liu et al., 2002b). Conversely, loading neurons with GTPγS (200 μm), an irreversible activator of G-protein, resulted in a persistent Orx-B-induced inward current (n = 8; data not shown), as well as a persistent inhibition of eEPSC amplitude (49 ± 5% of baseline; n = 8; p < 0.02; paired t test) (Fig. 4A1,A2). Theses results strongly suggest that Orx-B acts on postsynaptic OxRs located on 5-HT neurons and indirectly inhibits glutamate release through a retrograde signal.

Inhibition of postsynaptic G-protein signaling abolishes the Orx-B-induced depression of eEPSC. A1, Superimposed eEPSC traces recorded with an internal solution containing GTP (left traces), GDPβS (middle traces), and GTPγ S (right traces) before, during Orx-B (300 nm) application, and after washout. Calibration: 100 pA, 10 ms. A2, Summary plots of the time course and magnitude of the Orx-B-induced depression of eEPSC obtained with GTP (▵; n = 7), GDPβS (▪; n = 7), and GTPγ S (•; n = 7). Note that the intracellular infusion of GDPβS blocked the Orx-B-induced depression of EPSC, whereas injection of GTPγS resulted in an irreversible Orx-B-induced depression of eEPSCs.
The retrograde molecules that mediate the effect of Orx-B should have their specific receptors located on glutamatergic terminals to inhibit glutamate release. Previous studies have shown that 5-HT, acting at presynaptic 5-HT1B receptors, depresses glutamatergic EPSCs in several brain regions (Singer et al., 1996; Muramatsu et al., 1998; Laurent et al., 2002), including the median raphe nucleus (Li and Bayliss, 1998). As such, 5-HT per se could be retrogradely released after activation of OxRs and mediate the Orx-B induced inhibition of eEPSCs. However, we found that bath application of 5-HT (30 μm) had no effect on the amplitude of eEPSCs. Increasing the concentration of 5-HT (100 μm) induced only a marginal depression that did not reach statistical significance (93.03 ± 2.9% of control; n = 4; p > 0.05; paired t test). Similarly, no significant change in the amplitude of eEPSCs was observed in the presence of L 694,247 (30 μm), a selective 5-HT1B/1D receptor agonist (98.5 ± 2.01% of control; n = 3; p > 0.05; paired t test; data not shown). Together, these results show that 5-HT is unlikely to act as a retrograde messenger.
Because 5-HT neurons contain GABA (Belin et al., 1983; Millhorn et al., 1987; Stamp and Semba, 1995), we sought to determine whether GABA could be the retrograde signal that mediates the Orx-B-induced inhibition of eEPSCs. Bath application of the selective GABAB receptor agonist baclofen (10 μm) induced a robust depression of eEPSC amplitude (35 ± 6.8% of control; n = 5; p < 0.05; paired t test; data not shown), which was blocked by the selective GABAB receptors antagonist CGP 55845 (3 μm; 92 ± 3.4% of control; n = 4; p > 0.05; paired t test; data not shown). However, treatment of slices with CGP 55845 (3 μm) did not block the ability of Orx-B to inhibit the amplitude of eEPSCs (48.7 ± 3.7% of control in saline solution; 50.7 ± 3.5% of control in CGP 55845; n = 5; p > 0.05; unpaired t test). As such, it is unlikely that the release of GABA, acting at GABAB receptors, contributes to the Orx-B-induced depression of eEPSCs.
Previous studies have shown that endocannabinoids act as retrograde messengers that inhibit glutamate and GABA release in the CNS (Kreitzer and Regehr, 2001a; Maejima et al., 2001; Ohno-Shosaku et al., 2001; Wilson and Nicoll, 2001). Both CB1 cannabinoid receptors and the fatty acid amide hydrolase, an enzyme that hydrolyzes the endogenous cannabinoid receptor agonist, arachidonoyl ethanolamide (anandamide), are expressed in the DRN (Matsuda et al., 1993; Tsou et al., 1998; Egertova et al., 2003). We thus examined whether endocannabinoids could mediate the Orx-B-induced depression of glutamate release. Bath administration of anandamide (30 μm), induced a depression of eEPSC amplitude (65.7 ± 2.8% of control; n = 6; p < 0.05; paired t test) (Fig. 5A1,A2). Because anandamide also activates vanilloid receptors (Zygmunt et al., 1999; Smart et al., 2000), we also tested the effect of WIN 55,212-2, a selective cannabinoid receptor agonist, on the amplitude of eEPSCs. We found that bath administration of WIN 55,212-2 (10 μm) reduced the amplitude of eEPSCs (52.9 ± 1.2% of control; n = 6; p < 0.01; paired t test) (Fig. 5B1) but had no significant effect on the holding current and input resistance of DRN 5-HT neurons (n = 6; data not shown). Pretreatment of slices with AM 251 (3 μm), a selective CB1 receptor antagonist, totally blocked the inhibition of eEPSCs induced by WIN 55,212-2 (94.5 ± 3.8% of control; n = 6; p > 0.05; paired t test) (Fig. 5B1), indicating that this effect was mediated by CB1 cannabinoid receptors.

Cannabinoid receptor agonists mimic and occlude the depression of eEPSC induced by Orx-B. A1, Summary plot of the effect of anandamide on eEPSC amplitude (n = 5). Note that bath application of anandamide (30 μm) reduced the amplitude of eEPSCs. A2, Representative eEPSC traces collected during the course of the experiment as indicated by numbers in A1. Calibration: 100 pA, 10 ms. B1, Effect of the synthetic cannabinoid receptor agonist WIN 55,212-2 (10 μm) on eEPSC amplitude obtained in the absence (•; n = 7) and presence (○; n = 5) of the CB1 antagonist AM 251. B2, Plots of the corresponding change in PPR. Note that WIN 55,212-2 depressed the amplitude of eEPSC and increased the PPR. These effects were abolished by the CB1 receptor antagonist AM 251 (3 μm). B3, Sample of paired eEPSCs traces taken during the time indicated by number in B1. Calibration: 50 pA, 30 ms. C1, The Orx-B-induced depression of eEPSC is occluded by WIN 55,212-2 (10 μm; WIN). Orx-B (300 nm; left traces) and WIN 55,212-2 (10 μm; middle traces) induced inhibition of eEPSC amplitude obtained in the same neuron. In the presence of WIN 55,212-2, further addition of Orx-B had no additive effect on eEPSC amplitude (right traces). Calibration: 100 pA, 10 ms. C2, Summary graph of the mean changes in eEPSC amplitude in the presence Orx-B, WIN 55,212-2, and Orx-B plus WIN 55,212-2. Note that the effects of Orx-B and WIN 55,212-2 were not additive (p > 0.05; n = 7).
Similar to the effect of Orx-B, the depression of eEPSCs induced by WIN 55,212-2 was mediated by a decrease in glutamate release, as revealed by the increase in PPR (PPR control, 1.12 ± 0.15; PPR WIN, 1.96 ± 0.17; n = 6; p < 0.05; paired t test) (Fig. 5B2). The magnitude of the depression of eEPSCs induced by WIN 55,212-2 (10 μm) was also comparable with that induced by Orx-B (300 nm) (WIN 55,212-2, 51.9 ± 1.9% of control; Orx-B, 53 ± 2.7%; n = 7; p > 0.05; unpaired t test) (Fig. 5C2). In the presence of WIN 55,212-2 (10 μm), administration of Orx-B (300 nm) failed to induce an additional inhibition of eEPSC amplitude over that induced by WIN 55,212-2 alone (WIN 55,212-2, 51.9 ± 1.9% of control; WIN 55,212-2 plus Orx-B, 48.3 ± 4.5% of control; n = 6; p > 0.05; paired t test) (Fig. 5C1,C2). The observations that WIN 55,212-2 mimicked and occluded the inhibition of eEPSCs induced by Orx-B suggest that the two forms of eEPSC inhibition share the same receptors and/or presynaptic intracellular cascade.
If the depression of eEPSCs induced by Orx-B was indeed mediated by endocannabinoids acting at presynaptic CB1 receptors, blockade of these receptors should antagonize the effect of Orx-B. To test this idea, we examined the effect of the CB1 antagonist AM 251 on the Orx-B-mediated inhibition of eEPSCs. As illustrated in Figure 6A1, bath application of Orx-B (300 nm) in the absence of AM 251 induced a robust inhibition of eEPSC amplitude. Perfusion of slices with AM 251 (3 μm) for at least 10 min resulted in a complete blockade of the Orx-B-induced depression of eEPSC amplitude. On average, the amplitude of eEPSCs was reduced to 50.6 ± 2.4 and 99.5 ± 3.23% of control in the absence and presence of AM 251, respectively (n = 7; p < 0.02; paired t test) (Fig. 6A2). Because the Orx-B-induced depression of eEPSCs exhibited no obvious desensitization after two consecutive applications of Orx-B (first application, 49 ± 3.1% of control; second application, 50.1 ± 3.1% of control; n = 5; p > 0.05; paired t test) (Fig. 6A3), the blockade of the Orx-B effect obtained in AM 251 could not be attributed to a downregulation of OxRs. Together, these results strongly suggest that endocannabinoids acting at CB1 receptors mediate the Orx-B-induced depression of eEPSCs.

Orx-B-induced depression of glutamate release is blocked by CB1 cannabinoid receptor antagonist. A1, The CB1 receptor antagonist AM 251 (3 μm) abolished the Orx-B-induced depression of eEPSC amplitude. Bottom depicts the effect of Orx-B on eEPSC amplitude obtained in the absence and presence of AM 251. Top illustrates sample eEPSC traces collected during the course of the experiment as indicated by numbers. Calibration: 100 pA, 10 ms. A2, Summary graph of the mean changes in eEPSC amplitude induced by Orx-B in the absence and presence of AM251 (n = 6; **p < 0.01; paired t test). A3, The Orx-B induced depression of eEPSC exhibited no obvious desensitization. Bottom represents the effect of two consecutive applications of Orx-B (300 nm) on the amplitude of eEPSCs. Note that, in the absence of AM 251, the second application of Orx-B (300 nm) effectively depresses the amplitude of eEPSCs. Top illustrates average EPSC traces recorded during the course of the experiments as indicated by numbers. B1, The CB1 receptor antagonist AM 251 had no effect on the amplitude of Orx-B-induced inward current. Sample traces of Orx-B-induced inward current recorded in control condition (left trace) and in the presence of AM 251 (3 μm; right trace). Calibration: 50 pA, 2 min. B2, Summary graph of the average amplitude of Orx-B-induced current recorded in the absence and presence of AM 251 (n = 5; p > 0.05).
However, it remains possible that the cannabinoid receptor antagonist (AM 251) abolished the effect of Orx-B by blocking OxRs. To test for this possibility, we examined whether the inward current/membrane depolarization induced by OxRs activation in the same neurons is affected by AM 251. We found that AM 251 had no effect on the amplitude of Orx-B-induced inward current (Orx-B current, 65 ± 10 pA in control; Orx-B current, 58 ± 7 pA in AM 251; n = 4; p > 0.05; paired t test) (Fig. 6B1,B2), which indicates that OxRs were not blocked by cannabinoid receptor antagonist. Combined, these findings lead us to conclude that stimulation of OxRs on 5-HT neurons induces the release of endocannabinoids, which in turn activates presynaptic CB1 cannabinoid receptors and causes an inhibition of glutamate release.
Orx-B-induced endocannabinoid synthesis involves the phospholipase C/diacylglycerol pathway
We next examined the downstream mechanisms by which activation of OxRs induces endocannabinoid synthesis. A postsynaptic rise in intracellular calcium has been shown to play a crucial role in endocannabinoid synthesis and release (for review, see Piomelli, 2003). Such a calcium-dependent form of endocannabinoid release has been shown to induce transient depression of synaptic transmission at both glutamatergic (Kreitzer and Regehr, 2001a; Maejima et al., 2001) and GABAergic (Kreitzer and Regehr, 2001b; Wilson and Nicoll, 2001) synapses in the CNS. Because stimulation of OxRs increases intracellular calcium in a variety of neuronal preparations (van den Pol et al., 1998; Uramura et al., 2001), including DRN 5-HT neurons (Kohlmeier et al., 2004), we wondered whether a rise in postsynaptic intracellular calcium is required for Orx-B-induced depression of eEPSCs. To this end, neurons were recorded using a pipette solution with a high calcium-buffering capacity (25 mm BAPTA). Intracellular application of BAPTA (25 mm) for at least 10 min strongly reduced the amplitude of the calcium-activated potassium current responsible for afterhyperpolarization potential (IAHP) (n = 8) (Fig. 7A), which indicates an effective buffering of the rise in intracellular calcium. However, under this condition, bath application of Orx-B still induced a robust inhibition of eEPSC amplitude (51.9 ± 5.9% of baseline; p < 0.01; n = 8 cells; paired t test) (Fig. 7A). The magnitude of Orx-B-induced inhibition of eEPSCs obtained using intracellular solutions with high (25 mm BAPTA) and low (1 mm EGTA) calcium-buffering capacity were essentially identical (25 mm BAPTA, 51.9 ± 5.2% of baseline; 1 mm EGTA, 49.11 ± 6.8% of baseline; n = 8; p > 0.05; unpaired t test). These results indicate that the synthesis of endocannabinoids triggered by OxR activation does not require a rise in intracellular calcium in DRN 5-HT neurons.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Orx-B-induced endocannabinoid release is calcium independent but requires the activation of the PLC/DAG pathway. A, Intracellular application of BAPTA (25 mm) strongly reduced the amplitude of the outward tail current underlying the afterhyperpolarization potential (IAHP). The IAHP is triggered by a voltage steps from -70 to 0 mV (200 ms duration) delivered 1 min (left trace) and 10 min (left trace) after whole-cell accesses. Each trace depicts the average of 10 consecutive trials. Calibration: 50 pA, 300 ms. B, Intracellular administration of BAPTA (25 mm) had no effect on the Orx-B-induced depression of eEPSC amplitude. Bottom is a summary plot of the depression of eEPSC induced by Orx-B (300 nm) obtained with an internal solution containing 1 mm EGTA (•; n = 12) and 25 mm BAPTA (○; n = 7). Raising the calcium-buffering capacity of the pipette solution did not block the ability of Orx-B to inhibit eEPSC amplitude. Top depicts superimposed eEPSC traces recorded with an internal solution containing 1mm EGTA (left traces) and 25 mm BAPTA (right traces) and taken at the time points indicated by numbers. Calibration: 100 pA, 10 ms. C, Inhibition of PLC or DAG lipase abolished the Orx-B-induced inhibition of eEPSC amplitude. Bottom illustrates a summary graph of the effect of Orx-B (300 nm) on eEPSC amplitude obtained in slices treated with a PLC inhibitor (U 73122; 10 μm; n = 7 neurons; ○), its inactive analog (U 73343; 10 μm; n = 5; •), or a DAG lipase inhibitor (RHC-80287; 50 μm; n = 6; ⋄). Top illustrates averaged eEPSC traces (30 trials) recorded from slices treated with U 73343 (left traces), U 73122 (middle traces), or RHC-80287 (right traces) and collected during the time points indicated by the numbers. Calibration: 100 pA, 10 ms. D, DAG lipase inhibitor had no effect on the depression of eEPSCs induced by CB1 receptor agonist. Bottom illustrates the depression of eEPSC amplitude induced by WIN 55,212-2 (10 μm) obtained in control slices (○; n = 7) and in slices pretreated with 50 μm RHC-80287 (•; n = 5). Superimposed EPSC traces (top) obtained before and during WIN 55,212-2 application in control slices (left traces) and in slices treated with RHC-80287 (right traces). Calibration: 50 pA, 10 ms.
Several reports have provided evidence that the endocannabinoid 2-arachidonoyl-glycerol (2-AG) can be synthesized in response to the stimulation of phospholipase C (PLC) and the subsequent accumulation of diacylglycerol (DAG) (Stella et al., 1997; Stella and Piomelli, 2001). DAG is then converted into 2-AG by DAG lipase (Bisogno et al., 2003). We thus investigated whether pharmacological inhibition of the PLC/DAG pathway could affect the Orx-B-mediated inhibition of eEPSCs. As shown in Figure 7C, in slices pretreated for at least 1 h with the PLC inhibitor U 73122 (10 μm), administration of Orx-B (300 nm) failed to inhibit the amplitude of eEPSCs (96.6 ± 3.6% of control; p > 0.05; n = 8; paired t test). In contrast, the ability of Orx-B to inhibit the amplitude of eEPSC persisted in slices treated with U 73433 (10 μm), an inactive analog of U 73122 (52.04 ± 1.5% of control; n = 8; p < 0.05; paired t test) (Fig. 7C). Similar to the effect of PLC inhibitor, treatment of the slices with DAG lipase inhibitor RHC-80267 (50 μm) resulted in a total blockade of Orx-B-mediated inhibition of eEPSC amplitude (101.2 ± 2.4% of control; n = 8; p > 0.05; paired t test) (Fig. 7C). These results suggest that OxR-induced suppression of eEPSC requires the activation of PLC and DAG lipase pathways.
The inhibition of PLC or DAG lipase pathway could abolish the Orx-B-induced depression of eEPSCs by either blocking endocannabinoids synthesis or altering the signaling of presynaptic CB1 receptors. Therefore, we examined the effect of a DAG lipase inhibitor (RHC-80267) on the inhibition of eEPSCs induced by CB1 receptor agonist. We found that WIN 55,212-2 (10 μm) effectively inhibited the amplitude of eEPSCs in slices treated with RHC-80267 (50 μm) (n = 4 cells) (Fig. 7D). The depression of eEPSC amplitude induced by WIN 55,212-2 in slices treated with RHC-80267 was comparable with that obtained in control slices (control, 50.01 ± 4% of baseline; RHC-80267, 47.9 ± 6%; n = 4; p > 0.05; unpaired t test). As such, the blockade of Orx-B-induced suppression of eEPSCs observed in the presence of PLC or DAG lipase inhibitors could not be attributed to an alteration of CB1 receptor function but rather to a blockade of endocannabinoid synthesis. Together, these results indicate that stimulation of OxRs triggers the biosynthesis of endocannabinoids via PLC and DAG lipase enzymatic cascade and suggest that 2-AG is a potential retrograde messenger that mediates the inhibition of glutamate release caused by Orx-B in DRN 5-HT neurons.
Discussion
The present study shows that Orx-B inhibits glutamatergic synaptic transmission onto DRN 5-HT neurons by a presynaptic mechanism involving a decrease in glutamate release. More importantly, we have shown that the Orx-B-induced inhibition of glutamate release is not signaled by OxRs located on presynaptic terminals but rather by the activation by postsynaptic OxRs located on DRN 5-HT neurons via a retrograde messenger. This retrograde synaptic inhibition of glutamate release is likely mediated by endocannabinoids acting on presynaptic CB1 receptors. In addition, we provided evidence that OxRs-induced inhibition of glutamate release does not require a rise in postsynaptic intracellular calcium but depends on the activation of the PLC/DAG pathway. As such, our work reveals a novel mechanism by which activation of postsynaptic OxRs can lead to the depression of excitatory afferent input to DRN 5-HT neurons.
Mechanisms of Orx-B-induced inhibition of glutamatergic transmission to DRN 5-HT neurons
Several findings presented here support our conclusion that the Orx-B-induced inhibition of the amplitude of glutamatergic eEPSCs is predominantly caused by a decrease in glutamate release. First, Orx-B did not significantly change the amplitude of the inward current induced by exogenous application of glutamate, thereby excluding the possibility that the inhibition of eEPSC amplitude reflects a reduction in the sensitivity and/or the number of postsynaptic ionotropic glutamate receptors. Second, the depression of eEPSC amplitude is accompanied by a clear change in presynaptic parameters. These include an increase in PPR and CV, which usually indicate a reduction in the probability of neurotransmitter release. Finally, a decrease in glutamate release is further substantiated by the finding that Orx-B significantly reduces the frequency, but not the amplitude, of mEPSCs.
The conclusion that Orx-B inhibits glutamate release in the DRN is in contrast with previous findings that OxR activation increases the amplitude of evoked glutamate-mediated EPSCs in other brain areas (Burlet et al., 2002). Moreover, in the lateral hypothalamus and nucleus tractus solitarius, Orx-B increases the frequency of glutamate-mediated spontaneous EPSC and mEPSC (van den Pol et al., 1998; Li et al., 2002, Smith et al., 2002). The increase in glutamate-mediated synaptic activity observed in these studies is mainly attributed to the stimulation of OxRs presumably located on glutamatergic terminals and/or on local glutamate neurons. In the present study, however, we find that the inhibition of glutamate release in the DRN is mediated by the stimulation of postsynaptic OxRs via a retrograde endocannabinoid messenger (see below). As such, depending on whether OxRs are located on presynaptic terminals or on the target neurons, their activation can exert multifaceted effects on glutamatergic synaptic transmission in the CNS.
The experiments performed with the GTP analogs GDPβS and GTPγS have provided valuable information on whether the Orx-B-induced depression of glutamatergic input is signaled by presynaptic or postsynaptic OxRs. We have shown that intracellular infusion of GDPβS into a single postsynaptic 5-HT neuron completely blocks the Orx-B-induced depression of eEPSC amplitude. Furthermore, irreversible activation of G-protein with GTPγS causes a persistent Orx-B-induced inhibition of glutamatergic eEPSCs. Because GDPβS and GTPγS are membrane impermeable, the release of neurotransmitters from neighboring neurons is unlikely to contribute to the depression of glutamate release induced by Orx-B. As such, the most parsimonious interpretation of these results is that the suppression of glutamate release induced by Orx-B is signaled by postsynaptic OxRs located on the recorded neuron and involves a retrograde messenger.
Pharmacological analysis of the potential retrograde messengers involved in Orx-B-induced inhibition of glutamate release indicates that 5-HT is unlikely the retrograde messenger that mediates the effect of Orx-B. This is supported by the finding that 5-HT has no significant effects on glutamatergic eEPSCs. A similar conclusion can also be extended to the role of GABA. Indeed, although bath application of baclofen inhibits the amplitude of eEPSCs, blockade of GABAB receptors does not antagonize the Orx-B-induced depression of glutamate release.
The findings that cannabinoid receptor agonists mimic and occlude the Orx-B-induced inhibition of eEPSCs, along with the observation that blockade of CB1 receptors abolishes the effect of Orx-B, support the idea that the Orx-B-induced inhibition of glutamate release is mediated by retrograde endocannabinoid acting at presynaptic CB1 receptors. This conclusion is consistent with the report that CB1 receptor mRNAs are expressed in the DRN (Matsuda et al., 1993) and with their inhibitory effect on glutamate release in other brain areas (Shen et al., 1996; Auclair et al., 2000; Azad et al., 2003). It is interesting to note that other postsynaptic G-protein-coupled receptors have also been reported to inhibit glutamate release through retrograde endocannabinoid signal. In the cerebellum, activation of postsynaptic group I metabotropic glutamate receptors (mGluR1) has been shown to inhibit glutamate release at climbing fibers and Purkinje cell synapses through retrograde endocannabinoid release (Maejima et al., 2001; Brown et al., 2003). Similarly, an enhancement of endocannabinoid production has been reported in response to the stimulation of postsynaptic M1 and M3 muscarinic receptors located on CA1 pyramidal neurons of the hippocampus (Kim et al., 2002; Ohno-Shosaku et al., 2003; Fukudome et al., 2004). Interestingly, one cardinal feature of all of these receptors is their ability to activate Gq/G11 type G-proteins. As such, it is possible that retrograde endocannabinoid signaling may represent a widespread mechanism that enables postsynaptic Gq/G11-coupled receptors to control glutamate release at excitatory synapses throughout the brain.
Signaling pathway of Orx-B-induced endocannabinoid synthesis
We have shown that the inhibition of eEPSCs induced by the stimulation of OxRs is not prevented by loading postsynaptic neurons with a high concentration of the calcium chelator BAPTA (25 mm), which is fully effective in blocking calcium-dependent responses in these neurons. Hence, although stimulation of OxRs has been shown to increase intracellular calcium in DRN 5-HT neurons (Kohlmeier et al., 2004), our data indicate that the OxRs-induced endocannabinoid synthesis involves a calcium-independent biochemical pathway. A similar calcium-independent mechanism has been shown to mediate group I mGluR-induced endocannabinoids synthesis (Maejima et al., 2001; Chevaleyre and Castillo, 2003; Galante and Diana, 2004). Thus, our results are consistent with the notion that endocannabinoids can be synthesized and released independently of any increase in postsynaptic intracellular calcium.
The findings that PLC or DAG lipase inhibitors abolish the inhibition of eEPSC induced by Orx-B indicate that OxR-induced endocannabinoid synthesis requires the activation PLC and DAG lipase enzymatic cascades. The involvement of PLC in Orx-B-induced depression of eEPSC amplitude is in agreement with previous studies showing that OxRs activation stimulates PLC (Uramura et al., 2001; Zhu et al., 2003). Furthermore, because the Orx-B-induced inhibition of eEPSCs is blocked by inhibiting DAG lipase, which converts DAG into 2-AG, we propose that 2-AG is most likely the endocannabinoid that mediates the depression of glutamate release. It is worth noting that activation of PLC/DAG lipase pathways is also involved in group I mGluR-induced endocannabinoid synthesis and mediates retrograde inhibition of GABA release in both the hippocampus (Chevaleyre and Castillo, 2003) and the cerebellum (Galante and Diana, 2004). Thus, the present study supports and extends the idea that the PLC/DAG-driven endocannabinoids synthesis contributes to the retrograde synaptic communication in the CNS.
Possible functional implications
Previous studies have reported that activation of OxRs depolarizes and increases the activity of DRN 5-HT neurons (Brown et al., 2001; Liu et al., 2002b). In addition to these excitatory effects, we report for the first time that stimulation of postsynaptic OxRs also inhibits glutamatergic transmission to DRN 5-HT neurons via retrograde endocannabinoid messengers. One possible physiological role of retrograde synaptic modulation is to enable DRN 5-HT neurons to exert negative feedback control on their incoming glutamatergic inputs. As such, it could play a role in preventing excessive excitation of DRN 5-HT and maintaining the activity of these neurons within a desirable range, especially during higher levels of orexin release [e.g., during wakefulness (Kiyashchenko et al., 2002)]. In essence, the bidirectional effects exerted by Orxs on the excitability of DRN 5-HT may provide a homeostatic control that contributes to the stable firing activity of these neurons observed during wakefulness (McGinty and Harper, 1976; Trulson and Jacobs, 1979).
At the network level, DRN 5-HT neurons project to the wake-promoting orexin-containing neurons of the lateral hypothalamus (Hokfelt et al., 1987), which are inhibited by 5-HT (Yamanaka et al., 2003). Given that appropriate activation of both DRN 5-HT neurons and orexin-containing neurons is required for promoting and maintaining wakefulness (for review, see Saper et al., 2001), the bidirectional effects exerted by Orxs on the excitability of DRN 5-HT may play a role in maintaining a stable activation of these arousal-related neurons required for normal wakefulness. The loss of orexin signal (e.g., orexin deficiency) could lead to a disorganized activity in this and other arousal systems that underlie the fragmented wakefulness observed in narcolepsy (Mochizuki et al., 2004).
The finding that Orxs inhibit glutamatergic input to DRN 5-HT neurons via endocannabinoids also points to the potential interaction between Orxs and the endocannabinoid system in the modulation of the sleep-wake cycle. It is noteworthy that previous studies have shown that anandamide promotes sleep (Mechoulam et al., 1997) via the activation of CB1 receptors (Murillo-Rodriguez et al., 2001). However, future studies will be necessary to examine the role of the crosstalk between orexin and endocannabinoid systems in the modulation of the sleep-wake cycle.
Footnotes
- Received August 9, 2004.
- Revision received December 8, 2004.
- Accepted December 8, 2004.
This work was supported by National Institutes of Health Grant MH 62394 and by the Research Foundation of the State University of New York. R.-Y.S. was supported by National Institute on Alcohol Abuse and Alcoholism Grant AA 12435. We thank Drs. J. C. Beique and G. Connors for their helpful comments and suggestions.
Correspondence should be addressed to Dr. Samir Haj-Dahmane, Research Institute on Addictions, University at Buffalo, 1021 Main Street, Buffalo, NY 14203. E-mail: dahmane{at}ria.buffalo.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/250896-10$15.00/0