
Abstract
Many rhythmic behaviors are continuously modulated by endogenous peptides and amines, but whether neuromodulation is critical to the expression of a rhythmic behavior often remains unknown, particularly in mammals. Here, we address this issue in the respiratory network that was isolated in spontaneously rhythmic medullary slice preparations from mice. Under control conditions, the respiratory network generates fictive eupneic activity. We hypothesized previously that this activity depends on two types of pacemaker neurons. The bursting properties of one pacemaker rely on the persistent sodium current (INa(p)) and are insensitive to blockade of calcium channels with cadmium (CI-pacemakers), whereas bursting mechanisms of a second pacemaker are sensitive to cadmium (CS-pacemakers) and the calcium-dependent nonspecific cation current blocker flufenamic acid. During hypoxia, fictive eupneic activity is supplanted by the neural correlate of gasping, which is proposed to depend only on CI-pacemakers. Because CI-pacemakers require endogenous activation of 5-HT2A receptors, we tested the hypothesis that 5-HT2A receptor activation is critical for gasping. Here, we demonstrate that fictive gasping and CI-pacemaker bursting were selectively eliminated by the 5-HT2A receptor antagonist piperidine or ketanserin. Neither 5-HT2A antagonist eliminated bursting by CS-pacemakers and ventral respiratory group (VRG) population activity. However, this VRG activity was very different from eupneic activity. In the presence of 5-HT2A antagonists, VRG activity was eliminated by flufenamic acid and could not be reliably restored by adding substance P. These data support the hypothesis that two types of pacemaker bursting mechanisms underlie fictive eupnea, whereas only one burst mechanism is critical for gasping.
- substance P
- pre-Bötzinger complex
- hypoglossal
- rhythm generation
- serotonin
- 5-HT2A receptor
Introduction
Neuromodulators, such as peptides, amines, and pyridines, play important roles in many rhythmic behaviors including locomotion, whisker movements, and respiration (Hounsgaard and Kiehn, 1989; Gray et al., 2001; Frank et al., 2002; Viemari and Hilaire, 2002; Hattox et al., 2003; Crowell, 2004; McLean and Sillar, 2004). Here, we investigate the extent to which serotonin is essential for respiratory rhythm generation using an in vitro brain slice preparation containing a critical portion of the respiratory neural network, the pre-Bötzinger complex (PBC).
In vitro, the isolated PBC can generate the neural correlate of three respiratory rhythms observed in vivo: (1) eupneic activity (“fictive eupnea” in vitro); (2) augmented inspiratory efforts, called sighs (“fictive sigh” in vitro); and (3) under lowered oxygen conditions, eupnea and sigh rhythms are supplanted by another rhythm termed gasping (“fictive gasping” in vitro) (Lieske et al., 2000). Hypoxic gasping can be distinguished from eupnea because gasping has a shorter duration and more rapid inspiratory effort onset. In vivo, gasping is a major mechanism for autoresuscitation, failure of which may underlie sudden infant death syndrome (SIDS) (Sridhar et al., 2003).
In vitro studies suggest that two types of pacemakers contribute to respiratory rhythm generation during eupnea; these pacemakers can be distinguished by sensitivity of their bursting mechanism to cadmium, a general calcium channel blocker (Elsen and Ramirez, 1998; Thoby-Brisson and Ramirez, 2001). Cadmium-insensitive (CI) pacemakers are characterized by bursting that persists in the presence of cadmium and is blocked by the persistent sodium current blocker riluzole. Cadmium-sensitive (CS) pacemakers are characterized by bursting that is blocked by cadmium as well as the calcium-activated cationic (CAN)-current antagonist flufenamic acid (FFA) (Peña et al., 2004; Del Negro et al., 2005). Fictive eupneic activity in the in vitro slice preparation is blocked by combined application of riluzole and FFA (Peña et al., 2004), but neither by riluzole alone (Del Negro et al., 2002a; Peña et al., 2004) nor by FFA alone (Peña et al., 2004), suggesting that both types of pacemakers are necessary for eupneic rhythmogenesis (Peña et al., 2004).
In contrast to fictive eupneic activity, fictive gasping (Lieske et al., 2000) is eliminated by riluzole alone (Peña et al., 2004). During hypoxic conditions, synaptically isolated CS-pacemaker neurons cease endogenous bursting, whereas CI-pacemaker neurons, and the respiratory network, remain rhythmic (Thoby-Brisson and Ramirez, 2000; Peña et al., 2004). Thus, we hypothesized that gasping rhythmogenesis depends on CI-pacemakers (Peña et al., 2004), which we have previously shown require endogenous serotoninergic 5-HT2A receptor activation for proper bursting (Peña and Ramirez, 2002).
Here, we test the hypothesis that 5-HT2A receptor activation is critical for gasping rhythmogenesis. By blocking endogenous 5-HT2A receptor activation, we specifically eliminated CI-pacemaker activity and fictive gasping, suggesting that endogenous 5-HT2A receptor activation and CI-pacemaker activity is essential for fictive gasping rhythm generation in vitro.
Materials and Methods
All experiments conformed to the guiding principles for the care and use of animals approved by the National Institutes of Health and the Animal Care and Use Committees at the Medical College of Wisconsin, Cinvestav, and The University of Chicago.
Medullary brain slice preparation.
All experiments used the transverse, rhythmic medullary brain slice preparation taken from mice (Funk et al., 1994; Ramirez et al., 1996). Mice (5- to 10-d-old CD-1 outbred mice; Charles River Laboratories, Wilmington, MA) were deeply anesthetized with ether (delivered by inhalation) and quickly decapitated at the C3/C4 spinal level (Ramirez et al., 1996). The brainstem was dissected in ice-cold artificial CSF (ACSF) that was equilibrated with carbogen (95% O2 and 5% CO2, pH 7.4). Rhythmic 650-μm-thick slices containing the PBC (Ramirez et al., 1996) were obtained by slicing the medulla using a microslicer (VT1000S; Leica, Nussloch, Germany). Slices were submerged in a recording chamber (6 ml) under circulating ACSF (30°C; flow rate, 17 ml/min; total volume, 200 ml) containing the following (in mm): 118 NaCl, 3 KCl, 1.5 CaCl2, 1 MgCl2*6H2O, 25 NaHCO3, 1 NaH2PO4, and 30 d-glucose, equilibrated with carbogen (95% O2 and 5% CO2, pH 7.4). KCl was elevated from 3 mm to 8 mm over a span of 30 min before commencing recordings, to facilitate recording rhythmic population activity (Tryba et al., 2003). Note that raising [K+]o does not artificially introduce pacemaker bursting properties; pacemakers show similar bursting properties in 3 mm versus 8 mm [K+]o ACSF (Tryba et al., 2003). Furthermore, it should be noted that [K+]o rises during hypoxia in vivo (Acker and Richter, 1985), and both eupneic and gasping activities can be recorded in hypokalemic and hyperkalemic conditions in situ (St-John et al., 2005). All ACSF chemicals were obtained from Sigma (St. Louis, MO). Bath temperature was monitored and maintained at 30 ± 0.7°C using a Warner Instruments (Hamden, CT) TC-344B temperature regulator with an in-line solution heater (SH-27B); bath temperature at various locations within the bath was uniform. Fictive gasping was evoked by an anoxic stimulus whereby preparation bath-ACSF saturated with 95% O2/5% CO2 during control conditions was made hypoxic/anoxic by saturating it with 95% N2/5% CO2; we refer to this hypoxic/anoxic stimulus as “application of nonoxygenated ACSF.”
Electrophysiological recordings.
Extracellular recordings were obtained with glass suction electrodes positioned on the slice surface in the ventral respiratory group (VRG) near or on the PBC (see Fig. 1A,B) (Telgkamp and Ramirez, 1999). In some experiments, we also used glass electrodes positioned on the slice surface on the hypoglossal nucleus (XII) to record hypoglossal motor activity (Tryba and Ramirez, 2003). The VRG population bursting is dominated by inspiratory neurons such that integrated VRG (∫VRG) and PBC activity is in-phase with integrated XII (∫XII) activity (Tryba and Ramirez, 2003). Thus, VRG population bursts serve as a marker of fictive inspiration (Telgkamp and Ramirez, 1999). To block endogenous 5-HT2A receptor activation, we bath applied either one of two antagonists: (1) 20 μm 4-(4-fluorobenzoyl)-1(4-phenylbutyl)-piperidine (piperidine) (Tocris, Ellisville, MO) or (2) 40 μm ketanserin tautrate (ketanserin) (Tocris).

Mouse medullary slice preparations containing the neural network for respiration generate fictive gasping activity during application of nonoxygenated ACSF. A, Extracellular recording electrodes were placed on the surface of the brain slice preparation to record population activity from the VRG. B, Raw VRG population activity was integrated (∫); the integrated VRG activity is dominated by inspiratory neurons, giving rise to fictive inspiratory bursts in the integrated traces (∫VRG). C, During application of nonoxygenated ACSF, fictive eupneic ∫VRG bursts initially increase in frequency (Telgkamp and Ramirez, 1999); this respiratory activity is supplanted by fictive gasping during the last few minutes when nonoxygenated ACSF is applied for 10 min. Fictive gasping evoked during the first application of nonoxygenated ACSF is similar to a second application of nonoxygenated ACSF initiated 30 min later (n = 7). The onset of application of nonoxygenated ACSF begins at the origin. Fictive eupneic ∫VRG bursting frequency, 2 min before the second application of nonoxygenated ACSF, was similar to the first (between x = −2 min and x = 0). The VRG burst frequency at the onset of the second 10 min application of nonoxygenated ACSF (filled bars; between x = 0.5 and x = 2.5 min) was elevated relative to the first application of nonoxygenated ACSF (open bars). The asterisks above the bars indicate significant differences between VRG burst frequency between the first and second 10 min application of nonoxygenated ACSF. D, E, As is characteristic of gasping activity, VRG bursts from the slice preparation during the first (D) and second (E) 10 min application of nonoxygenated ACSF had a reduction in burst rise time and burst duration during the last 2 min of applying nonoxygenated ACSF (n = 7). The graphs in D and E plot the percentage of change in burst parameters relative to control bursts (100% is control rise time and duration, respectively).
Intracellular patch-clamp recordings were obtained with a MultiClamp 700B amplifier (Molecular Devices, Union City, CA), applying the blind-patch technique to VRG neurons in 650 μm brainstem slice preparations (Thoby-Brisson and Ramirez, 2000). Patch electrodes were manufactured from filamented borosilicate glass tubes (Clark G150F-4; Warner Instruments) and filled with an intracellular solution containing the following (in mm): 140 K-gluconic acid, 1 CaCl2*6H2O, 10 EGTA, 2 MgCl2*6H2O, 4 Na2ATP, and 10 HEPES.
Only inspiratory pacemaker neurons active in-phase with the ∫VRG population burst were recorded in this study. The discharge pattern of each cell type was first identified in the cell-attached mode and remained similar in the whole-cell configuration (Peña et al., 2004). Experiments were then performed in the whole-cell patch-clamp mode. The Vm values were corrected for liquid junction potentials as described by Neher (1992). In current clamp, neurons were isolated from chemical synaptic input using a mixture of glutamatergic, GABAergic, and glycinergic antagonists. These drugs were bath applied at the final concentrations of 20 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX; Tocris), 10 μm (RS)-3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP; Tocris), 1 μm strychnine (Sigma), and 20 μm bicuculline free-base (Sigma). Note that unlike bicuculline methiodide, the bicuculline free-base derivative is a specific GABA receptor antagonist that does not block apamin-sensitive, Ca2+-activated K+ currents (Johnson and Seutin, 1997; Debarbieux et al., 1998). All of the pacemakers met several criteria before being classified as inspiratory pacemaker neurons; the criteria used here were provided in detail previously (Tryba et al., 2003). Briefly, after isolation of the neuron from chemical synaptic input with bath-applied CNQX, CPP, strychnine, and bicuculline, pacemakers continued to burst in the absence of VRG population bursts. Second, isolated pacemakers exhibited voltage-dependent bursting properties. That is, brief depolarizing current injection could evoke a burst or hyperpolarizing current could terminate an ongoing burst; either of these reset the ongoing pacemaker bursting rhythm. Finally, depolarizing current injection increased, and injected hyperpolarizing current decreased, the bursting frequency. After synaptic isolation with CNQX, CPP, bicuculline, and strychnine, in some cases, cadmium (Cd2+), a calcium channel blocker, was used as a tool to discriminate between pacemaker neurons, the endogenous bursting mechanism of which is dependent on calcium versus sodium. Synaptically isolated pacemakers that continue to burst in the presence of voltage-gated calcium channel blockade with 200 μm Cd2+ are Cd2+-insensitive (CI) pacemakers, as described previously by Thoby-Brisson and Ramirez (2001), and typically riluzole sensitive (Peña et al., 2004). Isolated pacemakers that cease bursting in Cd2+ are Cd2+-sensitive (CS) pacemakers (Thoby-Brisson and Ramirez (2001) and FFA sensitive (Peña et al. 2004). Note that 200 μm Cd2+ ensures blockade of voltage-activated calcium currents in VRG neurons (Elsen and Ramirez, 1998, 2005). To test whether a synaptically isolated pacemaker was FFA sensitive, we bath applied 500 μm FFA; this concentration was used to ensure rapid and complete blockade of CS-pacemaker activity, as shown previously (Peña et al., 2004). We also coapplied 500 μm FFA and 40 μm ketanserin to block bursting in both CS- and CI-pacemakers to test whether the network population rhythm is dependent on these pacemakers. At this concentration, FFA may enhance K+ currents (Stumpff et al., 2001) and block gap junctions (Srinivas and Spray, 2003; Ye et al., 2003), which could have side effects on respiratory network activity. However, it should be noted that lanthanum, another ICAN antagonist, but with different pharmacological side effects than FFA, also abolishes respiratory network bursting that persists in the presence of INa(p) antagonists (Peña et al., 2004), suggesting FFA blockade of ICAN, and not its potential side effects, abolishes respiratory network rhythmic activity in the presence of INa(p) antagonists.
Data analysis.
All electrophysiological recordings were transferred to a personal computer using a Digidata 1332A (Molecular Devices) analog to digital conversion board. Data were stored using Axotape (version 2.0) and analyzed off-line using AxoScope (version 9.0; Molecular Devices) and Igor Pro (version 4.0; WaveMetrics, Lake Oswego, OR). Cursor values were used to make membrane potential (Vm) measurements. A cursor window was used to measure burst amplitude, burst duration, and frequency measurements under various experimental conditions that were automated with programs written in Igor Pro 4.0. Unless noted otherwise, measured parameter values are reported in the text as the mean ± SD. Statistical significance is considered at the 95% confidence level, and ANOVA, Student’s paired t tests, or the Mann–Whitney rank sum test were used for analysis as appropriate. Measurements of mean VRG population burst rise time and burst duration during anoxia were made for bursts in each preparation studied during the last 2 min of a 10 min anoxic stimulus episode. Having used the same time course for our measurements, we compared the VRG burst duration and rise-time measurements before and after treatment (e.g., application of nonoxygenated ACSF) using Student’s paired t tests. Measurements of VRG population burst rise time were made between 20 and 80% of the burst onset (Lieske et al., 2000). Burst duration was measured as the width at half-maximum amplitude. Both rise-time and duration measurements were performed in this manner to avoid potential problems arising from baseline fluctuations or peak burst shape. To analyze regularity of the VRG population or pacemaker bursting, we calculated an irregularity score (S), by applying a formula for consecutive cycle-length values: Sn = 100 × ABS(Pn − (Pn-1))/Pn-1, where Sn is the score of the nth cycle, Pn is its period, Pn-1 is the period of the preceding burst, and ABS is the absolute value (Peña et al., 2004). A high irregular score represents an irregular rhythm. Two-way ANOVA was used to detect changes in VRG bursting frequency, using data obtained after application of identical 10 min nonoxygenated ACSF stimuli. The Mann–Whitney rank sum test was used to test whether rhythm recovery duration (see Fig. 2) after application of nonoxygenated ACSF was prolonged in the presence of 5-HT2A antagonists; this test was used because it tests whether the medians of two different samples are significantly different, without assuming normally distributed samples. In this case, the two different samples were (1) preparations that received two identical applications of nonoxygenated ACSF, separated by 30 min (n = 6); and (2) preparations that received two identical applications of nonoxygenated ACSF, separated by 30 min, but additionally had piperidine bath applied 20 min before the second 10 min application of nonoxygenated ACSF (n = 6).

Blocking 5-HT2A receptor activity prevents fictive gasping. A, After blockade of 5-HT2A receptors with piperidine, expected fictive gasping is prevented during a second 10 min application of nonoxygenated ACSF. B, Histogram showing fictive respiratory activity was continuous during the first 10 min application of nonoxygenated ACSF (control; open bars). After bath application of 20 μm piperidine, unlike Figure 1C in which piperidine was not applied, fictive gasping was not evoked during the late stages of a second 10 min application of nonoxygenated ACSF in the presence of piperidine (filled bars; n = 6). The asterisks above the bars indicate significant differences between control burst frequency and following piperidine application. The onset of a 10 min application of nonoxygenated ACSF begins at the origin. The ∫VRG bursting frequency, 2 min before the second application of nonoxygenated ACSF, was similar to the first (between x = −2 min and x = 0 min). C, After a 10 min application of nonoxygenated ACSF (filled bar, ending at // marks) and restoration of control oxygenation levels (Reoxygenation; open bar), the respiratory rhythm (fictive gasping) is depressed. Fictive eupneic activity then recovers, and its frequency is typically elevated for some time after reoxygenation (Blitz and Ramirez, 2002). We measured the duration it took for the respiratory rhythm to again generate VRG bursts after a posthypoxic depression initiated by restoration of control oxygenation levels (←Recovery Duration→). D, Plot of mean normalized recovery duration (±SD) after a second 10 min application of nonoxygenated ACSF normalized to recovery after the first 10 min application of nonoxygenated ACSF (solid horizontal line; n = 6 control; n = 6 in piperidine).
Results
Fictive gasping during anoxic conditions
To examine the possible effect of serotonergic neuromodulation on the respiratory network hypoxic response, it was first necessary to obtain baseline data and characterize the responses of the respiratory network during hypoxic conditions. Fictive gasping activity was evoked by an anoxic stimulus whereby bath-ACSF saturated with 95% O2/5% CO2 during control conditions was made hypoxic/anoxic by saturating it with 95% N2/5% CO2. Applying this nonoxygenated ACSF to the slice preparation was shown previously to induce fictive gasping in vitro (Lieske et al., 2000). In the present study, gasping activity in vitro is defined, as in vivo, as having a rhythmic respiratory activity during hypoxia that has a shorter inspiratory duration and more rapid inspiratory rise time than fictive eupneic activity (Lieske et al., 2000) (for review, see Ramirez and Lieske, 2003).
VRG population bursting was recorded throughout a 10 min application of nonoxygenated ACSF (control) (Fig. 1A–C). VRG population bursting measured during the last 2 min of a 10 min application of nonoxygenated ACSF had a significant decrease in burst rise time and duration (Fig. 1C,D) (p ≤ 0.05; Student’s paired t test; n = 7), confirming studies by Lieske et al. (2000). Distinct from fictive eupneic-like bursts, the gasp-like reduction in fictive inspiratory burst rise time and duration during application of nonoxygenated ACSF is shown in Figure 1C (inset). Thus, we refer to this activity pattern during hypoxic/anoxic conditions as fictive gasping (Lieske et al., 2000). Note also in Figure 1C (inset) the change in inspiratory burst shape from augmenting, bell-shaped (eupneic like) to decrementing (gasp like) that bears resemblance to the switch from augmenting inspiratory activity during eupnea to decrementing inspiratory activity during hypoxic gasping, which is seen in vivo (for review, see Ramirez and Lieske, 2003).
In one set of experiments, after a 10 min application of nonoxygenated ACSF, VRG population bursting frequency was allowed to recover to baseline in ACSF saturated with 95% O2/5% CO2 for 30 min (Fig. 1C). This recovery period was chosen because Blitz and Ramirez (2002) showed that application of nonoxygenated ACSF, as done here, can lead to a postanoxic increase in VRG bursting frequency. This frequency increase returns to baseline after 30 min “recovery” in ACSF saturated with 95% O2/5% CO2. After this recovery period, we applied nonoxygenated ACSF a second time, again for 10 min. Note that during hypoxia, the respiratory network typically responds with an initial increase in burst frequency, followed by a decline (Telgkamp and Ramirez, 1999). During a second application of nonoxygenated ACSF, the initial frequency augmentation of the hypoxic/anoxic response was larger than during the first application of nonoxygenated ACSF (Fig. 1C) (p ≤ 0.05; two-way ANOVA). However, throughout the remainder of the second episode of nonoxygenated ACSF, VRG population bursting activity continued at a similar frequency as during the first anoxia (Fig. 1C) (p ≥ 0.05; ANOVA) and also had a decrease in burst rise time and duration (p ≤ 0.05; Student’s paired t test) that was qualitatively similar to that observed during the first nonoxygenated ACSF application (Fig. 1D,E) (n = 7).
As described by Blitz and Ramirez (2002), after each 10 min application of nonoxygenated ACSF, during reoxygenation of bath ACSF with 95% O2/5% CO2, there was an initial cessation of respiratory bursting activity that was followed by a recovery of VRG population bursting. This recovery of VRG population bursting after reoxygenation after the second 10 min application of nonoxygenated ACSF was slightly longer than VRG population bursting recovery that followed the first 10 min application of nonoxygenated ACSF (difference in means, 0.7 ± 0.5 min; p = 0.032; Student’s paired t test; n = 7).
Blockade of 5-HT2A receptors with piperidine eliminates fictive gasping activity
Having established above in seven preparations that two 10 min applications of nonoxygenated ACSF, separated by a 30 min recovery in oxygenated ACSF, evokes fictive gasping in a similar manner, we next examined the effect of the 5-HT2A antagonist piperidine on fictive gasping activity in six preparations that were not previously exposed to nonoxygenated ACSF. Naive slice preparations were used to avoid potential hysteresis from repetitive exposure to hypoxic/anoxic conditions (Millhorn et al., 1980; Kinkead and Mitchell, 1999; Fuller et al., 2001; Mitchell et al., 2001; Blitz and Ramirez, 2002; Bocchiaro and Feldman, 2004). In six naive preparations, we applied nonoxygenated ACSF for 10 min to evoke fictive gasping in all examined preparations. We then bath applied the 5-HT2A receptor antagonist, 20 μm piperidine, during the last 20 min of a 30 min recovery period (during which the ACSF was reoxygenated with 95% O2/5% CO2). By the end of the 30 min recovery, VRG bursting frequency was similar to controls (Fig. 2A,B). In all six preparations, after blockade of 5-HT2A receptors, fictive gasping activity was absent during the second application of nonoxygenated ACSF (Fig. 2A,B) (n = 6). After restoration of ACSF gas saturation with 95% O2/5% CO2, VRG population bursting returned, despite continuous application of piperidine (Fig. 2C). However, the recovery duration after blockade of 5-HT2A receptors was 4.3 ± 1.0 min longer than the recovery after the first control application of nonoxygenated ACSF (p = 0.003; paired t test; n = 6) (Fig. 2D). This recovery duration was also longer (p = 0.008; Mann–Whitney rank sum test) than expected after two identical applications of nonoxygenated ACSF, separated by the same amount of time, but without bath application of piperidine, which had an average difference in recovery duration of 0.7 ± 0.5 min (see above).
In another set of experiments, 20 μm piperidine was bath applied 20 min before application of nonoxygenated ACSF for 10 min (to slices that had not been exposed previously to nonoxygenated ACSF). In all of these cases, application of nonoxygenated ACSF for 10 min did not evoke continuous VRG population bursting, indicating piperidine eliminated expected fictive gasping activity (Fig. 3A,B) (n = 7).

Piperidine, a 5-HT2A antagonist, blocks fictive gasping. A, Exposure to 20 μm piperidine prevented fictive gasping expected during a 10 min application of nonoxygenated ACSF. As shown in Figures 1 and 2, fictive gasping normally is evoked during a 10 min application of nonoxygenated ACSF to naive slice preparations. B, Fictive gasping was not observed during a 10 min application of nonoxygenated ACSF in naive slices pre-exposed to 20 μm piperidine for 20 min before the application of nonoxygenated ACSF (n = 7). The onset of a 10 min application of nonoxygenated ACSF begins at the origin. ∫VRG bursting activity continued in piperidine before application of nonoxygenated ACSF (shown between x = −2 and x = 0 min). C, The VRG population bursting that occurs after application of 20 μm piperidine is irregular, slower, and lower in amplitude than during control conditions (Peña and Ramirez, 2002). The VRG rhythmic bursting that persists in the presence of piperidine is blocked by the addition of FFA (n = 7).
Fictive eupneic activity can be eliminated by blocking 5-HT2A receptors with piperidine and FFA coapplication
As described previously, in ACSF saturated with 95% O2/5%CO2, VRG population bursting in vitro persists in the presence of 20 μm piperidine, albeit at a lower frequency and with less regularity than during controls (Peña and Ramirez, 2002). In all cases, the VRG population bursting that persisted in the presence of 20 μm piperidine was eliminated, within 7.7 ± 2.6 min, after the addition of 500 μm FFA, a blocker of the CAN current (n = 7) (Fig. 3C).
Ketanserin application confirms the effects of piperidine and blocks VRG and hypoglossal population bursting during application of nonoxygenated ACSF
The VRG synaptically drives hypoglossal (XII) motoneuron bursting during inspiration (Telgkamp and Ramirez, 1999; Tryba and Ramirez, 2003). We examined whether blockade of 5-HT2A receptors eliminates both VRG and XII population activity during application of nonoxygenated ACSF. Moreover, to confirm that the observed effects were attributable to a blockade of the 5-HT2A receptors and not attributable to a nonspecific effect caused by piperidine, we performed these experiments with another 5-HT2A antagonist, ketanserin. Population bursting from the VRG and XII was recorded simultaneously before and during a 10 min application of nonoxygenated ACSF. VRG and XII population bursting remained rhythmic throughout a 10 min application of nonoxygenated ACSF, generating fictive gasping during the last few minutes of nonoxygenated ACSF application (Fig. 4A) (n = 5). After bath application of 40 μm ketanserin for 20 min, neither VRG nor XII population bursting continued throughout a second 10 min application of nonoxygenated ACSF (Fig. 4B) (n = 5). Fictive gasping, during a 10 min application of nonoxygenated ACSF, was also abolished in three additional preparations in which we recorded only VRG population bursting activity (without XII), after a 20 min ketanserin application. Thus, consistent with the results obtained for piperidine, fictive gasping activity during a 10 min application of nonoxygenated ACSF was also blocked by ketanserin.

The 5-HT2A receptor antagonist ketanserin also eliminates fictive gasping activity recorded from the VRG and XII. A, Both ∫VRG and ∫XII population activity remained rhythmic during fictive gasping induced by application of nonoxygenated ACSF. B, Fictive gasping activity was blocked after bath application of the 5-HT2A antagonist ketanserin at 40 μm.
As was shown above for piperidine, bath application of ketanserin to the slice preparation alone for 20 min did not block VRG population bursting in ACSF saturated with 95% O2/5% CO2 (n = 8 of 8). But, in all examined preparations, additional application of FFA eliminated the VRG population rhythm within 4.03 ± 1.7 min (n = 8 of 8). In some of these preparations (n = 5 of 8), we had simultaneously recorded VRG and XII population bursting, both of which were eliminated by the combined application of ketanserin and FFA (n = 5 of 5).
Application of nonoxygenated ACSF and the 5-HT2A antagonist ketanserin differentially modulate respiratory pacemaker bursting activity
As already mentioned in the Introduction, the respiratory network in the PBC contains pacemaker neurons with two types of bursting properties that are differentially affected by hypoxic conditions (Thoby-Brisson and Ramirez, 2000; Peña et al., 2004). However, previous studies examined only the responses of synaptically isolated pacemakers to hypoxic conditions via application of nonoxygenated ACSF. Here, we show that application of nonoxygenated ACSF also has differential effects on respiratory pacemaker bursting when they are embedded in the VRG neuronal network (n = 10).
In the slice preparation containing the functional VRG respiratory network, 60% of the recorded pacemakers remained rhythmic throughout a 10 min application of nonoxygenated ACSF that induced fictive gasping activity (n = 6 of 10) (Fig. 5A,B). Each of these neurons was subsequently identified as a CI-pacemaker (Fig. 5C). Before adding Cd2+, we verified that each of these neurons was a pacemaker using the following experimental procedures: we first blocked chemical synaptic transmission with (in μm) 20 CNQX, 10 CPP, 20 bicuculline, and 1 strychnine (Fig. 6A,B) and identified pacemakers using criteria described in Materials and Methods and by Tryba et al. (2003); we then bath applied 200 μm Cd2+ to block voltage-dependent calcium currents (Fig. 6C). Each of the isolated CI-pacemakers continued to generate pacemaker activity throughout a second 10 min application of nonoxygenated ACSF (Fig. 6D,E) (n = 6). Subsequent bath application of 40 μm ketanserin eliminated CI-pacemaker bursting (Fig. 6F) (n = 6).

CI-pacemaker neurons remain rhythmic during fictive gasping. A, B, VRG population activity and inspiratory neuron activity during fictive eupnea under control conditions (A) and during fictive gasping during a 10 min application of nonoxygenated ACSF (B). The inset contains an overlay of fictive eupneic (red) and fictive gasp bursts to show that population gasping activity has both a faster rise time and shorter duration. C, The endogenous bursting properties of pacemaker neurons that remained rhythmic during a 10 min application of nonoxygenated ACSF were not blocked in the presence of 200 μm cadmium, a calcium channel antagonist.

Bursting properties of cadmium insensitive (CI) pacemakers remain rhythmic during a 10 min application of nonoxygenated ACSF but are blocked by ketanserin. A, B, Inspiratory pacemaker neurons embedded in the respiratory neural network retain endogenous bursting properties after blocking chemical synaptic transmission with 20 μm CNQX, 10 μm CPP, 20 μm bicuculline (Bic), and 1 μm strychnine (Str) (B). C, The bursting mechanism of some pacemakers is insensitive to 200 μm cadmium application [Cd2+-insenstive (CI) pacemakers]. D, E, CI-pacemakers remain endogenously rhythmic during a 10 min application of nonoxygenated ACSF and endogenously burst throughout application of a 10 min application of nonoxygenated ACSF (E; n = 6). The record in D was taken during the last minute of a 10 min application of nonoxygenated ACSF. One hundred percent in the histogram of E is frequency within the first minute of hypoxia. F, Endogenous bursting properties of CI-pacemakers are blocked by the 5-HT2A antagonist ketanserin (40 μm). G, Histogram of CI-pacemaker bursting after additional bath application of 40 μm ketanserin (initiated at the origin), which eliminated endogenous CI-pacemaker bursting within 16 min (n = 6). The black numbers inset into the histogram bars indicate the number of the total of the six synaptically isolated CI-pacemakers that retained their bursting properties in ketanserin at the time shown. As in F, loss of bursting properties in each case was confirmed by current injection. The percentage of control frequency (G) was calculated based on mean frequency 1 min before bath application of ketanserin.
We showed above that bath application of 40 μm ketanserin for 20 min was sufficient to block fictive gasping (Fig. 4B). To establish the time course within which we could expect ketanserin application to block CI-pacemaker bursting, we examined the bursting properties of CI-pacemakers after bath application of 40 μm ketanserin for 20 min and found it eliminated the CI-pacemaker bursting properties. Pacemaker bursting could not be restored even when injecting depolarizing current (Fig. 6F) within ∼16 min after ketanserin addition (Fig. 6G). Measurements taken from 20 consecutive pacemaker bursts per neuron, starting 1 min before the cessation of bursting properties, revealed 40 μm ketanserin application significantly increased pacemaker bursting irregularity (mean percentage of change in irregularity, 173.7 ± 40.7%; n = 6); a similar increase in irregularity was reported by Peña and Ramirez (2002).
Embedded in the slice VRG network, 40% of the examined pacemaker neurons did not remain rhythmic during fictive gasping, evoked by applying nonoxygenated ACSF for 10 min (Fig. 7A–C) (n = 4 of the total of 10 pacemakers examined). During application of nonoxygenated ACSF, these neurons hyperpolarized by −6.1 ± 2.1 mV (p = 0.01; paired t test) and ceased bursting within ∼7 min from the onset of the application of nonoxygenated ACSF (Fig. 7C) (n = 4). After recovery of their bursting in reoxygenated ACSF, we synaptically isolated these neurons and blocked VRG population bursting with (in μm) 20 CNQX, 10 CPP, 20 bicuculline, and 1 strychnine. After synaptic isolation, we subsequently bath applied 40 μm ketanserin for 20 min. This 20 min ketanserin application did not eliminate endogenous pacemaker bursting in any of the pacemakers that ceased bursting during hypoxia (Fig. 7D) (n = 4). Typical for CS-pacemakers, their pacemaker bursting properties were blocked within 2.3 ± 2.1 min after subsequent additional bath application of 500 μm FFA (n = 4) (Fig. 7E).
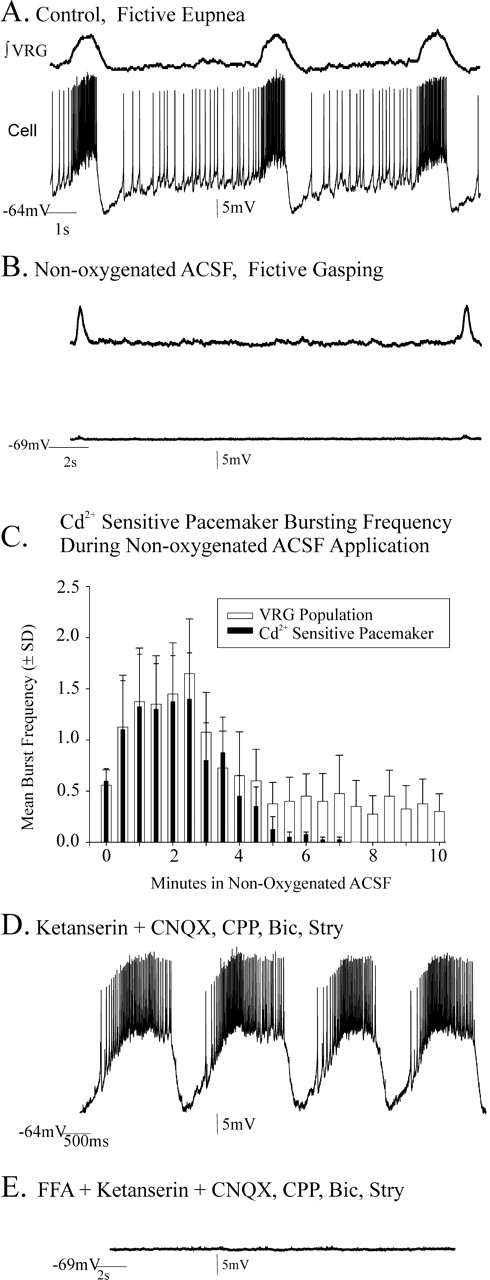
CS-pacemakers cease bursting during a 10 min application of nonoxygenated ACSF and remain rhythmic in 40 μm ketanserin. A, B, Inspiratory CS-pacemaker activity under control conditions (A) and during fictive gasping (B). The data shown are taken during the last minute of a 10 min application of nonoxygenated ACSF to the slice. C, Histogram of a CS-sensitive pacemaker and VRG bursting frequency 30 s before (x = 0 min) and during a 10 min application of nonoxygenated ACSF. Note that VRG bursting remains rhythmic throughout the 10 min application of nonoxygenated ACSF, whereas CS-pacemakers stop pacemaker bursting within 7 min of applying the nonoxygenated ACSF onset (n = 4). D, After bath application of the 5-HT2A antagonist ketanserin (40 μm), for 20 min, synaptically isolated CS-pacemakers remained rhythmic. Before bath application of ketanserin, pacemakers were isolated from glutamatergic and inhibitory synaptic transmission by bath application of 20 μm CNQX, 10 μm CPP, 20 μm bicuculline, and 1 μm strychnine. E, Subsequent addition of FFA (500 μm) eliminated CS-pacemaker bursting activity in synaptically isolated pacemakers, the pacemaker bursting of which stopped during fictive gasping (B). Bic, Bicuculline; Stry, strychnine.
Can substance P restore respiratory rhythmic activity?
Del Negro et al. (2005) suggested that substance P application (1–2 μm) could restore rhythmicity to the VRG network even after blocking both CI- and CS-pacemaker bursting. Here, we extended their interesting observation. Confirming the observation by Peña and Ramirez (2002), we show that 40 μm ketanserin, when bath applied for 20 min, eliminated Cd2+-insensitive pacemaker bursting that is dependent on INa(p) (Peña et al., 2005). If VRG population bursting depends on two types of pacemakers and, as we show, ketanserin eliminates CI-pacemakers, whereas FFA blocks CS-sensitive pacemakers, then coapplication of ketanserin and FFA should block both CI- and CS-pacemaker bursting and VRG rhythmicity.
After blocking VRG population bursting by combined application of 40 μm ketanserin (Fig. 8A) (for 20 min) and 500 μm FFA (Fig. 8B) (for 4 additional min), the subsequent addition of 1–2 μm substance P >20 min did not restore VRG bursting to seven of eight examined preparations (Fig. 8C,D). In one preparation (12.5%) in which bath application of both ketanserin and FFA eliminated VRG and XII population activity (Fig. 9A,B), the subsequent addition of 1 μm substance P temporarily restored population bursting for 3.01 min, and this rhythm never resumed during recordings made for >20 min even after increasing the substance P concentration to 2 μm (Fig. 9C,D).

Coapplication of ketanserin and FFA eliminated VRG population activity in most preparations; VRG bursting was not restored by excitation of the network by additionally applying substance P. A, Integrated VRG population bursting (∫VRG) remains rhythmic after bath application of 40 μm ketanserin. B, The ∫VRG population rhythm expressed in the presence of 40 μm ketanserin is blocked by additional application of 500 μm FFA. C, D, In 9 of 10 preparations, this ∫VRG population rhythm is not recovered by the additional application of the excitatory neuropeptide substance P at concentrations of 1 μm (C) and 2 μm (D).

After blocking VRG population bursting with coapplication of ketanserin and FFA, subsequent application of substance P temporarily restored rhythmic bursting in 1 of 10 preparations examined. A, Integrated VRG population bursting (∫VRG) and integrated hypoglossal (∫XII) motor nuclei population bursting remains rhythmic after bath application of 40 μm ketanserin. B, The ∫VRG population rhythm expressed in the presence of 40 μm ketanserin is blocked by additional application of 500 μm FFA. C, D, In 1 of 10 preparations, this ∫VRG population rhythm was temporarily recovered by the additional application of the excitatory neuropeptide substance P at the concentration of 1 μm (C); this rhythm ceased for several minutes and was not reinitiated when substance P was increased to a concentration of 2 μm (D). Downward arrows indicate the time when 1 μm substance P was added or when it was increased to 2 μm.
Can substance P restore pacemaker rhythmicity after blockade of bursting mechanisms?
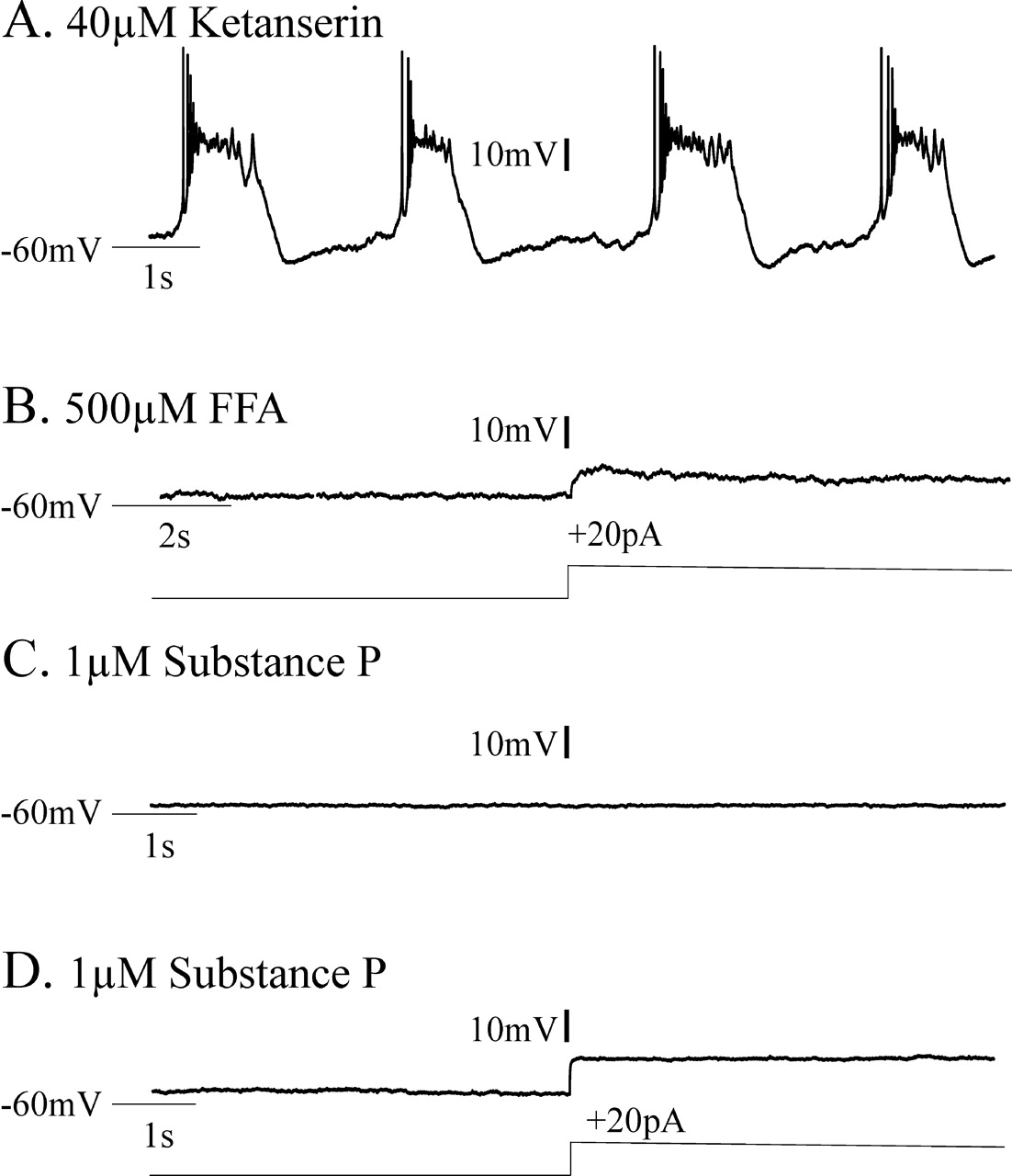
We considered whether bath application of 1–2 μm substance P might be able to enhance pacemaker bursting even in experimental conditions in which ketanserin and FFA coapplication should eliminate endogenous pacemaker bursting. This might be possible, because substance P can enhance the excitability of inspiratory neurons via a TTX-insensitive sodium current (Peña and Ramirez, 2004), whereas ketanserin blocks the TTX-sensitive INa(p) (Peña and Ramirez, 2002) and FFA is proposed to block ICAN. Thus, we additionally tested whether CI-pacemaker bursting, having been blocked by 40 μm ketanserin application for 20 min, could be restored after subsequent bath application of 1–2 μm substance P. Five of six synaptically isolated CI-pacemaker neurons, the bursting of which had been eliminated after 20 min of 40 μm ketanserin application, did not become rhythmic after additional bath application of 1–2 μm substance P for up to 30 min (Fig. 10A–D). However, one of six CI-pacemakers (16.7%), the bursting of which was eliminated by ketanserin application, temporarily became rhythmic after the subsequent addition of 1 μm substance P (Fig. 11A–C). This bursting activity ceased after 2.1 min and did not return within 30 min even after increasing the concentration of substance P to 2 μm (Fig. 11D). After their bursting properties were blocked by FFA, none of the ketanserin-insensitive CS-pacemakers (Fig. 12A,B) became rhythmic after additional bath application of 1–2 μm substance P (Fig. 12C,D) (n = 4 of 4).

CI-pacemaker bursting properties were eliminated by application of the 5-HT2A antagonist ketanserin. CI-pacemaker bursting was typically not restored by subsequent additional application of substance P. A, CI-insensitive pacemakers remained rhythmic in 200 μm Cd2+. Note that 20 μm CNQX, 10 μm CPP, 20 μm bicuculline, and 1 μm strychnine were also bath applied before adding 200 μm Cd2+; this was done to initially synaptically isolate the neuron and test for pacemaking properties. B, Adding 40 μm ketanserin blocked CI-pacemaker bursting, even during depolarizing current injections. C, D, In five of six CI-pacemakers, endogenous bursting was not recovered by the additional application of the excitatory neuropeptide substance P at concentrations of 1 μm (C) and 2 μm (D), but single action potentials were elicited during depolarizing current injections.

CI-pacemaker bursting properties were eliminated by application of the 5-HT2A antagonist ketanserin. In one of six CI-pacemakers, CI-pacemaker bursting was temporarily restored after subsequent additional application of substance P. A, CI-pacemakers remained rhythmic in 200 μm Cd2+. Before adding cadmium, 20 μm CNQX, 10 μm CPP, 20 μm bicuculline, and 1 μm strychnine were bath applied to block synaptic transmission and synaptically isolate the pacemaker. B, Adding 40 μm ketanserin blocked CI-pacemaker bursting, even during depolarizing current injections (+25 pA). C, D, In this particular CI-pacemaker, endogenous bursting was temporarily recovered by the additional application of the excitatory neuropeptide substance P at a bath concentration of 1 μm (C), but this rhythmic bursting ceased and was supplanted by tonic spiking despite increasing the substance P concentration to 2 μm (D).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cadmium-sensitive (CS) pacemaker bursting is not eliminated by application of 5-HT2A antagonists, is blocked by FFA, and is not restored by subsequent addition of substance P. A, CS-pacemakers remained rhythmic in 40 μm ketanserin. A, Before adding ketanserin, 20 μm CNQX, 10 μm CPP, 20 μm bicuculline, and 1 μm strychnine were bath applied to block synaptic transmission and synaptically isolate the pacemakers. B, Additional bath application of 500 μm FFA blocked CS-pacemaker endogenous bursting, even during depolarizing current injections (+20 pA). C, D, None of the CS-pacemakers examined recovered endogenous bursting properties after the additional application of the excitatory neuropeptide substance P at a bath concentration of 1 μm (C) or 2 μm (D), despite +DC current injections (20 pA).
Discussion
Our data indicate that endogenous 5-HT2A receptor activity is required for the generation of fictive gasping activity by the VRG and hypoglossal motoneurons (XII) within the transverse brainstem slice preparation of mice (Figs. 2⇑–4). Although not tested here, the brainstem slice preparation used in these studies contains the raphe nuclei, which may serve as an endogenous source of 5-HT within the slice preparation. In vivo studies indicate that serotonin levels increase during hypoxia. Endogenous 5-HT profoundly affects the isolated respiratory network in vitro, as it was shown previously that 5-HT2A antagonists significantly reduce the frequency, regularity, and amplitude of fictive eupneic bursting generated by the VRG (Peña and Ramirez, 2002). In these cases, fictive VRG population bursting is perturbed but not eliminated by blockade of 5-HT2A receptor activity. Here, we show that the role of an endogenous neuromodulator during respiratory activity is state dependent, because during application of nonoxygenated ACSF, endogenous activation of 5-HT2A receptors is required to generate fictive gasping. The recovery of VRG population bursting after application of nonoxygenated ACSF was also significantly delayed after blockade of 5-HT2A receptors (Fig. 2D). These data suggest that endogenous 5-HT2A receptor activity may also play an important role in restoring network rhythmicity and eupneic activity after hypoxic conditions.
Our data support the hypothesis that gasping is generated by CI-pacemakers because riluzole (Peña et al., 2004), piperidine, and ketanserin block these pacemakers (Peña and Ramirez, 2002; Peña et al., 2004) and block fictive gasping (Peña et al., 2004) (Figs. 2, 3, 6). Note also that the elimination of fictive gasping by ketanserin application is unlikely the result of blocking CS-pacemaker bursting (Fig. 7D), or blocking synaptic transmission, because both the VRG and XII remained rhythmic and continued to burst in-phase in the presence of ketanserin (Fig. 9A). Fictive gasping is also not likely to be driven by CS-pacemakers (Fig. 7C), the bursting of which was not blocked by ketanserin (Figs. 7D, 12A). In medullary slice preparations, bursting in synaptically isolated CS-pacemakers typically ceases within about 6 min after application of nonoxygenated ACSF (Peña et al., 2004). Here, we showed that CS-pacemaker bursting activity ceased ∼7 min after applying nonoxygenated ACSF to the VRG network contained in the slice (Fig. 7C). Thus, intrinsic bursting in CS-pacemakers would not be expected to contribute to fictive gasping evoked during the last 3 min of a 10 min application of nonoxygenated ACSF (Peña et al., 2004).
Whereas caution must be used when extrapolating in vitro findings to the situation in vivo, our data may have important implications for understanding the failure of autoresuscitation in SIDS victims. A leading hypothesis for explaining SIDS is the failure of appropriate gasping as a mechanism of autoresuscitation (Sridhar et al., 2003). Along these lines, gasping is disrupted in SIDS victims, as is the serotonergic system in the brainstem respiratory areas (Ozawa and Takashima, 2002; Kinney et al., 2003; Weese-Mayer et al., 2003a,b; Kinney, 2005). Thus, our data suggest an intriguing possibility that, in SIDS victims, disturbances in the serotonergic system may underlie the failure to gasp appropriately and autoresuscitate despite increased release of 5-HT during hypoxia (Erickson and Millhorn, 1994; Teppema et al., 1997; Richter et al., 1999; Kinkead et al., 2001). Note also that during rapid eye movement (REM) sleep, 5-HT neuron activity levels decrease (Fornal and Jacob, 1998), perhaps leaving at-risk victims [who spend more time in REM sleep between 2:00 and 5:00 A.M. (Cornwell et al., 1998)] more vulnerable to resuscitation failure. Although this hypothesis is consistent with our in vitro data, extensive in vivo experiments would be necessary to validate this hypothesis in the intact animal.
In contrast to fictive gasping, bath application of the INa(p) antagonist riluzole alone does not block eupneic activity in vitro (Del Negro et al., 2002; Peña et al., 2004). Fictive eupnea in vitro is blocked after combined application of riluzole and FFA (Peña et al., 2004). Here, we also found that bath application of either piperidine or ketanserin, which selectively blocks CI-pacemaker activity, was not sufficient to eliminate VRG bursting during control conditions (Figs. 3, 4, 6, 8⇑–10) (Peña and Ramirez, 2002). Consistent with the hypothesis that eupneic activity in vitro is dependent on at least two types of pacemakers, CI- and CS-pacemakers (Peña et al., 2004), VRG activity that remained after blockade of endogenous 5-HT2A receptor activity alone was eliminated after additional bath application of FFA (Figs. 8, 9). Along these lines, studies by Del Negro et al. (2005) confirmed the finding (Peña et al., 2004) that neurons with CI- and CS-pacemaker properties coexist in the VRG. However, it must be emphasized that although pacemaker bursting mechanisms can be blocked by either INa(p) or ICAN antagonists, both INa(p) and FFA-sensitive currents may be expressed within the same neuron (Peña et al., 2004; Del Negro et al., 2005).
Pacemaker activity and network properties have been proposed to underlie rhythm generation in the respiratory neural network. Under conditions in which we showed ketanserin blocks CI-pacemakers and FFA eliminates CS-pacemaker bursting, VRG and XII bursting are also eliminated (Figs. 6, 7, 9) (Peña and Ramirez, 2002; Peña et al., 2004), suggesting pacemakers underlie rhythmogenesis in vitro. In one of eight preparations, 1 μm substance P application temporarily restored VRG bursting that had been blocked by combined application of ketanserin and FFA (Fig. 9). These data suggest the possibility that the rhythm generated under these conditions is not dependent on endogenous pacemaker bursting, but rather the emergent property of network activity, as suggested by Del Negro et al. (2005). Indeed, substance P can increase excitability via augmenting TTX-insensitive INa and NMDA-dependent rhythmic bursting (Peña et al., 2004). An alternative, parsimonious explanation for the VRG population rhythmicity in the presence of ketanserin, FFA, and substance P is that, in that particular experiment, pacemaker bursting properties were not fully blocked, and substance P enhanced the bursting properties enough to underlie rhythmic bursting. This likely possibility is suggested by one of our experiments on synaptically isolated pacemakers. After blockade of bursting of either CI- or CS-pacemaker activity, 1 of 10 pacemakers became temporarily rhythmic in 1 μm substance P (Figs. 11, 12). Regardless of the mechanism, the temporary nature of the VRG rhythmic bursting generated in these experiments (Figs. 9, 11), after substance P application, suggests that stable respiratory rhythmogenesis is not observed under conditions in which pacemaker bursting mechanisms had been blocked.
In addition to modulating pacemaker bursting, altered serotonin levels may facilitate the change in respiratory patterns during and after hypoxia. For example, during hypoxic depression, serotonin levels increase, and 5-HT1A receptor activation may contribute to the cessation of eupneic activity (Richter et al., 1999). 5-HT2A modulation has also been proposed to play a critical role in long-term facilitation of phrenic activity after intermittent hypoxia (Millhorn et al., 1980; Kinkead and Mitchell, 1999; Fuller et al., 2001; Mitchell et al., 2001; Baker-Herman and Mitchell, 2002; Blitz and Ramirez, 2002; Baker-Herman et al., 2004).
Serotonin acting via 5-HT2A receptors can modulate the respiratory rhythm generating network in vitro (Peña and Ramirez, 2002) and respiratory motor neuron activity, including the recruitment (Lindsay and Feldman, 1993; Hilaire et al., 1997) and maturation (Bou-Flores et al., 2000) of phrenic motoneurons during development. Hypoglossal (XII) motoneuron activity can also be modulated by 5-HT receptors both in vivo and in vitro (Okabe and Kubin, 1996; Ladewig et al., 2004). In vitro, 5-HT2 receptor activation can depolarize XII motoneurons into a range in which they fire action potentials that in turn determine global [Ca2+]i changes (Ladewig et al., 2004). Moreover, mRNA for 5-HT2A receptors (as well as 5-HT1B, 5-HT2C, 5-HT3, and 5-HT7) has been detected in XII motoneurons (Okabe et al., 1997). Thus, both the central pattern generator and motor components of respiration are subject to 5-HT2A modulation. However, the specific effects of 5-HT2A activation on XII motor neuron activity remains to be investigated and may have important implications not only for SIDS but also sleep apnea, in which a reduction in 5-HT release during sleep (Fornal and Jacob, 1998) may decrease motor tone and patency of the upper airways, possibly by modulation of hypoglossal inspiratory activity (Kubin et al., 1996; Okabe and Kubin, 1996). Our findings in the isolated slice respiratory network, containing the PBC, have intriguing implications for the generation of these pathologies. However, neurons located more rostral to the PBC may also contribute to respiratory rhythm generation, and some have pacemaker properties (Ballanyi et al., 1999; Mellen et al., 2003; Onimaru and Homma, 2003). These and other respiratory-related neurons are not part of our slice preparation; thus, we do not know how they might contribute to the final pattern of respiratory activity when the PBC is part of a more extensive brainstem respiratory network. Despite this caveat, note that the medullary brainstem slice preparation used here contains sufficient respiratory network properties to capture and identify a neural correlate of three distinct respiratory activities, including fictive eupnea, fictive sighs, and fictive gasping (Lieske et al., 2000), which provides important mechanistic insights into their dependence on neuromodulators.
Footnotes
- Received October 1, 2005.
- Revision received January 19, 2006.
- Accepted January 23, 2006.
This work was supported by National Institutes of Health (NIH) Grant R01-HL 079294 and a Parker B. Francis Fellowship (A.K.T.), Conacyt-42870 (F.P.), and NIH Grants RO1-HL 60120 and RO1-HL 68860 (J.-M.R.).
- Correspondence should be addressed to Dr. Andrew K. Tryba, Department of Physiology, Medical College of Wisconsin, 8701 Watertown Plank Road, Milwaukee, WI 53226. Email: atryba{at}mcw.edu