
Abstract
Microglia exist under physiological conditions in a resting state but become activated after neuronal injury. Recent studies have highlighted the reciprocal role of neurons in controlling both the number and activity of microglia. In this study, microglia derived from newborn rat cortices were cultured and activated by interferon-γ (IFNγ) treatment, then exposed to recombinant Sema3A or conditioned medium derived from stressed embryonic cortical neurons. We found that activation of microglia by IFNγ induced differential upregulation of the semaphorin receptors Plexin-A1 and Neuropilin-1. This result was confirmed by Northern blotting, reverse transcription-PCR, and Western blotting. Furthermore, recombinant Sema3A induced apoptosis of microglia when added to the in vitro culture, and a similar result was obtained on activated microglia when Sema3A was produced by stressed neurons. Using an in vivo model of microglia activation by striatal injection of lipopolysaccharide demonstrated a corresponding upregulation of Plexin-A1 and Neuropilin-1 in activated microglia and enhanced production of Sema3A by stressed adult neurons. These results suggest a novel semaphorin-mediated mechanism of neuroprotection whereby stressed neurons can protect themselves from further damage by activated microglia.
- microglia
- neuron
- semaphorin
- plexin
- neuropilin
- neuroprotection
Introduction
Microglia are resident immune effector cells of the CNS. Under physiological conditions, they exist in resting conditions but become activated in response to neuronal injury. Activation alters both morphological and functional properties of microglia. It is a feature of many inflammatory and degenerative neurological diseases (Kreutzberg, 1996; Benveniste, 1997; McGeer and McGeer, 1999). The phagocytic, antigen-presenting, cytotoxic, and trophic effects of activated microglia are thought to facilitate repair (Elkabes et al., 1996; Polazzi et al., 2001; Harada et al., 2002). Activation has been associated with neuronal death in vitro. It may therefore also contribute to neuronal injury in the context of pathological processes (Chao et al., 1992; Giulian, 1999; Kingham et al., 1999; Liberatore et al., 1999; Streit et al., 1999; Golde et al., 2002). Furthermore, recent studies have highlighted the influence of neuron-derived signaling effects on microglial function, suggesting that controlled elimination of activated microglia may provide a method of limiting the inflammatory response (Neumann and Wekerle, 1998; Liu et al., 2001; Polazzi and Contestabile, 2003).
Semaphorins are a family of secreted and cell-bound signaling molecules defined by the presence of a common 500 aa Sema domain. They are best characterized in relation to axon guidance during development of the nervous system (Tessier-Lavigne and Goodman, 1996; Bagnard et al., 1998; Kolodkin, 1998). Neuropilin and plexin represent the two families of semaphorin receptors that interact to form a coreceptor complex for secreted semaphorins, including Sema3A. Although much remains to be learned about the biological activities and specificities of semaphorins, the functions of Sema3A are mediated primarily through binding to the Neuropilin-1 (Npn-1) and Plexin-A1 coreceptor complex (Takahashi et al., 1999; Tamagnone and Comoglio, 2000). Neuropilins lack a signaling-competent cytoplasmic domain and ensure semaphorin binding, whereas the transmembrane receptor plexin mediates the intracellular response (Kolodkin, 1998; Winberg et al., 1998; Tamagnone et al., 1999).
An emerging body of evidence suggests that semaphorins subserve diverse functions, many unrelated to axon guidance. These include immune modulation, organogenesis, angiogenesis, and cell migration (Behar et al., 1996; Comeau et al., 1998; Gagliardini and Fankhauser, 1999; Miao et al., 1999; Spriggs, 1999; Spassky et al., 2002; Cohen et al., 2003). Sema3A has also been implicated as a mediator of embryonic neuronal apoptosis. Taken with the recent identification of Sema3A in the adult and injured brain, this suggests a possible role for semaphorin in influencing neural repair (Shirvan et al., 1999; Fujita et al., 2001; Pasterkamp and Verhaagen, 2001). The observation that neurons secrete Sema3A and that Npn-1 and Npn-2 are expressed by activated microglia after middle cerebral artery occlusion suggests the potential for reciprocal interaction between neurons and microglia in association with tissue injury (Fujita et al., 2001).
In the present study, we characterized the differential expression profile of neuropilin and plexin in resting and activated microglia. Activated microglia upregulate Npn-1 and Plexin-A1 in vitro and in vivo. We provide evidence that stressed neurons express Sema3A and mediate microglial apoptosis. The death effect of stressed neuron conditioned medium (SNCM) is blocked by inhibition of Npn-1 binding. Together, these findings suggest a novel mechanism whereby neurons exploit the microglial expression of Npn-1 and Plexin-A1, protecting themselves from potentially threatening effects of activated microglia by secreting Sema3A.
Materials and Methods
Microglial cell culture.
Mixed glial cultures were prepared following the protocol of McCarthy and de Vellis (1980). Briefly, the forebrains of newborn rat pups were removed, and the meninges were stripped before mechanical and enzymatic dissociation. The resulting cell suspension was plated onto poly-l-lysine-coated 75 cm2 tissue culture flasks. Culture medium, DMEM supplemented with 10% fetal calf serum (FCS; Sigma, Poole, UK), was changed at 24 h and twice weekly thereafter. After 8–10 d, a confluent monolayer of astrocytes was apparent, on top of which oligodendrocyte precursors and a loosely attached layer of phase-bright microglia could be seen. Microglia were harvested by shaking for 20 min and were resuspended in 2% B27 (Invitrogen, Gaithersburg, MD)-supplemented DMEM before plating onto sterile 8-well chamber slides (∼40,000 cells per well; Labtek; Fisher Scientific, Loughborough, UK) or plastic 6-well plates (∼2 million cells per well; Nunclon, Naperville, IL). Contaminating cells were removed by allowing the cell suspension to adhere to the surface for 10 min at 37°C before washing in two changes of medium. Adherent microglia were incubated for 24 h to allow them to become “quiescent” before culturing for an additional 24 h in conditioning medium [insulin-free Sato-supplemented DMEM containing bovine serum albumin (100 μg/ml), transferrin (100 μg/ml), progesterone (0.06 μg/ml), putrescine (16 μg/ml), selenite (0.04 μg/ml), thyroxine (0.4 μg/ml), and triiodothryonine (0.04 μg/ml)] with or without interferon-γ (IFNγ; 100 U/ml) to produce highly enriched populations of activated or resting microglia.
Neuronal cell culture.
Neuronal cell cultures were prepared from the cortices of embryonic day 16 (E16) rat embryos (Wilkins et al., 2001). After enzymatic and mechanical dissociation, cells were plated onto freshly prepared poly-l-lysine-coated 13 mm glass coverslips (1.5 × 103 neurons per coverslip or 2.5 × 103 neurons per coverslip) or poly-l-lysine-coated plastic 6-well plates (2 million cells per well) in 2% B27-supplemented medium or insulin-free Sato-supplemented DMEM.
Preparation of SNCM.
Neuronal cultures were prepared in poly-l-lysine-coated 6-well plates (2 million cells per well) and cultured for 1 or 5 d in 2% B27-supplemented medium. Cells were then washed in DMEM and cultured in conditioning medium (insulin-free Sato) for up to 72 h. After centrifugation at 1000 rpm for 5 min and passage through a 45 μm filter, the resulting supernatant was stored at −20°C and diluted 1:1 with fresh medium for experiments, unless stated otherwise.
Sema3A-conditioned medium was generated from 293T cells transfected with chicken Sema3A using myc-his tag and culture in 1% FCS-supplemented DMEM for 24 h (Koppel et al., 1997). Sema3A activity of conditioned medium was validated using the standard collapse unit assay on E15 rat dorsal root ganglia explants. At a 1:250 dilution, Sema3A-conditioned medium induced 50% growth cone collapse after 30 min (Luo et al., 1993). For experimental procedures, Sema3A-conditioned medium was used at a 1:10 dilution with fresh medium (unless stated otherwise) for 24 h at 37°C. Control conditioned medium was derived from green fluorescent protein-transfected 293T cells.
For caspase inhibition experiments, 50 μm of the pan-caspase inhibitor Z-VAD-FMK (R & D Systems, Abingdon, UK) was added to the incubation medium for 24 h. Function-blocking anti-Npn-1 antibody (a gift from A. Kolodkin, Johns Hopkins, Baltimore, MD) was used at 100 μg/ml.
Immunocytochemistry.
Immunocytochemistry was used to identify cell phenotypes. Cells were stained “live” or after fixation in 4% paraformaldehyde. The primary antibodies against oligodendrocyte cell-surface markers were galactocerebroside (GalC; 1:4) and A2B5 (1:4) (both derived from hybridoma lines; European Collection of Cell Cultures, Salisbury, UK). Intracellular markers were stained after permeabilization of fixed cultures with 100% methanol for 10 min at −20°C: β4-isolectin (1:50), OX6 (1:200), and neuronal β3-tubulin (1:200) (all from Sigma); and astrocyte GFAP (1:200; Dako, High Wycombe, UK). Flourescence-conjugated secondary antibodies were used to visualize primary antibody staining, and Hoechst 33258 (1:5000 bisbenzamide; Sigma) was added for the final 10 min of incubation for nuclear identification. Analysis was undertaken using a terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling kit (Boehringer, Bagnolet, France)
Lipopolysaccharide lesion model.
Adult rats were killed and perfused with 4% paraformaldehye 24 h after basal ganglia microinjection of lipopolysaccharide (LPS; 5 μg) or saline (both in 0.5 μl volume) using a fine pulled siliconized glass micropipette. After a 1 h blocking step in 3% normal goat serum (NGS), 30 μm sections were incubated overnight with primary antibody in Tris-buffered saline (TBS) containing 0.2% Triton X-100 (Sigma) with 1% NGS. The following antibodies were used: OX42 (1:400; Serotec, Oxford, UK), Plexin-A1 culture supernatant (a gift from H. Fujisawa, National Institute of Genetics, Mishima, Japan) Npn-1 (1:200) (Kolodkin et al., 1997; Pasterkamp et al., 1999), Sema3A (1:250; a gift from A. Barzilai, Tel Aviv University, Tel Aviv, Israel) (Shirvan et al., 2002), and NeuN (1:200; Chemicon, Hampshire, UK). After washing in TBS, sections were incubated in biotinylated secondary antibodies (1:200) for 3 h at room temperature. After three washes in TBS, sections were incubated in a streptavidin-biotinylated horseradish peroxidase (HRP) complex (ABC kit; Dako) for 2 h before incubation in chromagen diaminobenzidine (0.5 mg/ml; BDH Chemicals, Poole, UK) or SG peroxidase substrate (Vector Laboratories, Peterborough, UK) for visualization.
Reverse transcription-PCR.
Total RNA was isolated from microglia conditioned for 24 h with or without IFNγ, postnatal day 1 (P1) rat cortices, or cortical neuron cultures using Trizol (Invitrogen, Paisley, UK), and poly(A+) RNA was then purified using the Oligotex mRNA mini kit (Qiagen, Hilden, Germany). cDNA was generated by Oligo(dT) primed reverse transcription using the SuperScript II RT-PCR kit (Invitrogen) according to the manufacturer’s protocol. Rat Plexin-A1, Plexin-A2, Plexin-A3, Npn-1, and Npn-2 were amplified using primers derived from nucleotide sequences unique for their respective isoforms but conserved between human and mouse plexins and neuropilins (Kameyama et al., 1996a,b; Maestrini et al., 1996). Multiple cDNAs derived from at least three independent amplifications were generated using thermostable Taq polymerase (Promega, Southampton, UK) and sequenced to identify potential errors that may have been introduced during amplification. Primer sequences were as follows, 5′ to 3′ (forward and reverse; all from Sigma Genosys, Haverhill, UK): Plexin-A1, GTGTGTGGATAGCCATCA and CCAGCCTCTCGAACACT; Plexin-A2, TGTGCCCTGCACAACATGT and TGCAGCTCTAGGCCAAACCA; Plexin-A3, GGTGACACCGAACTGGACTT and GACATATTCGGTGGGAATGG; Npn-1, GGCCTCCTGCGATTCGTTACTGCT and CTTAGCCTTGCGCTTGCTGTCATC; Npn-2, TGGATGTACGACCGTGCCAAGTGG and CTGATACTCCATGTCATAG CTGGG; Sema3A, GAAGTTCGACATCATCCTGAGGAC and CTCCATAGACAATTGGATTTTTAGGATC. Control primers used were as follows: actin, AGAAGAGCTATGAG CTGCCTGACG and TACTTGCGTCAGGAGGAGCAATG. Primers were annealed at 60°C, and 30 cycles of amplification were performed.
Northern blot analysis.
Poly(A+) RNA was isolated from resting and activated microglia or P1 whole rat brain. For Northern analysis, 3 μg of poly(A+) RNA was separated on a 1% formaldehyde-agarose gel and capillary blotted to Hybond N membrane (Amersham Biosciences, Uppsala, Sweden). Gene-specific 32P-labeled probes were synthesized (from PCR products) by random priming and purified through a Sephadex-50 column before hybridization to membranes in 5× saline-sodium phosphate-EDTA, 0.5% SDS, and 5× Denhardt’s solution (100× Denhardt’s solution: 2% BSA, 2% Ficoll, and 2% polyvinyl pyrrolidone) with 250 μg/ml sheared salmon sperm DNA at 65°C for 16 h. The membrane was washed in increasing dilutions from 0.5× to 2× SSC/0.1% SDS at 60°C and exposed to Biomax MS film (Eastman Kodak, Rochester, NY) at −80°C in the presence of an intensifying screen for up to 7 d.
Western blot analysis.
Resting and activated microglia were cultured at high density (2 × 106 cells per well) for 24 h, lysed in 150 mm Tris-HCl, 8 m urea, 2.5% w/v SDS, 20% v/v glycerol, 10% v/v 2-mercaptoethanol, 3% w/v DTT, and 0.1% bromophenol blue, pH 6.8, and stored (−20°C) until use. Equal volumes of cell lysates (equivalent to equal amounts of total protein as determined by protein quantification using the BCA protein assay kit; Pierce, Cheshire, UK) were separated on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membrane (Hybond C-super; Amersham Biosciences). Membranes were blocked in 4% (v/v) powdered milk TBS–Tween [50 mm Tris, pH 7.4, and 150 mm NaCl containing 0.05% (v/v) Tween 20] for 1 h at room temperature, followed by incubation with primary antibodies in blocking solution overnight at 4°C. The antibodies used were as follows: Plexin-A1, Plexin-A2, and Plexin-A3 (all from Santa Cruz Biotechnology, Santa Cruz, CA); Sema3A (a gift from A. Barzilai); and actin (Sigma). Immunoreactivity was visualized using HRP-conjugated secondary antibodies with enhanced chemoluminescence (Renaissance ECL reagent; NEN, Boston, MA).
Cell counting.
Cells were viewed using a Leitz microscope. Five consecutive random fields (using a grid) were counted for each coverslip. Results are expressed as mean ± SEM from at least three different coverslips and at least three separate experiments, unless stated otherwise. Statistical analysis (ANOVA with Newman–Keuls post hoc) was conducted using Prism 2.0 software.
Results
Activated microglia express Plexin-A1 and Npn-1
Enriched populations of resting and activated microglia from neonatal rat cortex mixed glial cultures were characterized. Microglia (identified by β4-isolectin-positive staining) represented 95.5 ± 1.3% (n = 3) of cells at 24 h after plating; 3.1 ± 0.5% were GFAP+ astrocytes, and the remainder were A2B5+ oligodendrocyte precursor cells. Cells were allowed to become quiescent in 2% B27-supplemented medium for 24 h before culture for an additional 24 h with or without IFNγ to produce resting or activated microglia. Resting microglia adopted a characteristic ramified morphology compared with the amoeboid appearance of activated microglia, which were OX6 positive with increased inducible nitric oxide synthase expression (Fig. 1).

Characterization of the activation state of microglia. Microglia were purified from mixed glial cultures, plated for 24 h to become quiescent, and cultured for an additional 24 h with (right) or without (left) IFNγ, representing activated and resting microglia, respectively. A–D, Representative photomicrographs of microglia under phase-contrast microscopy (A, B) or immunolabeled using an antibody against the microglial marker β-isolectin (C, D) show the distinct morphology of resting (A, C) and activated (B, D) microglia. E–H, Resting microglia are characterized by ramified morphology in contrast to an amoeboid appearance after activation. Immunomicrographs show OX6-positive activated microglia after treatment with IFNγ (G, >) with corresponding phase-contrast images (E, G). I, Western blot demonstrating upregulation of inducible nitric oxide synthase (iNOS) after IFNγ-mediated activation of microglia (representative of three independent experiments). Equal loading is confirmed by actin. R, Resting microglia; A, activated microglia.
Initial studies to examine the effect of activation on gene expression in microglia using differential display identified an unknown cDNA, the expression of which was significantly upregulated after activation (data not shown). Homology with corresponding human and mouse sequences identified this transcript as Plexin-A1 (Fig. 2 ). In view of the fact that, together with Npn-1, Plexin-A1 forms part of the high-affinity receptor complex for Sema3A, we next studied the expression profile of other members of the semaphorin receptor family in resting and activated microglia by Northern analysis (Fig. 2A). Upregulation of Npn-1 (and Plexin-A1) after microglial activation was confirmed by reverse transcription-PCR (RT-PCR) and by Western blot analysis (Fig. 2B,C). The other members of the neuropilin–plexin family were not differentially expressed after microglia activation.

Differential expression of plexin and neuropilin RNA dependent on the activation state of microglia. A, B, RNA isolated from microglia cultured with or without IFNγ for 24 h was analyzed for expression of plexin and neuropilin isoforms by Northern blot (A) and RT-PCR (B). Both techniques show upregulation of Plexin-A1 and Npn-1 after activation (representative gels of three independent experiments). C, Immunoblots of entire cell lysates from resting and activated microglia blotted for antibodies against Plexin-A1 and Plexin-A2 show upregulation of Plexin-A1 after activation. Equal loading is demonstrated by actin. Results are based on three independent experiments. R, Resting microglia; A, activated microglia.
Dose-dependent death effect of Sema3A on microglia
After the demonstration of increased Plexin-A1 and Npn-1 in activated microglia, we next examined the influence of Sema3A on both resting and activated microglia. We exposed resting and activated microglia to Sema3A-conditioned medium for 24 h, followed by fixation and immunocytochemistry. Contingent on the activation state, microglia showed a dose-dependent death effect, and, in particular, activated cells were more susceptible (Fig. 3 A). No effect on survival was observed using control conditioned medium. β-Lectin and Hoechst counts were comparable, consistent with the purity of the cultures; >98% were β-lectin positive cells at this time point. Interestingly, at a 1:10 and 1:20 dilution, there was a significant differential death effect, and activated microglia were more susceptible to death at each dilution (p < 0.01 and p < 0.05, respectively).
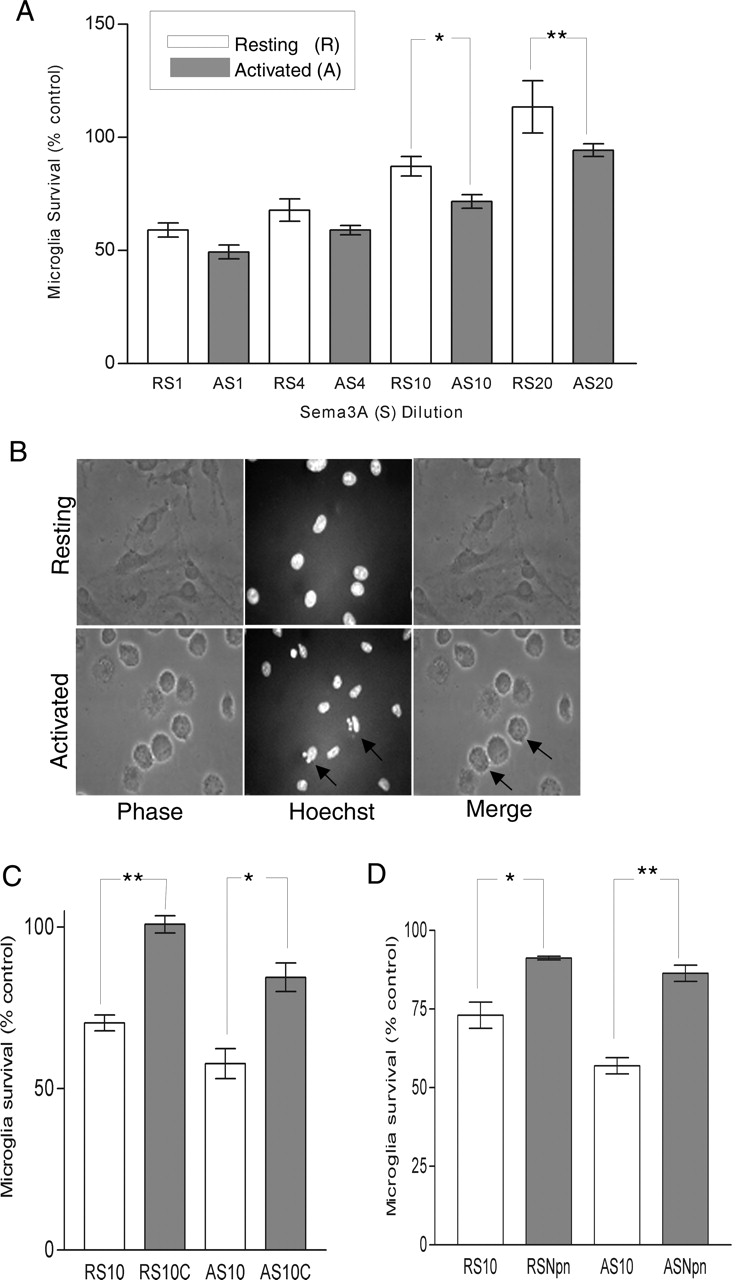
Semaphorin-mediated death of microglia. A, Graph showing the dose-dependent death effect of Sema3A-conditioned medium on microglia, contingent on the activation state. Death is significantly greater in activated (A) microglia than resting (R) cultures at a conditioned medium dilution of 1:10 (S10) and 1:20 (S20). Cell viability was determined at 24 h after treatment and is expressed as a percentage with respect to surviving non-Sema3A-treated control cells exposed to an identical amount of 1% FCS alone. In view of the fact that Sema3A-conditioned medium contains 1% FCS, appropriate controls were required for each dilution of Sema3A (n = 4; *p < 0.01; **p < 0.05). B, Semaphorin-mediated death is apoptotic. Phase and Hoechst morphological analysis of nuclei in resting (top row) and activated (bottom row) microglia cultures exposed to a 1:10 dilution of Sema3A-conditioned medium for 24 h. Fragmented and condensed nuclei typical of apoptosis are apparent after Hoechst staining (arrows). C, Inhibition of caspase activity reduces the overall death effect of Sema3A. Significant attenuation of the Sema3A death effect of conditioned medium on resting and activated microglia in the presence (filled bars) compared with the absence (open bars) of pan-caspase inhibitor Z-VAD-FMK (50 μm) is observed (n = 3; *p < 0.01; **p < 0.001). D, Function-blocking antibody to Npn-1 inhibits the death effect of Sema3A. Significant attenuation of the Sema3A death effect of conditioned medium on resting and activated microglia in the presence (filled bars) compared with the absence (open bars) of Npn-1 blocking antibody (100 μg/ml) is observed. Microglia were cultured under test conditions for 24 h (n = 3; *p < 0.01; **p < 0.001). Error bars indicate SEM. RS, Resting microglia; AS, activated microglia.
Morphological analysis of Hoechst-labeled nuclei and DNA ladder assay (data not shown) demonstrated characteristic fragmented condensed nuclei and DNA cleavage after Sema3A exposure. Moreover, this, together with the finding that the addition of the pan-caspase inhibitor Z-VAD-FMK significantly reduces the death effect of Sema3A after microglia (p < 0.001) (Fig. 3C), indicates that Sema3A causes apoptosis of microglia. The addition of anti-Npn-1 function-blocking antibody to microglia cultures significantly inhibited the death effect of Sema3A (p < 0.001) (Fig. 3D). Control experiments in non-Sema3A-treated cells showed no discernible effect of anti-Npn-1 on microglial survival (data not shown).
Stressed neurons produce Sema3A
We next examined neuronal expression of semaphorin (Fujita et al., 2001). E16 rat cortical neurons were cultured in 2% B27-supplemented medium for 5 d and stressed by replacement of medium with insulin-free Sato for up to 3 d. Neuronal cultures were phenotypically characterized at 5 d in vitro. βIII-Tubulin+ neurons represented 94.6 ± 1.6% (n = 3) of the cells; the remainder were predominantly astrocytes, with 0.4% O4+ oligodendrocyte precursors and 0.2% GalC+ oligodendrocytes. Immune characterization at 3 d after supplement replacement revealed 75% neuronal death. RT-PCR demonstrated Sema3A expression by remaining neurons at 24, 48, and 72 h after the induction of neuronal stress (Fig. 4 A).

Stressed neurons express Sema3A. A, Expression of Sema3A mRNA in E16 rat cortical neurons plated for 5 d in vitro (Div) in 2% B27-supplemented medium, followed by 0, 24, 48, or 72 h in insulin-free Sato-supplemented medium. Total RNA was isolated from neurons at each time point and used to perform RT-PCR against Sema3A. Negative control reactions were performed on total RNA isolated from neurons 72 h after supplement deprivation, omitting reverse transcriptase (-rt). Total RNA derived from whole P1 rat cortex was used as a positive control template (P1). Results are based on representative gels from three independent experiments. B, SNCM induces death in microglia dependent on the activation state. Quantification of the death effect of SNCM dilutions 1:2 (CM2) and 1:10 (CM10) on resting (open bars) and activated (filled bars) microglia is shown. Activated microglia are more vulnerable to the death effect of SNCM at a 1:2 dilution. Cell viability was determined at 24 h after treatment and is expressed as a percentage of surviving nontreated control cells (n = 3; *p < 0.05). C, Function-blocking anti-Npn-1 antibody (100 μg/ml) inhibits the death effect of SNCM when used at a 1:2 dilution, on microglia cultured under test conditions for 24 h (n = 3; *p < 0.05; **p < 0.01). Error bars indicate SEM.
SNCM mediates microglia death
We next asked whether SNCM could influence microglia survival. SNCM was derived from E16 rat cortical neurons, cultured for 5 d in 2% B27-supplemented medium, followed by an additional 3 d in Sato-only-supplemented medium. Analysis of cultures 24 h after the addition of SNCM to both resting and activated microglia revealed a significant death effect by SNCM (Fig. 4B). Activated microglia were significantly more susceptible (p < 0.05).
To determine whether death induced by SNCM is attributable to Sema3A present in conditioned medium, we used function-blocking anti-Npn-1 antibody. Anti-Npn-1 (α-Npn-1) resulted in a significant reduction in SNCM-mediated microglial death (p < 0.01), suggesting that Sema3A is in part responsible for SNCM-mediated microglial cell death (Fig. 4C). A limited effect was also observed in the resting microglia. This is likely to be attributable to the presence of some activated microglia as a result of culturing, even within the resting cultures.
In vivo expression of Sema3A, Plexin-A1, and Npn-1 in adult brain
To determine whether the identification in vitro of differential Plexin-A1 and Npn-1 expression contingent on the microglia activation state also occurs in vivo, we studied adult rat brains that were killed at 24 h after the induction of focal injury using LPS. Immunohistological characterization confirmed that LPS injection resulted in an acute inflammatory response characterized by microglial activation (Fig. 5 A–E). Contralateral control lesions of vehicle alone did not produce an acute inflammatory response (Fig. 5A,B,D). Immunohistochemistry for OX42 demonstrated ramified resting microglia in control lesion cortex and amoeboid-appearing activated microglia in LPS lesions. Significantly, Plexin-A1 and Npn-1 immunoreactivity was present only in LPS-mediated inflammatory lesions, colocalized with activated microglia (Fig. 5F–I). Sema3A expression was upregulated in the LPS-treated region compared with control lesions (Fig. 6 A–D). Furthermore, Sema3A expression was evident in NeuN-positive neurons (Fig. 6E–G). Western blot analysis of brain sections after LPS or vehicle lesions confirmed significantly enhanced Sema3A expression in LPS-treated tissue (Fig. 6H,I). Consistent with our in vitro findings, in vivo nuclear morphological analysis provides evidence that Sema3A-mediated microglial death is apoptotic (Fig. 7).

In vivo expression of Plexin-A1 and Npn-1 by microglia. Immunohistochemical analysis of rat brain 24 h after vehicle (left) compared with LPS (right) injections is shown. A–C, Representative low-magnification overview of staining with OX42 showing widespread activation in LPS lesion hemisphere compared with the small area of activation, confined to the injection site, in the control saline hemisphere (A, arrow). D, E, Higher magnification of B and C detailing increased numbers of OX42+-reactive microglia (E, arrow), characterized by hypertrophied cell bodies with distinct morphology and heavy accumulation of staining. Resting microglia with a typical ramified appearance (D, arrow) are present in the control lesion hemisphere. F–I, Double-labeled immunohistochemistry using OX42 and Plexin-A1 or Npn-1 antibodies. Neither Plexin-A1 nor Npn-1 is detected on microglia in the control lesion (F, H). After LPS injection, Npn-1- and Plexin-1-positive staining is present in activated microglia (G, I). Microglia are shown in black (insets, arrows). Brown (diaminobenzidine) identifies Plexin-A1 (G, inset, arrowhead) or Npn-1 (I, inset, arrowhead). Negative controls, omitting the primary antibody, showed no staining (data not shown).

In vivo expression of Sema3A by neurons. A, B, Immunohistochemical analysis of rat brain 24 h after vehicle (left) compared with LPS (right) injections. A representative overview of Sema3A staining in cell bodies showing upregulation in both cells and surrounding tissue after LPS lesion (A) compared with vehicle injection (B) is shown. C, D, Higher magnification of A and B showing Sema3A localized to cells with characteristic neuronal morphology. Neurons in the LPS lesion area (D) show marked upregulation of Sema3A compared with vehicle (C). E, F, Representative sections immune-labeled using NeuN show distinct nuclear staining in neurons in both vehicle (E) and LPS-treated (F) sections. Negative controls, omitting the primary antibodies, showed no staining (data not shown). G, Double staining using antibodies against NeuN and Sema3A shows colocalization of NeuN-positive nuclei (brown) and Sema3A-positive cytoplasm (blue) after LPS treatment. H, Immunoblots of entire hemisphere lysates 24 h after vehicle (saline) compared with LPS injections show upregulation of Sema3A after LPS lesion. (Equal loading is confirmed using an antibody against actin.) I, Relative abundance of Sema3A expression quantified by computerized densitometry of immunoblots (*p < 0.005). Results are based on three independent experiments. Error bars indicate SEM.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In vivo apoptotic morphology of microglia: spatial localization with Sem3A neurons. Immunohistochemical analysis of rat brain 24 h after LPS injection. A, B, A representative image showing Sema3A staining in neuronal cell bodies (A, brown) and microglia (A, OX42, black) with corresponding Hoescht staining (B). Microglia adjacent to Sema3A-positive neurons have an apoptotic morphology (arrow) in contrast to healthy-appearing microglia in the absence of Sema3A neurons (arrowhead).
Discussion
Here, we show for the first time that Sema3A can mediate neuronal–microglia interactions. We demonstrate that, contingent on the activation state, microglia differentially express the semaphorin receptors Plexin-Al and Npn-1 in vitro and in vivo. We confirm neuronal expression of Sema3A in vitro and in vivo and provide evidence that stressed neurons are able to mediate microglial death by production of Sema3A.
Differential expression of Plexin-A1 and Npn-1 by microglia
The profile of neuropilin and Sema3A has been analyzed in a cerebral model of ischemia, demonstrating microglial expression of Npn-1 and Npn-2 (Fujita et al., 2001). Our findings confirm and extend these observations, highlighting the differential expression profile of Plexin-A1 and Npn-1 between resting and activated microglia. Importantly, the other members of the plexin–neuropilin family were not found to be differentially expressed after microglia activation. We provide evidence in vitro and in vivo for upregulation of both Plexin-A1 and Npn-1 after activation of microglia. Recent reports also implicate semaphorin-mediated neuronal and neuronal progenitor apoptotic cell death (Gagliardini and Fankhauser, 1999; Shirvan et al., 1999, 2002; Bagnard et al., 2001). We therefore examined whether semaphorin has a similar effect on microglia. Exposure to Sema3A-conditioned medium resulted in dose-dependent death of microglia, with activated microglia proving significantly more vulnerable. Death was apoptotic as determined by nuclear morphology, caspase inhibition, and DNA ladder assay analysis. The addition of anti-Npn-1 antibody significantly attenuated the death effect, consistent with the conclusion that Sema3A can mediate apoptosis of microglia.
Microglia–neuron interaction: a role for semaphorin
An emerging concept is the idea of cross talk between microglia and neurons, an interaction that can be regarded as harmful or beneficial depending on context. In vitro studies demonstrate microglia neurotoxicity through various mechanisms including nitric oxide and NMDA-mediated toxicity (Piani et al., 1991; Chao et al., 1992; Kingham et al., 1999). Activated microglia have been implicated in demyelinating and neurodegenerative diseases (Itagaki et al., 1989; Lucchinetti et al., 2000; McGeer and McGeer, 2003). There is some experimental support for the proposal that blocking microglia activation may be neuroprotective, for example through the production of trophic factors including NGF and NT3 (Mallat et al., 1989; Thanos et al., 1993; Elkabes et al., 1996; Rabchevsky and Streit, 1997; Lim et al., 2000; Wu et al., 2002; Zhang et al., 2003). Furthermore, the observation that TNF-α null mice show increased neuronal death after ischemia in association with reduced microglia recruitment is consistent with microglia-mediated neuroprotection (Bruce et al., 1996). Studies highlighting the reciprocal relationship between microglia and neurons emphasize the dynamic nature of microglia–neuron interaction (Zietlow et al., 1999; Polazzi and Contestabile, 2003). The precise identity of the protein signals underlying neuron-mediated apoptosis of activated microglia remains unclear (Polazzi and Contestabile, 2003).
Increasing recognition of the importance of semaphorin-mediated signaling in roles other than developmental axon guidance, such as the response to injury, offers a possible biological explanation for the sequence of microglial neuropilin and plexin expression, followed by an apoptotic response to Sema3A. Adult semaphorin expression contributes to the nonpermissive environment of glial scars (Pasterkamp et al., 1999; Niclou et al., 2003). In addition, reciprocal expression in the ischemic adult brain of neuronal Sema3A and microglial neuropilin suggests semaphorin-mediated microglia–neuron interaction (Fujita et al., 2001). Our observation that the effect of medium conditioned by stressed neurons on microglia is significantly blocked by anti-neuropilin antibody suggests that neuron-derived Sema3A mediates microglial apoptosis. Additional evidence is provided by the upregulation of neuronal Sema3A, and Plexin-A1 and Npn-1, by activated microglia after LPS-mediated activation in the adult rat brain. Together, these findings indicate that semaphorin is capable of mediating cell death (Shirvan et al., 1999; Bagnard et al., 2001).
Tanaka and Koike (2002) have identified upregulation of a novel microglia gene (mrf-1) after neuronal death that is also implicated in cross talk between microglia and damaged neurons. Our study identifies a novel mechanism of microglia–neuron interaction that may inform the emerging concept of neuronal control of microglia. Because activated microglia are implicated in many disease processes, a biological context for these in vitro and in vivo findings can be readily envisaged (Chao et al., 1992; Giulian, 1999; Kingham et al., 1999; Liberatore et al., 1999; Streit et al., 1999; Neumann, 2001).
In summary, we demonstrate differential expression by activated microglia of the semaphorin receptors neuropilin and plexin. Stressed neurons mediate death of microglia by a semaphorin-dependent mechanism indicating reciprocal regulation of microglia. This may represent a mechanism for containing the inflammatory response to tissue injury whereby the survival of stressed but viable neurons is ensured without compromising the removal by microglia of neurons that have already sustained a lethal injury.
Footnotes
- Received May 31, 2004.
- Revision received November 11, 2005.
- Accepted November 14, 2005.
This work was supported by a PhD studentship (H.H.M.), a Clinician Scientist Fellowship (S.C.), and a Co-operative Group Grant (A.C. and M.G.S.) from the Medical Research Council, United Kingdom. We thank A. Kolodkin (anti-Npn-1), H. Fujisawa (anti-Plexin-A1), and A. Barzilai (anti-Sema3A) for the generous gifts of antibodies.
*H.H.M. and S.C. contributed equally to this work.
- Correspondence should be addressed to Dr. Alastair Compston, Department of Clinical Neurosciences and Centre for Brain Repair, University of Cambridge, Forvie Site, Robinson Way, Cambridge CB2 2PY, UK. Email: alastair.compston{at}medschl.cam.ac.uk